
Abstract
The therapeutic effects of conventional treatments for advanced colorectal cancer with colorectal peritoneal carcinomatosis (CRPC) and malignant ascites are not very encouraging. Vascular endothelial growth factor-A/vascular permeability factors (VEGF-A/VPF) play key roles in the formation of malignant ascites. In previous work, we demonstrated that pigment epithelium-derived factor (PEDF) antagonized VEGF-A and could repress tumor growth and suppress metastasis in several cancer types. Thus, PEDF may be a therapeutic candidate for treating malignant ascites. Mesenchymal stem cells (MSCs) are promising tools for delivering therapeutic agents in cancer treatment. In the study, MSCs derived from bone marrow were efficiently engineered to secrete human PEDF by adenoviral transduction. Then, intraperitoneal Ad-PEDF-transduced MSCs were analyzed with respect to CRPC and malignant ascites in a CT26 CRPC model. MSCs engineered to secrete PEDF through adenoviral transduction significantly inhibited tumor metastasis and malignant ascites formation in CT26 CRPC mice. Antitumor mechanisms of MSCs-PEDF (MSCs transduced with Ad-PEDF: MOI 500) were associated with inhibiting tumor angiogenesis, inducing apoptosis, and restoring the VEGF-A/sFLT-1 ratio in ascites. Moreover, MSC-mediated Ad-PEDF delivery reduced production of adenovirus-neutralizing antibodies, prolonged PEDF expression, and induced MSCs-PEDF migration toward tumor cells. As a conclusion, MSCs engineered to secrete PEDF by adenoviral transduction may be a therapeutic approach for suppressing tumor metastasis and inhibiting malignant ascites production in CRPC.
Introduction
C
Pigment epithelium-derived factor (PEDF) is a 50 kDa secreted glycoprotein that belongs to the serpin superfamily of serine protease inhibitors, which was first identified in a conditioned medium of cultured primary human fetal retinal pigment epithelial cells. 6 PEDF is widely expressed in human tissues and a multifunctional protein that has antiangiogenic, antitumorigenic, and neurotrophic functions. 7,8 In recent years, its antiangiogenic function has gained attention. PEDF is a more potent inhibitor of angiogenesis than other endogenous angiogenic inhibitors, including endostatin, angiostatin, and thrombospondin. 9 Tumor angiogenesis, or the proliferation of blood vessel network, is a vital process facilitating tumor growth, survival, and metastases. 10 Previously, we have reported that PEDF could repress tumor growth and suppress metastasis in several types of cancers. 11 –13 Antitumor effects of PEDF contribute to inhibition of tumor angiogenesis. VEGF-A plays a key role in both angiogenesis and formation malignant ascites. 3,14 However, no reports of PEDF (VEGF-A antagonist) for malignant ascites treatment are available.
Mesenchymal stem cells (MSCs) are multipotent nonhematopoietic cells that can differentiate into various cell types. 15 Stromal stem cells are of interest because of their ability to home to the tumor site. 16 Furthermore, MSCs are protected from cytotoxic lymphocyte-mediated lysis in vivo because of their hypoimmunogenic properties and production of immunosuppressive molecules. 17 The tumor microenvironment is known to provide a preferential niche for MSC tumor-homing and survival. 18 Thus, MSCs may be promising tools for delivering therapeutic agents in cancer treatment. Although significant advances in cancer gene therapy have been made, clinical trials confirm limited antitumor efficacy of these agents because of lack of tumor tropism of vectors and stimulation of an immune response. 15 Many studies suggest that MSCs are viable platforms for cell-based gene therapy. 19 –21
In the present study, MSCs derived from bone marrow were efficiently engineered to secrete human PEDF by adenoviral transduction. Then, Ad-PEDF-transduced MSCs in CRPC and malignant ascites in a CT26 CRPC model were studied.
Materials and Methods
Cell lines and animals
A CT26 murine colon carcinoma cell line and a 293A human embryonic kidney cell line were purchased from the American Type Culture Collection (ATCC) and cultured in RPMI1640 and DMEM (Gibco) containing 10% fetal bovine serum (FBS; Gibco) and 100 μg/ml amikacin, respectively, maintained in a 37°C incubator with a humidified 5% CO2 atmosphere. Primary human umbilical vein endothelial cells (HUVECs) were isolated from human umbilical cord veins according to a standard procedure, 13 and grown in EBM-2 medium with SingleQuots (Lonza) containing VEGF and other growth factors. HUVECs at passages 2–5 were used for experiments.
Female BALB/c mice aged 4–8 weeks were purchased from Vital River Laboratories and maintained in pathogen-free conditions. All procedures were reviewed and approved by the institute's Animal Care and Use Committee.
Isolation, culture, and identification of bone marrow MSCs
Female BALB/c mice (4–6 weeks old) were sacrificed and MSCs were isolated as described previously. 22 Briefly, MSCs were isolated from femur bone marrow mononuclear cells under pathogen-free conditions and were separated by centrifugation using Ficoll-Hypaque separation medium. Then, monocular cells were suspended in low-glucose DMEM containing 10% FBS and 10 U/ml bFGF and maintained in a 37°C incubator with a humidified 5% CO2 atmosphere. Two days later, nonadhesive cells were discarded after washing with PBS and adherent MSCs were preserved for further expansion. Medium changes were performed twice per week for a 14-day period of culture before the first passage. Cells of 4–5 passages were used in the experiments. MSCs had been proved to express a panel of specific antigens. 23 Therefore, the MSCs were further analyzed by flow cytometry using CD11b, CD29, CD31, CD45, CD105, and Sca-1 antibodies (BD Biosciences) for phenotype characteristics of MSCs. Isotypic control analyses were conducted in parallel. Flow cytometry was performed using a FACScalibur flow cytometer (BD Biosciences), and data were analyzed with the CellQuest Pro software (BD Biosciences).
Adenoviral vector, adenoviral transduction of MSCs, and PEDF expression in vitro
A PEDF-expressing adenovirus was created using an AdEasy system and was amplified in HEK293 cells and purified on a CsCl gradient as described previously. 13 To monitor adenoviral transduction efficiency in MSCs, cells were seeded in 6-well plates and grown to ∼95% confluency. Medium was replaced with 0.5 ml fresh DMEM supplemented with 2% FBS with adenovirus expressing GFP diluted to the indicated multiplicity of infection (MOI). After 2 hr, another 2 ml fresh DMEM containing 10% FBS was added. At 48 hr after transduction, the percentage of GFP-positive cells was analyzed using a Nikon TE2000-U inverted fluorescent microscope. To measure PEDF expression in vitro, MSCs were transduced with adenovirus expressing PEDF as mentioned above. PEDF secreted in culture supernatants was measured with a sandwich ELISA kit for the human PEDF protein (R&D Systems) according to manufacturer's protocol.
Tube formation assay
A tube formation assay was performed as described previously. 24 Briefly, prechilled Matrigel (BD Biosciences) was added to each well of a 96-well plate and polymerized at 37°C for 30 min. Then, 2 × 104 HUVECs were seeded onto a Matrigel-coated well. Cells were treated with DMEM or conditioned medium collected from MSCs, MSCs-GFP (MSCs transduced with Ad-GFP: MOI 500), MSCs-PEDF (MSCs transduced with Ad-PEDF: MOI 500). After 6 hr of incubation, HUVECs were observed and photographed under an inverted microscope. Five fields were viewed, and tubes were counted and data were averaged.
In vitro MSCs migration assay
To measure MSCs' ability to home to tumors, a migration assay was performed as described previously. Briefly, 2 × 104 CT26 or 293A cells were plated on a 24-well plate and allowed to attach overnight. Cells were then washed three times with PBS and incubated with 600 μl fresh DMEM without FBS. Twenty-four hours later, the Transwell chambers with 8 μm pore filters (Millipore) were placed on the cell culture plates. Next, 1 × 104 MSCs-GFP or MSCs-PEDF were suspended in 200 μl fresh DMEM without FBS and seeded on the top chamber of the Transwell filter. After incubation for 6 hr, nonmigrated cells were removed from the upper surface of the membrane and cells that migrated to the lower surface were fixed with methanol for 15 min and stained with crystal violet (Sigma-Aldrich). Finally, the number of migrated cells from five random fields was counted under a light microscope. 25 All experiments were performed in triplicate.
Animal study
For the CRPC and malignant ascites murine model, 2 × 105 CT26 cells were injected intraperitoneally (ip). Mice were divided into five groups randomly (N = 20 per group). Then, 3 days later, mice were given 0.9% NaCl (normal saline, NS, ip), 108 pfu Ad-PEDF, 2.0 × 105 MSCs, 2.0 × 105 MSCs-GFP, or 2.0 × 105 MSCs-PEDF every 4 days 4 times. Tumor growth and survival rate were monitored every 3 days. To evaluate the therapeutic effect, mice were dissected and the tumor node numbers and weights in each group (n = 10) were measured on day 18 after tumor cell injection. Meanwhile, the ascetic fluid was collected from the peritoneal cavity. The volume was measured using a graduated conical centrifuge tube. Also, mouse survival time was monitored for each group (n = 10).
TUNEL assay
Tumor nodes were fixed in 4% paraformaldehyde. Tumor tissues were embedded in paraffin and cut into 3–5 μm sections. Apoptosis was measured within tumor tissues, and terminal dUTP nick-end labeling (TUNEL) staining was performed with ApoBrdU Red DNA Fragmentation Kit (BioVision) following manufacturer's protocol. Apoptosis was calculated as a ratio of the number of apoptotic cells to total tumor cells in the field (5 high power fields/slide).
Microvessel density assay
Neovascularization of tumor tissues was measured via a microvessel density (MVD) assay as described previously. 26 Briefly, frozen sections of tumor tissues were fixed in acetone and stained using rat antimouse CD31 polyclonal antibody (BD Pharmingen), followed by incubation with a rhodamine-conjugated second antibody (Abcam). MVD was measured by counting the number of microvessels per high power field under a fluorescent microscope.
ELISA
Ascites and sera from mice were collected and centrifuged on day 18 after tumor cell injection, and the supernatant was subjected to ELISA to measure antiadenovirus antibody as described previously. 27 Briefly, Ad-Null was diluted to 1 × 109 IU/ml by 50 mM carbonate coating buffer (pH 9.6). Then, 100 μl diluted Ad-Null was added to each well of 96-well of the ELISA plate and incubated overnight at 4°C. The plates were blocked with 150 μl 1% BSA at 37°C for 1 hr. Then, 100 μl diluted ascites was added to each well and incubated at 37°C for 90 min. After washing, 100 μl horseradish peroxidase-labeled goat antimouse antibody (1:5000) was added and incubated at 37°C for 1 hr. After washing, TMB was added and incubated for 20 min at 37°C, and the absorbance was read immediately at 450 nm in a microplate reader (Bio-Rad). To measure human PEDF, murine VEGF-A, and murine soluble fms-like tyrosine kinase-1 (sFLT-1) in ascites, ELISA was performed according to the manufacturer's protocol with a commercially available Quantikine Immunoassay kits (R&D Systems).
Statistical analysis
SPSS program (version 15.0; SPSS Inc.) was used for statistical analysis. Comparison of tumor nodes was performed with one-way analysis of variance (ANOVA). Survival curves were generated based on the Kaplan–Meier method. Significance was defined as p < 0.05.
Results
Characteristics of MSCs, adenoviral transduction efficiency, and PEDF expression in MSCs in vitro
Analysis of phenotype characteristics showed that isolated bone marrow-derived cells shared classical immunophenotype of MSCs, including positivity for CD29, CD105, and Sca-1, but negativity for CD11b, CD31, and CD45 (Fig. 1A). Thus, as the data showed, we successfully obtained abundant MSCs from mouse bone marrow. MSCs have few coxsackie-adenoviral receptors (CARs), which diminishes adenoviral transfection efficiency. 15 Here we investigated adenoviral transduction efficiency of MSCs. At 48 hr after adenovirus transduction, the percentage of GFP-positive MSCs was quantified under a fluorescent microscope. Figure 1B shows that more than 90% of MSCs expressed GFP at 48 hr after transduction with Ad-GFP (MOI 500 or 1000). Although almost all MSCs expressed GFP after transduction with Ad-GFP (MOI 1000), some MSCs died. PEDF secreted from MSCs transduced with Ad-PEDF at MOI 500 and 1000 was 120.74 ± 3.9 ng/ml and 119.19 ± 4.0 ng/ml, respectively (Fig. 1C). So, 500 was selected as an optimal MOI.
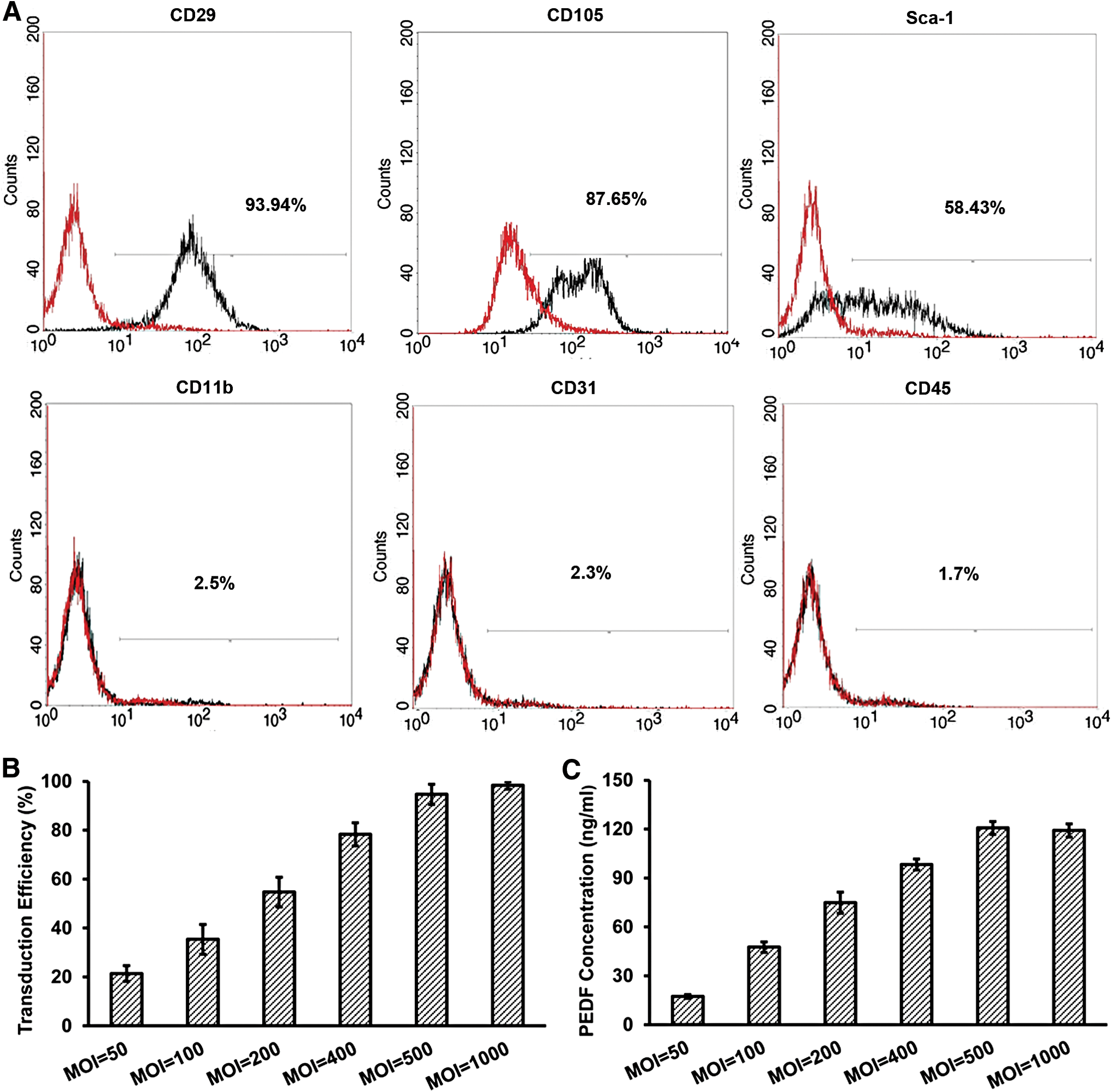
Characteristics of MSCs, adenoviral transduction efficiency, and PEDF expression in MSCs in vitro.
PEDF produced by MSCs-PEDF inhibits the tube formation in vitro
Antiangiogenic activity of PEDF produced by MSCs-PEDF was assessed by tube formation of human HUVECs in vitro. Tube formation of vascular endothelial cells is a key step of angiogenesis. When vascular endothelial cells are cultured in Matrigel, they rapidly align and form tube-like structures. Figure 2 shows that conditioned medium derived from MSCs-PEDF significantly inhibited tube formation. However, treatment with DMEM or conditioned medium from MSCs or MSCs-GFP did not affect tube formation.
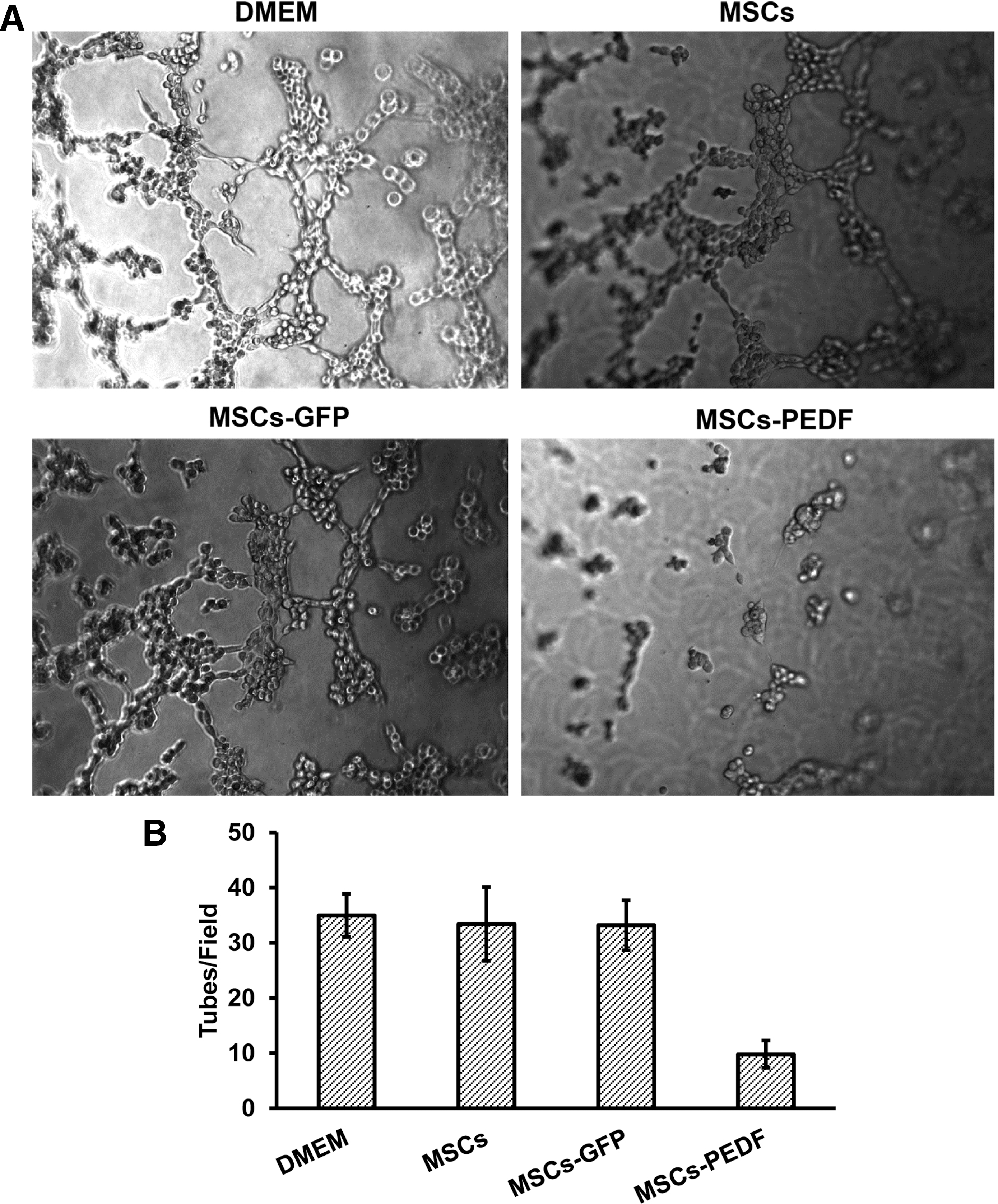
PEDF produced by MSCs-PEDF (MSCs transduced with Ad-PEDF: MOI 500) inhibits tube formation in vitro. Conditioned medium derived from MSCs-PEDF significantly inhibited tube formation compared with controls (p < 0.05).
Migratory capacity of MSCs toward tumor cells in vitro
As shown in Fig. 3, MSCs-GFP and MSCs-PEDF that migrated toward CT26 cells were significantly higher than MSCs-GFP and MSCs-PEDF that migrated toward 293A cells (p < 0.05). Migration assays revealed a directed migratory ability of MSCs to CT26 cells. However, there was no difference between the MSCs-GFP and MSCs-PEDF group (p > 0.05). The result suggested that there was no influence on homing ability of MSCs after being modified by Ad-PEDF. The migratory ability of MSCs toward tumor cells strongly supports the targeted effect of MSCs as a delivery system.
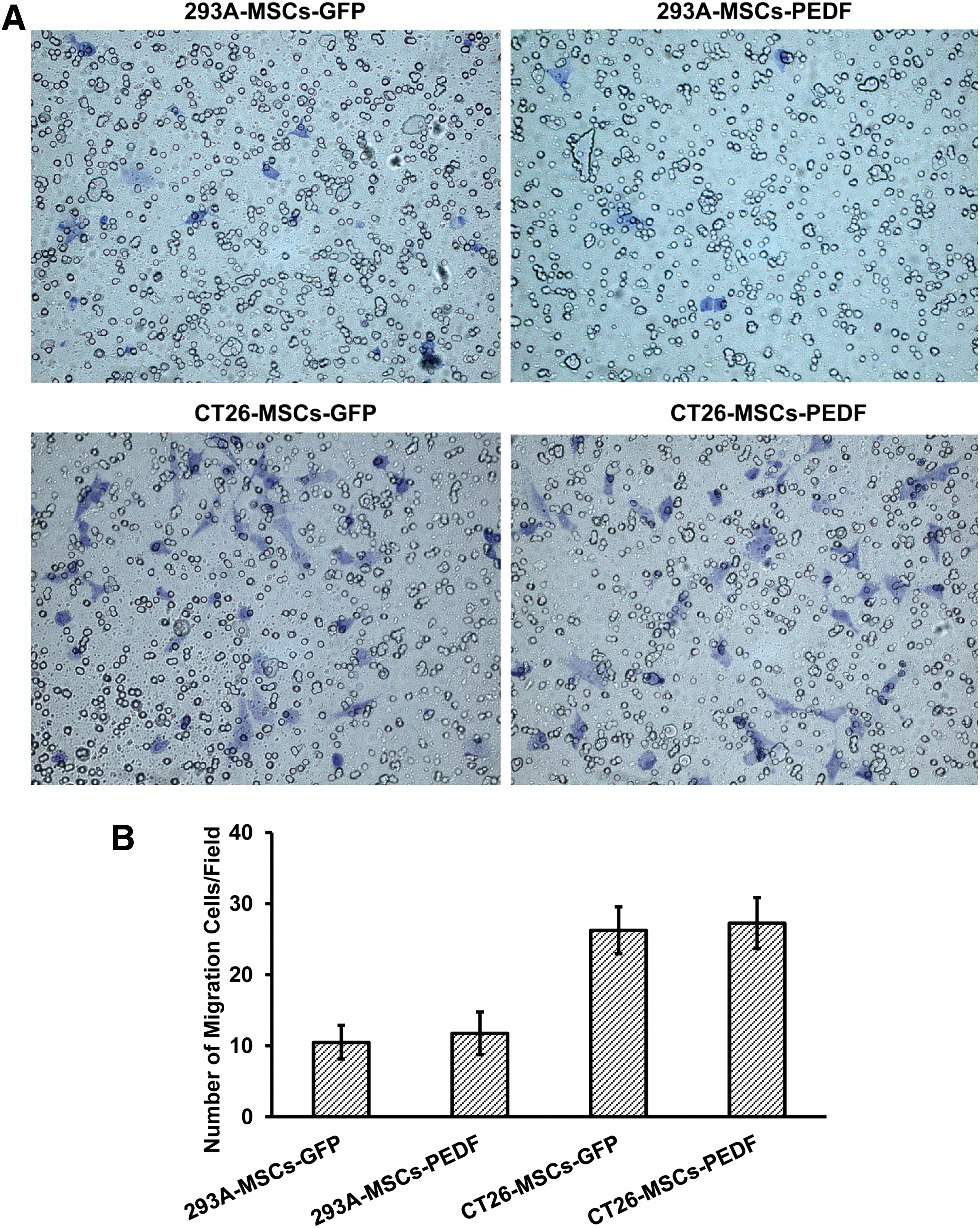
Migratory capacity of MSCs toward tumor cells in vitro. MSCs-GFP and MSCs-PEDF that migrated toward the conditioned medium derived from CT26 cells were significantly higher than those of MSCs-GFP and MSCs-PEDF that migrated toward conditioned medium derived from 293A cells (p < 0.05), but there was no difference between the MSCs-GFP and MSCs-PEDF group (p > 0.05).
Inhibition of tumor metastasis and formation of malignant ascites with MSCs-PEDF in mice
We evaluated the therapeutic effect of MSCs-PEDF in CRPC mice. On day 18 after cell inoculation, tumor node number and weights in the MSCs-PEDF-treated group were dramatically decreased compared with those of control groups (p < 0.01) (Fig. 4A and 4B). Moreover, ascites volume in the MSCs-PEDF-treated group was significantly smaller than that in other control groups, including the Ad-PEDF group (p < 0.05) (Fig. 4C).
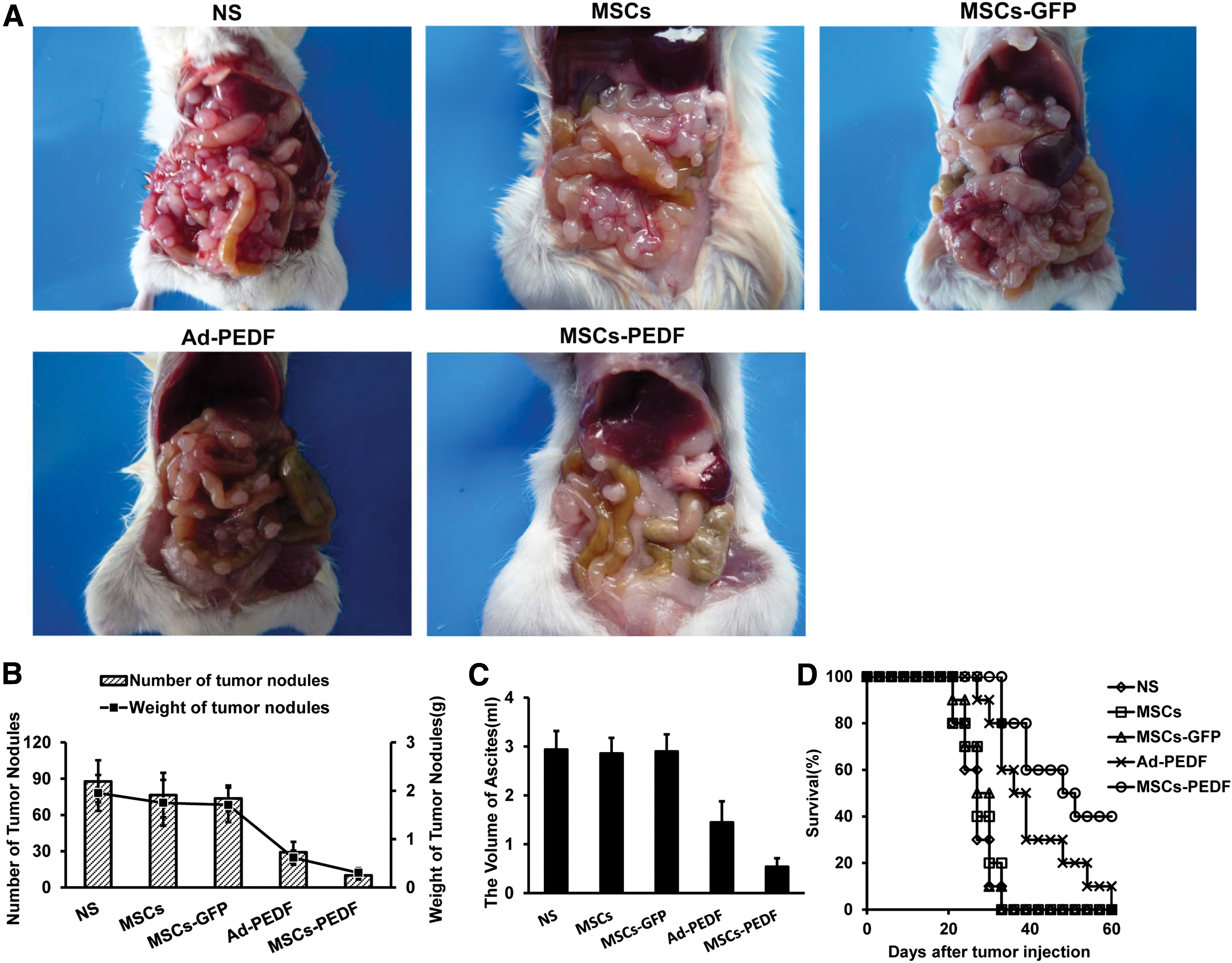
MSCs-PEDF inhibits tumor metastasis and formation of malignant ascites in CRPC mice.
To assess the potential of MSCs-PEDF as a therapeutic agent for CRPC, mouse survival was observed over 60 days (Fig. 4D). All animals from NS, MSCs, and MSCs-GFP groups died by day 33. Animals from the Ad-PEDF group died by day 60. However, 40% of mice in the MSCs-PEDF-treated group survived to day 60. Survival time of the MSCs-PEDF group was significantly longer than other groups, including the Ad-PEDF group (p < 0.05).
MSCs-PEDF inhibits tumor angiogenesis and induces apoptosis in mice
To explore the mechanisms of tumor metastatic suppression mediated by MSCs-PEDF, angiogenic blood vessels within tumor tissue were measured using CD31 immunohistochemistry. Figure 5A shows that treatment with MSCs-PEDF apparently reduced the number of vessels compared with other control groups (p < 0.05). To investigate the role of MSCs-PEDF treatment in tumors in vivo, tumor resections were subjected to immunofluorescent TUNEL staining assays to measure apoptosis. MSCs-PEDF-treated tumors were more apoptotic (with red nuclei) than tumors from other control groups. Apoptosis quantification within tumor sections revealed a significant increase in apoptosis in tumors treated with MSCs-PEDF (p < 0.05) (Fig. 5B).
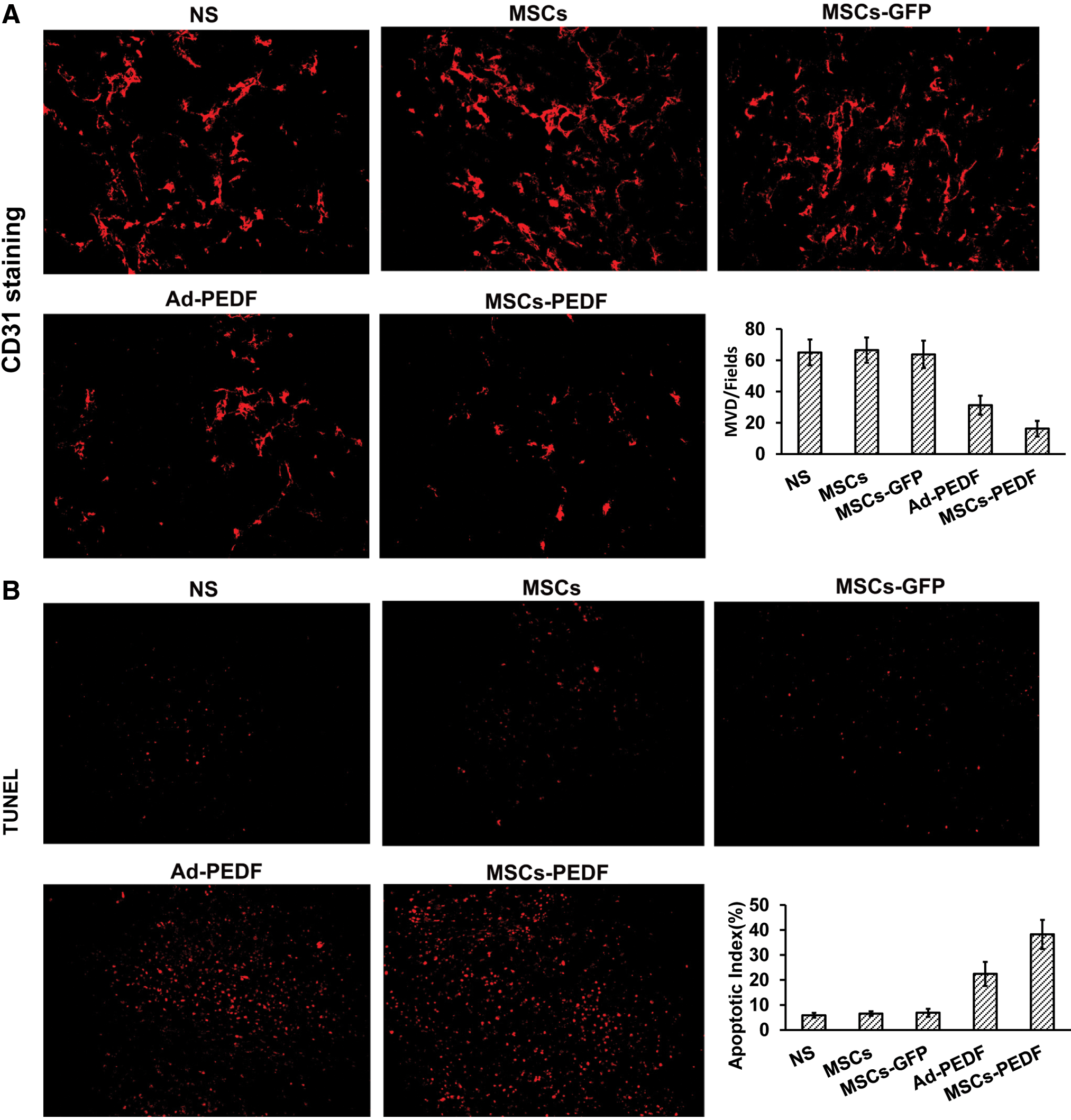
MSCs-PEDF inhibits tumor angiogenesis and induces apoptosis in mice.
MSC-mediated Ad-PEDF delivery reduces the production of adenoviral neutralizing antibodies and prolongs PEDF expression
To measure adenoviral neutralizing antibodies, we collected ascites and sera from Ad-PEDF- and MSCs-PEDF-treated animals, respectively, and measured the neutralizing antibody concentrations. Figure 6A shows that on day 18 after CT26 cell injection, adenoviral neutralizing antibodies in the Ad-PEDF treatment group in ascites and sera were significantly higher than that of the MSCs-PEDF treatment group (p < 0.05). Meanwhile, PEDF in ascites from the MSCs-PEDF treatment group was significantly higher than that of the Ad-PEDF treatment group (p < 0.05). However, there was no difference in PEDF in sera between Ad-PEDF and MSCs-PEDF groups (p > 0.05) (Fig. 6B). Thus, MSC-mediated Ad-PEDF delivery reduced adenoviral neutralizing antibody production and prolonged PEDF expression.
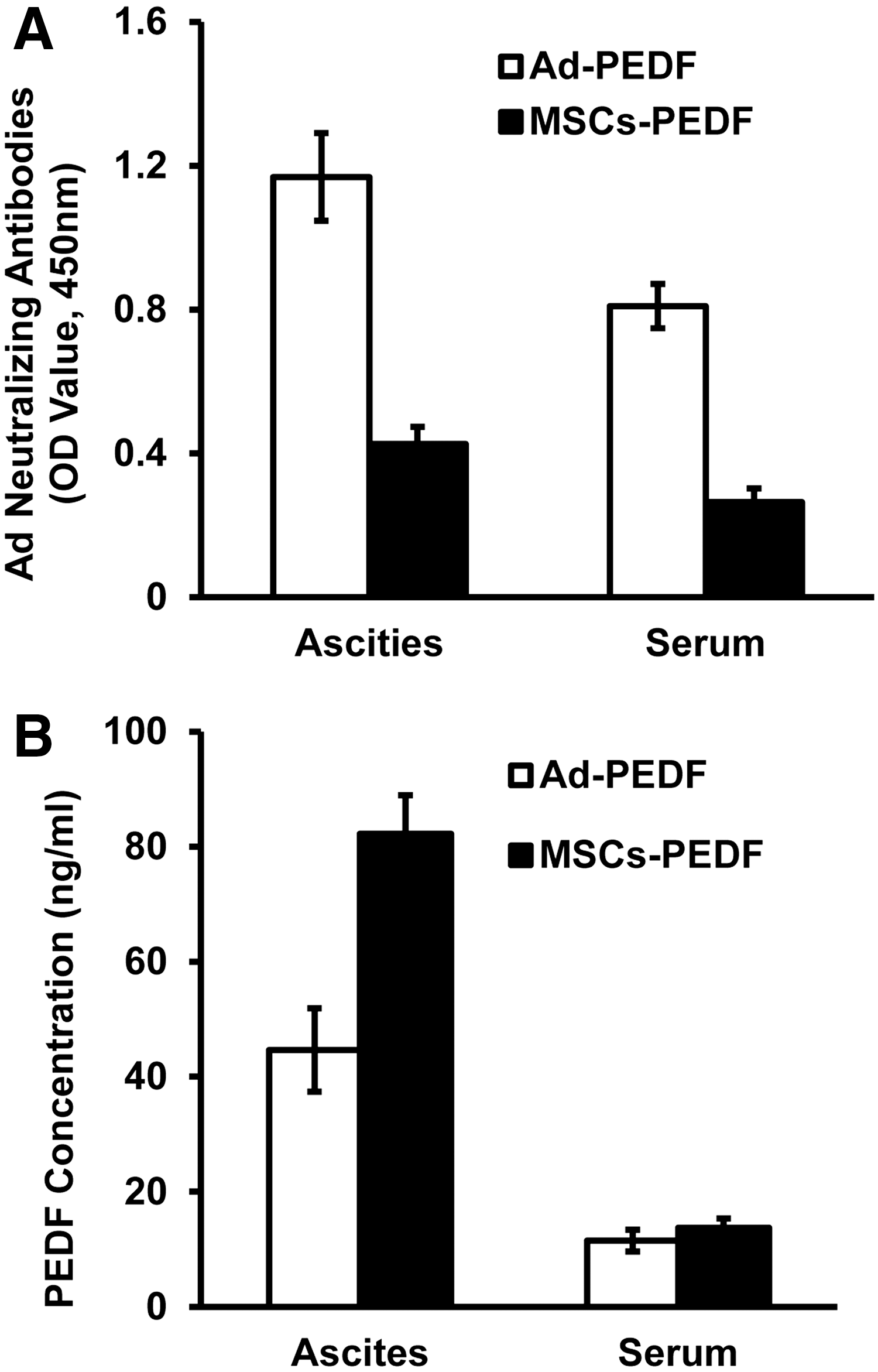
MSC-mediated Ad-PEDF delivery reduces adenoviral neutralizing antibodies and prolongs PEDF expression.
MSCs-PEDF balances the VEGF-A/sFLT-1 ratio in ascites
Figure 7 shows that VEGF-A accumulated in ascites but not in sera from CRPC mice 18 days after tumor inoculation. Treatment with Ad-PEDF and MSCs-PEDF reduced VEGF-A in ascites compared with other control groups, especially treatment with MSCs-PEDF (p < 0.05). Interestingly, sFLT-1 in ascites from mice treated with Ad-PEDF and MSCs-PEDF was higher than that of controls, especially treatment with MSCs-PEDF (p < 0.05). However, treatment with Ad-PEDF and MSCs-PEDF did not significantly change VEGF-A and sFLT-1 in sera compared with other control groups.

MSCs-PEDF balances the VEGF-A/sFLT-1 ratio in ascites.
Discussion
Surgical resection, chemotherapy, and radiotherapy are conventional and widely used therapeutic strategies for cancer. However, they fail to offer complete therapeutic effects for advanced colorectal cancer with CRPC and malignant ascites. 28 Recently, studies have reported that increases in vascular permeability are associated with tumor metastasis and malignant ascites. 29 –31 VEGF, a potent growth factor that stimulates vascular endothelial cell growth and blood vessel formation, was studied for its role in tumor angiogenesis as well as tumor growth, invasion, and metastasis. 32 Recently, VEGF was defined to be an important factor affecting vascular permeability, a key component in ascites formation. 3 High VEGF is often documented in malignant effusions of ovarian, colorectal, and breast cancer patients. 33 VEGF has also been confirmed to be the most potent permeability factor (50,000 times more potent than histamine). 5 Thus, anti-VEGF therapy may offer promise as an innovative treatment approach for of malignant ascites.
PEDF, an antagonist of VEGF, is notable for potent endogenous inhibition of angiogenesis. Previously, we demonstrated that PEDF could repress tumor growth and suppress metastasis by inhibiting tumor neoangiogenesis in several cancer types. 11 –13 Here, we report that MSC-engineered PEDF via adenoviral transduction could significantly inhibit tumor metastasis and malignant ascites formation in CT26 CRPC mice (Fig. 4). The antitumor mechanisms of PEDF are associated with inhibition of tumor angiogenesis and induction of apoptosis in CT26 CRPC mice (Fig. 5). Moreover, PEDF inhibits the synthesis of VEGF-A (an important vascular permeability factor) and reduces vascular permeability as well as malignant ascites formation.
Interestingly, we also found that PEDF could increase sFLT-1 in ascites (Fig. 7). Previous study demonstrated that the VEGF-A/sFLT-1 ratio was increased in ascites of patients with ovarian cancers. 34 Here, we reported that PEDF could restore the VEGF-A/sFLT-1 ratio balance in ascites. sFlt-1 is an endogenously expressed antagonist of VEGF. sFlt-1 inhibits VEGF activity through sequestering VEGF and forming inactive heterodimers with VEGF receptors in a dominant negative fashion. 35 Accumulating evidence suggested that sFlt-1 could inhibit tumor growth and suppress malignant ascites production. 35,36 Thus, the antitumor mechanisms of PEDF may be partly attributed to the increasing expression of sFlt-1. Further investigations are needed to clarify how PEDF regulates the expression of sFlt-1.
Adenoviral vectors use either the CAR or CD46 receptor and can infect diverse cell types. These vectors achieve greater transgene expression, 37 and so they are a widely utilized vehicle for delivering therapeutic genes. However, because of preexisting neutralizing antibodies and cellular immunity of adenovirus and its targeting cellular receptor dependency, clinical efficacy is often significantly compromised. 38,39 Apart from preexisting immunity, adenoviral injection can induce neutralizing antibodies that can hinder repeat administration. 27 Cell-mediated gene therapy can overcome adenoviral problems, however. Recently, MSCs as a cellular vehicle for delivery of antitumoral agents in tumor gene therapy have gained attention. 19,20 A significant advantage of MSCs as a cellular vehicle is their accessibility for genetic manipulation in vitro. MSC-mediated adenoviral delivery can protect the adenovirus from immunologic surveillance. 21 Moreover, the poorer immunogenicity of MSCs enables them to escape from immunologic surveillance and survive for a long time in the host. 15 Here, we found that MSC-mediated Ad-PEDF delivery reduces production of adenoviral neutralizing antibodies and prolongs PEDF expression in vivo (Fig. 6). Another significant advantage of MSCs as a cellular vehicle is that MSCs can home to malignant tissues. We confirmed that MSCs-PEDF could migrate toward tumor cells as well (Fig. 3).
MSCs are a class of adult stem cells predominantly isolated from bone marrow and multiple tissues. Unlike embryonic stem cells, MSCs can be easily isolated and expanded with minimal safety or ethics concerns. 15 Their regenerative properties, diverse immune functions, and homing abilities make MSCs an appealing choice as cellular vehicle for drug delivery. 40 Thus, MSCs are emerging as candidates for gene and drug delivery to treat various diseases and can be progressed toward clinical trials. To date, thousands of MSC-based clinical trials for representative diseases, including inflammation, cancer, and autoimmune diseases, are being conducted. 41
However, although some results of the MSC-based clinical trials are encouraging, the potential role of MSCs in tumourigenesis is a significant concern that must be addressed. The role remains a topic of continued debate. Because MSCs are immune suppressor cells, the MSCs could favor tumor growth through protection of cancer cells from immune surveillance. 42 Conversely, MSCs could also suppress tumor growth in some cancer types. 43,44 Therefore, we suggest that patients treated with MSCs should be followed because the potential side effects of immunosuppression induced by MSCs cannot be ignored. Although the immunosuppressive effects of MSCs have to be considered in further clinical studies, the usefulness of MSCs for treatment of various diseases still remains of great interest.
In conclusion, MSCs engineered to secrete PEDF by adenoviral transduction may offer a therapeutic approach for suppressing tumor metastasis and inhibiting malignant ascites production in a CRPC model. Antitumor activity mediated by MSCs-PEDF is associated with inhibition of tumor angiogenesis and restoring the VEGF-A/sFLT-1 ratio.
Footnotes
Acknowledgments
This study was supported by the Fundamental Research Funds for the Central Universities (Grant No. LZUJBKY-2012-162), Gansu Province Science and Technology Major Special Funds (Grant No. 1302FKDA029) and the Joint Project of Science and Technology Department of Gansu Province and the First Hospital of Lanzhou University (Grant No. GWGL2010-25).
Author Disclosure
No competing financial interests exist.