
Abstract
Cystic fibrosis (CF) is a genetic disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene, resulting in a deficiency in chloride channel activity. In this study, extracellular vesicles (EVs), microvesicles, and exosomes were used as vehicles to deliver exogenous CFTR glycoprotein and its encoding mRNA (mRNAGFP-CFTR) to CF cells to correct the CFTR chloride channel function. We isolated microvesicles and exosomes from the culture medium of CFTR-positive Calu-3 cells, or from A549 cells transduced with an adenoviral vector overexpressing a GFP-tagged CFTR (GFP-CFTR). Both microvesicles and exosomes had the capacity to package and deliver the GFP-CFTR glycoprotein and mRNAGFP-CFTR to target cells in a dose-dependent manner. Homologous versus heterologous EV-to-cell transfer was studied, and it appeared that the cellular uptake of EVs was significantly more efficient in homologous transfer. The incubation of CF15 cells, a nasal epithelial cell line homozygous for the ΔF508 CFTR mutation, with microvesicles or exosomes loaded with GFP-CFTR resulted in the correction of the CFTR function in CF cells in a dose-dependent manner. A time-course analysis of EV-transduced CF cells suggested that CFTR transferred as mature glycoprotein was responsible for the CFTR-associated channel activity detected at early times posttransduction, whereas GFP-CFTR translated from exogenous mRNAGFP-CFTR was responsible for the CFTR function at later times. Collectively, this study showed the potential application of microvesicles and exosomes as vectors for CFTR transfer and functional correction of the genetic defect in human CF cells.
Introduction
M
EVs participate in normal biological processes such as tissue repair, 8,9 immune surveillance, 10 blood coagulation, 11 and stem cell maintenance, 12 by delivering effectors such as transcription factors, small and large noncoding regulatory RNAs, as well as mRNAs. They act as mediators of intercellular communications via the transfer of proteins, lipids, and nucleic acids. 3,6,10 They are also associated to several diseases such as tumorigenesis, 13,14 Alzheimer's and Parkinson's diseases, 15,16 the spread of the prion protein PrPc, 17,18 the dissemination of HIV-1, 19 or the reactivation of latent HIV-1. 20 The EVs have a natural ability to transfer membrane, cytoplasmic, and genetic material to neighboring and distant cells, and thus have been exploited as vehicles for the delivery of therapeutic molecules such as bioactive proteins, lipids, and nucleic acids. 8 Several protocols of EV-based vaccination and antitumor therapy are already at the preclinical or clinical stage. 21
Cystic fibrosis (CF) is an autosomal recessive genetic disease caused by mutations in a single gene, the CF transmembrane conductance regulator (CFTR) gene. The CFTR is a type III membrane glycoprotein, with large intracytoplasmic N- and C-terminal domains and 12 membrane-spanning domains constituting a transmembrane, cAMP-dependent chloride channel. 22,23 It is present at the apical surface of epithelial cells, and localized in the lipid raft microdomains of the plasma membrane. 24 –26 In a previous study, we showed that MVs and EXos were able to achieve the transfer of the human CFTR mature glycoprotein and CFTR-encoding mRNA molecules (mRNACFTR) in a Chinese hamster ovarian (CHO) cell model, resulting in the gain in CFTR chloride channel function by MV- and EXo-recipient cells. 27
In the present study, we investigated the feasibility of MV- and EXo-mediated delivery of the exogenous CFTR glycoprotein and CFTR-encoding mRNA molecules to CFTR-deficient cells derived from CF patients (CF cells) to restore the CFTR chloride channel function. We used a GFP-CFTR fusion construct, which has been shown to be biologically active and capable of correcting the transepithelial chloride transport, 28,29 to track the exogenous CFTR protein in EV-recipient cells. We found that both MVs and EXos were capable of packaging GFP-tagged CFTR and mRNAGFP-CFTR, and transferred both types of molecules to target CF cells, resulting in the correction of the CFTR defect in CF cells. The CFTR chloride channel activity newly acquired by EV-transduced CF cells was measurable until day 3 after the EV-mediated transfer, and still detectable at day 5. Experimental results suggested that the maintenance of this function was performed by newly synthesized CFTR molecules translated from exogenous mRNAGFP-CFTR. This is, to our knowledge, the first demonstration that EVs can be exploited as therapeutic vectors for a genetic disease.
Materials and Methods
Cells
A549 and HEK-293 cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA). HEK-293 (ATCC CRL-1573) and A549 cells (ATCC CCL-185), adenocarcinomic human alveolar basal epithelial cells, were maintained as monolayers in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS; Life Technologies), penicillin (200 U/ml), and streptomycin (200 μg/ml) at 37°C and 5% CO2. Calu-3 cells, derived from human bronchial submucosal glands, were obtained from the laboratory of M.J. Welsh. 30,31 Calu-3 cell monolayers were grown at 37°C and 5% CO2 in a combined medium made up of ¾ DMEM and ¼ Ham's F12 (Life Technologies), supplemented with FBS, penicillin, and streptomycin at the same doses as above. CF15 cells, human nasal epithelial cells homozygous for the DeltaF508 CFTR mutation, 30,32 were obtained from the laboratory of M. Chanson. 32,33 CF15 were grown as monolayers in collagen I-coated flasks (BioCoat; BD Biosciences) at 37°C and 5% CO2 in CNT-17 medium (CellNTec), supplemented with penicillin and streptomycin.
Adenoviral vectors
Human adenovirus type 5 (HAdV5)-derived vectors HAdV5-GFP and HAdV5-GFP-CFTR have been described and characterized previously. 28,29 HAdV5-GFP-CFTR encoded the wild-type allele of the CFTR gene, fused to the 3′ end of the GFP gene. Vector stocks were produced and titrated on HEK-293 cell monolayers, as described previously. 28,29,34,35
Isolation of microvesicles and exosomes from extracellular vesicles
EVs were recovered from the culture medium of Calu-3, A549, and A549 transduced by HAdV5-GFP or HAdV5-GFP-CFTR, using a previously published protocol. 27 In brief, cells grown to 80–90% confluency in T150 flasks (5 × 107 cells/flask) were rinsed 3 times with phosphate buffered saline (PBS), and fresh culture medium, preclarified by ultracentrifugation at 100,000 × g for 16 hr at 4°C, was added to the cells. The EVs in the culture medium were then recovered at 24, 48, and 72 hr, by two successive steps of ultracentrifugation. The culture medium was first centrifuged for 5 min at 13,000 × g and 4°C to remove the cell debris, before the first ultracentrifugation step at 30,000 × g and 4°C for 2 hr, to obtain the first pellet originally called MP30 (now identified as MVs). The supernatant was then subjected to a second round of ultracentrifugation at 100,000 × g and 4°C for 2 hr, which gave the second pellet called MP100 (now identified as EXos). The MV and EXo pellets were resuspended in 200 μl PBS with gentle mixing, and stored at 4°C. They were used within 10 days in our experiments of EV-mediated cell transfer assays.
Protein assay
Aliquots of each EV sample were kept for protein estimation using the Bradford Reagent (Sigma-Aldrich Corp.). Triplicates of MV and EXo preparations, usually diluted 1:10 to 1:20 in PBS, were lysed with 0.01% SDS at room temperature for 30 min, and then reacted with the Bradford reagent, according to manufacturer's recommendations. Values were extrapolated from a standard curve of serially diluted bovine serum albumin (BSA) samples.
Titration of microvesicles and exosomes by nanoparticle tracking analysis
The concentrations of MV and EXo preparations were subjected to NTA, using the NanoSight LM10 system (Malvern Instruments Ltd.), equipped with a 405 nm laser and a high-sensitivity digital camera (Scientific CMOS trigger camera). Videos were collected and analyzed using the NTA software (version 2.3), with the minimal expected particle size, minimum track length, and blur setting, all set to automatic. Camera sensitivity and detection threshold were set to maximum (16) to detect small particles. Ambient temperature was recorded manually, ranging from 22°C to 27°C. Each sample was diluted in sterile PBS (Life Technologies) to obtain concentration range between 108 and 109 EVs/ml, and the number of completed tracks per sample was between 500 and 1500. The screen gain was 6 for EXos and 12 for MVs, with a capture time of 60 sec.
Gel electrophoresis and Western blot analysis
Polyacrylamide gel electrophoresis of SDS-denatured protein samples (SDS-PAGE), and immunoblot analysis have been described in previous studies. 36 –39 Briefly, samples were denatured in SDS/beta-mercaptoethanol-containing loading buffer at 100°C for 2 min, and proteins electrophoresed in SDS-denaturing 10% polyacrylamide gel and electrically transferred to nitrocellulose membrane (Hybond-C-extra; GE Healthcare Life Sciences). Blots were blocked in 5% skimmed milk in Tris-buffered saline (TBS) containing 0.05% Tween-20 (TBS-T), rinsed in TBS-T, and then successively incubated with primary antibody and peroxidase-labeled secondary antibodies. Blots were reacted with SuperSignal West Pico chemiluminescence substrate (Thermo Fisher Scientific Inc.), and exposed to autoradiographic film (Hyperfilm MP; GE Heathcare Bio-Sciences). The apparent molecular weights of proteins were estimated by comparison with prestained protein markers (Precison Plus Protein Standards, Dual Color; Bio-Rad Laboratories, Inc.).
Antibodies and reagents
Mouse monoclonal antibody against the human CFTR C-terminus (mouse IgG2A; Clone 24-1; MAB25031) was from R&D Systems. Goat polyclonal anti-CD63 antibody was purchased from Santa Cruz Biotechnology (Catalog No. SC-15363). Mouse monoclonal anti-GFP antibody (mixture of clones 7.1 and 13.1) was from Roche Applied Science. Peroxidase-coupled monoclonal anti-β-actin antibody was purchased from Sigma-Aldrich (Catalog No. A3854). Peroxidase-labeled rabbit antigoat IgG antibody (Catalog No. A5420) and sheep antimouse IgG antibody (Catalog No. A5906) were also purchased from Sigma-Aldrich.
Metabolic inhibitors of endocytic pathways included two inhibitors of the clathrin-dependent endocytosis, chlorpromazine and the lysosomotropic agent chloroquine; NH4Cl, an inhibitor of the acidification of the endosomes; Bafilomycin A1, a proton-pump inhibitor that blocks the vacuolar ATPase and the endosome/lysosome fusion; the isoflavone drug genistein and the polyene macrolide antibiotic filipin III, two inhibitors of the clathrin-independent, caveolin-mediated endocytosis; cytochalasin B (CytB), nocodazole (NDZ), and latrunculin A (LatA), which all disorganize the cell cytoskeleton, and have individual preferential negative impacts on endocytosis (CytB), intracytoplasmic trafficking (NDZ), and macropinocytosis (LatA); and the proteasome inhibitor MG132. All drugs were purchased from Sigma-Aldrich and prepared according to the manufacturer's instructions. They were used under sterile conditions, and at concentrations that did not decrease the cell viability to levels lower than 75%, as determined by the MTT assay (not shown). For testing the inhibitor effects, cell monolayers, taken at 80% confluence, were pretreated with each drug for 1 hr at 37°C before MVsGFP or EXosGFP addition. After a 6 hr incubation at 37°C with EVs (2 × 104 EVs/cell) in the presence of the drug, the cells were collected, fixed, and analyzed by flow cytometry.
EV-mediated transfer of GFP and GFP-CFTR into target cells
In a typical experiment, samples of GFP-loaded EVs (MVsGFP and EXosGFP) and GFP-CFTR-loaded EVs (MVsGFP-CFTR and EXosGFP-CFTR) in PBS were mixed with an equal volume of prewarmed serum-free medium, and 50–100 μl aliquots added to A549 or CF15 cell monolayers (2 × 105 cells/well). The total input (50–100 μl) contained 5–50 μg protein and 2 × 109 to 10 × 109 EVs, with transducing doses ranging from 5 × 103 to 5 × 104 EVs/cell. The cells were further incubated at 37°C, and the GFP signal was monitored in live cells using a Zeiss Axiovert-135 inverted microscope equipped with an AxioCam digital camera. Cells were harvested at different time periods, and GFP-positive or GFP-CFTR-positive cells were quantitated using flow cytometry. CFTR-associated chloride channel activity was measured using an iodide-selective microelectrode, as described below.
DNA and RNA isolation, and PCR analyses
DNA from cells and EVs was extracted using the QIAamp DNA Mini Kit (Qiagen). Total RNA was extracted from cells using the Nucleospin RNA II kit (Macherey Nagel), and from EVs using the QIAamp viral RNA Mini kit (Qiagen). Aliquots (100 ng) of RNA were reverse transcribed using the Iscript DNA synthesis kit (BioRad). Real-time PCR (RT-PCR) was performed using the KAPA Sybr Fast qPCR kit (KAPA Biosystems) and the StepOnePlus apparatus (Thermofisher). Quantitative RT-PCR (qRT-PCR) analysis of RNA was performed using the protocol described in ref. 40 and the ribosomal protein RPL27 RNA transcript as internal standard, amplified with the following primers: sense 5′-ATCGCCAAGAGATCAAAGATAA-3′; antisense 5′- TCTGAAGACATCCTTATTGACG-3′. For the CFTR gene, a PCR-amplified fragment of 415 nucleotides (nt) in length, overlapping exons 3 and 5 of the CFTR gene, was obtained with the following primers 41 : sense 5′-AGAATGGGATAGAGAGCTGGCTTC-3′ (exon 3); antisense 5′-TTCATCAAATTTGTTCAGGTTGTTG 3′ (exon 5). In the case of the GFP-CFTR fusion, a fragment of 196 nt in length, overlapping the GFP and CFTR junction sequence, 28,29 was obtained using a forward primer designed from the 3′ end of the GFP gene (nucleotide position 621; 5′-AACGAGAAGCGCGATCACATG), and an antisense primer designed from the 5′ end of the CFTR gene (nucleotide position 78 in the CFTR gene; 5′-GCGCTGTCTGTATCCTTTCCTCAA). For the GFP gene, a fragment of 99 nt within the GFP gene was obtained using the following primers: sense 5′- ATGGTGAGCAAGGGCGAGGA-3′; antisense 5′-CTCGCCGGACACGCTGAACT-3′. For the HAdV5 hexon gene, a fragment of 465 nt was obtained using the following primers: forward 5′- GCTGTATTTGCCCGAC-3′; reverse 5′-CATGGCCTCAAGCGTG-3′, corresponding to the genome nt 20,276–20,291 and 20,726–20,741, respectively.
Iodide efflux assay
The iodide efflux assay, which reflected the activity of the CFTR channel, was measured using an iodide-selective microelectrode. 42 The experimental setup included the MicroIodide Ion Electrode (LIS-146ICM; Lazar Research Laboratories), connected to an ORP-meter (ORP Model 62; Jenco Instruments Inc.), and the protocol described by Aleksandrov et al. 43 Replicates of cell monolayers were loaded with sodium iodide by incubation for 30 min at 37°C in prewarmed iodide-loading buffer (ILB:136 mM NaI, 3 mM KNO3, 2 mM Ca[NO3]2, 11 mM glucose, 20 mM HEPES, pH 7.4 with NaOH). Cells were then washed 10 times with efflux buffer (EB; 136 mM NaNO, 3 mM KNO3, 2 mM Ca[NO3]2, 11 mM glucose, 20 mM HEPES, pH 7.4 with NaOH) to completely remove extracellular iodide. The CFTR stimulation mixture (10 μM forskolin, 100 μM dibutyryl-cAMP, 1 mM 3-isobutyl-1-methylxanthine [IBMX] as enhancer of intracellular cAMP) was added at time 0, to activate efflux through CFTR channels. The values of the voltage generated by iodide in solution were extrapolated from the standard curve generated by measuring the voltages of standard NaI solutions. Iodide efflux values, expressed in nmol/min, represented the mean ± SEM of the amount of iodide released from the cells during each 1 min interval (n = 3). At the end of each assay, efflux buffer containing 0.1% NP-40 was added to release the whole bulk of intracellular iodide.
Flow cytometry
Cells were seeded in complete media in 24- or 48-well plates to obtain a confluency of 70–80% the following day. The cells were then incubated with MVs or EXos at 37°C and further incubated for different lengths of time. At the end of the incubation periods, the cells were rinsed with PBS, and detached with PBS containing 0.05% trypsin and 1 mM Na2EDTA. After centrifugation, the cells were fixed in PBS containing 1% paraformaldehyde, and analyzed in an LSRII flow cytometer (Becton Dickinson Biosciences).
Cell imaging
Fluorescence microscopy
Cell monolayers were fixed with 1% paraformaldehyde in PBS, and permeabilized with 0.1% (v/v) Triton X-100 in PBS. Cells were then blocked in 1% BSA in PBS for 1 hr at room temperature and incubated with primary monoclonal antibodies against Rab5 (clone C8B1; Cell Signalling), Rab4a (D-20; Santa Cruz Biotech), Rab11 (clone H-87; Santa Cruz Biotech), and Lamp1 (or CD107a; BD Biosciences), following dilutions recommended by the manufacturer. Samples were then rinsed in PBS and incubated with Alexa Fluor 568-conjugated goat antimouse IgG antibody, diluted 1:1000 (Molecular Probes, Invitrogen) for 1 hr at room temperature, rinsed, and treated with DAPI before being mounted on slides. Observations were performed using an Axioimager Z1 epifluorescence microscope (Carl Zeiss France) and a confocal microscope (LSM710; Carl Zeiss). Images were analyzed using the Zen software (Carl Zeiss).
Electron microscopy
For negative staining of MVs and EXos, samples were adsorbed on gold grids, and negatively stained with 2% uranyl acetate before EM observation. 44 For ultrathin sections, cell samples incubated with MVs or EXos were fixed with 2% glutaraldehyde in 0.1 M sodium cacodylate buffer pH 7.4, pelleted, and postfixed with osmium tetroxide (1% in 0.1 M cacodylate buffer, pH 7.4). The specimens were dehydrated and embedded in Epon resin, sectioned, and processed as previously described. 27,38 Ultrathin sections were examined under a JEM-1400 Jeol electron microscope equipped with an Orius-Gatan digital camera (Gatan France, 78113 Grandchamp).
Statistics
Results were expressed as mean ± SEM of n observations. Comparison between two samples was performed using the Student's t test (Excel); multiple samples were compared using the one-way ANOVA test. Differences were considered statistically significant when p < 0.05.
Results
Isolation, characterization, and nomenclature of EVs recovered from human cells
Human epithelial cell lines Calu-3 (human bronchial submucosal glandular cells) and A549 (human adenocarcinoma alveolar basal epithelial cells) were tested as EV-producing cells. Calu-3 cells are considered as a pertinent in vitro model of respiratory functions and chloride channel activity. 45 On the other hand, A549 cells are permissive to human adenovirus serotype 5 (HAdV5), and to the adenoviral vectors HAdV5-GFP or HAdV5-GFP-CFTR, which express the green fluorescent protein (GFP) or GFP-CFTR, a GFP-fused version of the human CFTR glycoprotein. 28,29 EVs that are naturally released in the culture supernatant of A549 and Calu-3 cells were separated into two populations, using a two-step ultracentrifugation procedure. The first fraction, MP30, was recovered in the pellet after centrifugation at 30,000 × g, and the second fraction, MP100, was pelleted after a second round of centrifugation at 100,000 × g. The two EV populations could be distinguished by their morphology under the EM, their size, and protein surface markers.
As exemplified by EV samples from A549 cells, MP30 consisted of large particles characterized by their size heterogeneity, ranging from 200 to 1200 nm in diameter (Fig. 1a, left panel). The MP100 population, however, showed more homogenous regular particles, rather spherical and relatively small in size, ranging from 15 to 60 nm (Fig. 1a, right panel). Size measurements were performed using dynamic light scattering (DLS), and representative DLS patterns obtained with EVs from Calu-3 cells are shown in Fig. 1b. A similar range of values as those estimated by EM was found for both types of EVs: 100–1000 nm for the diameter of MP30 (320 ± 280 nm; mean ± SEM; n = 2,272 k counts/s), and 20–200 nm (65 ± 24 nm; n = 1,770 k counts/s) for MP100 (Fig. 1b, compare the left and right panels).

Morphological, biophysical, and biochemical characterization of extracellular vesicles (EVs) isolated from human cells.
MP30 and MP100 issued from Calu-3 cells were also analyzed with respect to their CD63 content, a specific marker of the exosomal compartment. 46 Only the MP100 fraction, and not MP30, was found to contain CD63 (Fig. 1c, left panel), indicating that MP100 fraction was essentially composed of exosomes, whereas the MP30 fraction contained a majority of MVs. Thus, for the rest of this study, the terminology of microvesicles (abbreviated MVs) and exosomes (abbreviated EXos) was used instead of MP30 and MP100, respectively. The term “extracellular vesicles” (EVs) included both MVs and EXo populations.
Production and recovery of extracellular EVs
Culture supernatants of Calu-3 or A549 cells were collected three times over a period of 72 hr, successively at 24, 48, and 72 hr. After pooling the three samples, the recovery of MVs and EXos was assayed by nanoparticle tracking analysis (NTA). From 2.5 × 108 EV-producing cells, we obtained 4.92 ± 0.96 × 1010 MVs and 10.8 ± 2.1 × 1010 EXos with A549, and 4.46 ± 0.59 × 1010 MVs (mean ± SEM; n = 6) and 8.20 ± 1.35 × 1010 EXos with Calu-3 cells. The yields did not significantly differ for the two types of EV-donor cells, which corresponded to about 200 MVs per cell and 300–400 EXos per cell, over a 72 hr period culture. After resuspension in 200 μl PBS, the EV titer was ∼2.5 × 108/μl for MVs, and 5 × 108/μl for EXos. The protein concentrations ranged between 0.25 and 0.5 μg/μl, that is, a ratio of 2 × 108 MVs or EXos per μg protein. This ratio was consistent with the values reported in the literature for exosomes preparations obtained by ultracentrifugation and titrated by NTA (6 × 108 EXos/μg; see ref. 47 ).
EV-mediated delivery of chloride channel activity to human CF cells
In an earlier study, we generated a CHO cell line that stably expressed the GFP-CFTR protein and released MVs and EXos containing the GFP-CFTR glycoprotein and GFP-CFTR-encoding mRNA (mRNAGFP-CFTR). We showed that both types of molecules were transferable to target CHO cells. 27 Because Calu-3 cells are known to naturally express CFTR at relatively high levels, 45 we assumed that MVs and EXos isolated from Calu-3 would contain high amounts of CFTR and mRNACFTR. This was confirmed by SDS-PAGE and Western blot analysis, which showed that CFTR glycoprotein was detectable in both MVs and EXos, but in higher amounts in MVs compared with EXos (Fig. 1c, right panel). RT-PCR performed on MV and EXo samples normalized to the same EV titer showed that both types of EVs contained mRNACFTR, but MVs had an apparent higher mRNACFTR content, compared with EXos (Fig. 2a).
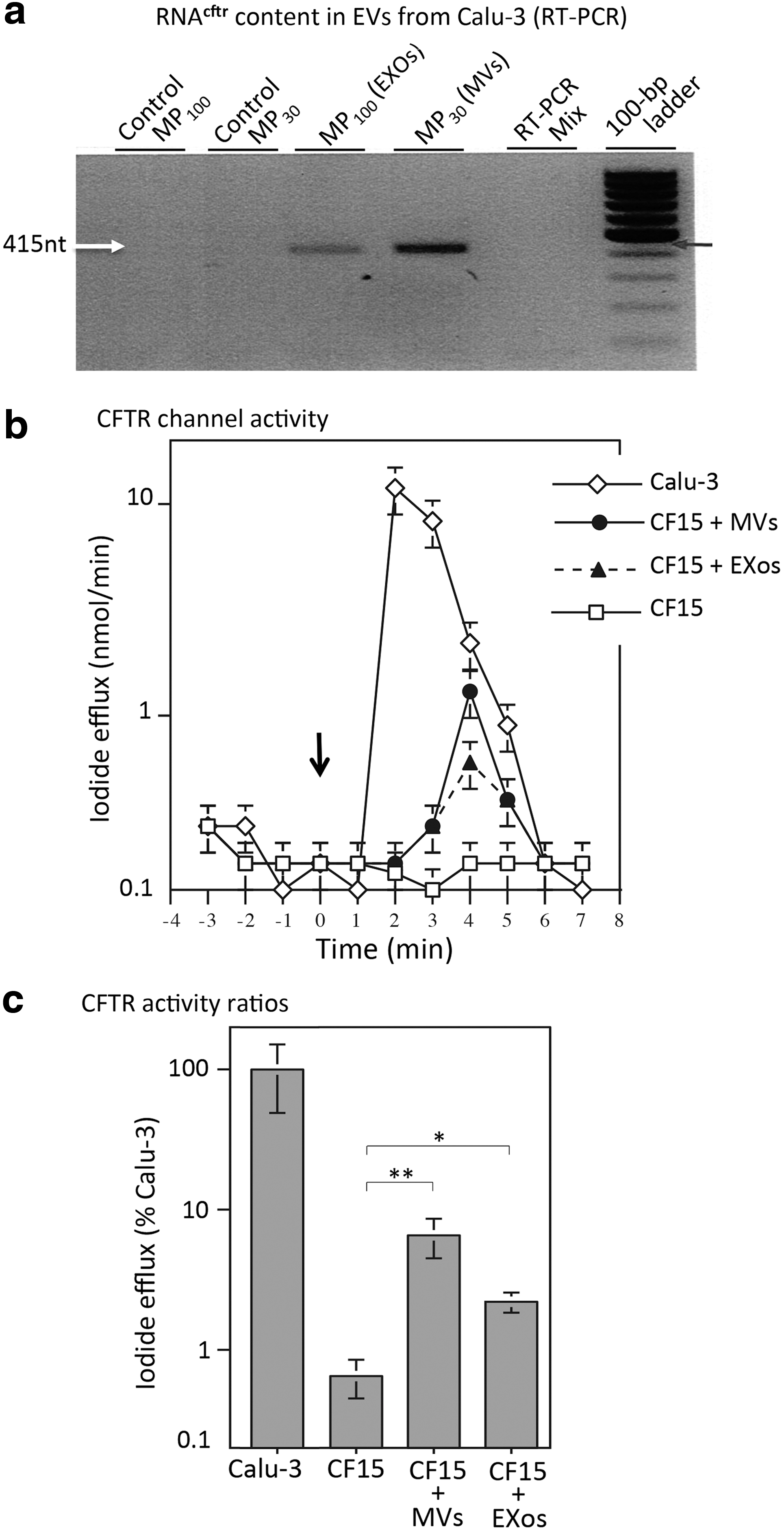
Characterization of Calu-3 as EV-donor cells.
We next determined whether MVs and/or EXos isolated from the Calu-3 cell culture medium (referred to as [Calu-3]MVs and [Calu-3]EXos) were capable of conferring the CFTR-associated chloride channel function to CF15 cells, a human nasal epithelial cells homozygous for the ΔF508 CF mutation. 30,32,33 CF15 cells were incubated for 6 hr at 37°C with [Calu-3]MVs or [Calu-3]EXos at the transducing dose of 200 EVs/cell. Fresh culture medium was then added, and cells were further incubated for 72 hr at 37°C. At the end of this period, the medium was removed, and the cells were tested for the cAMP-dependent CFTR chloride channel activity, measured by the iodide efflux assay using an iodide-selective microelectrode. 42,43
Curves representative of the cyclic cAMP-stimulated iodide efflux response in EV-transduced CF15 cells were compared with the curves of untreated CF15 cells (negative control), and of Calu-3 cells (positive control). A peak of CFTR channel activity was detected in EV-treated CF15 cells, suggesting the restoration of the CFTR function in these target cells (Fig. 2b). The CFTR chloride channel activity measured in EXo-treated CF15 cells was slightly lower, compared with that of MV-treated CF15 cells (Fig. 2b, c). Interestingly, the peak of channel activity was obtained at 4 min in EV-treated CF15 cells, a slight delay compared with the peak of Calu-3 that occurred at 2–3 min (Fig. 2b, c). Even though the levels of CFTR channel activity observed in EV-treated CF15 cells were 10- to 20-fold lower than that of the Calu-3 cells, this positive result incited us to pursue our studies, in an attempt to improve the efficiency of the CFTR transfer in human cells. The factors that could influence the transfer of the CFTR function included the nature of the EV-donor and EV-recipient cells, the EV cargo and the EV doses, and the experimental conditions of EV-cell interaction. These different parameters were investigated in the following experiments.
Characterization of EV-donor cells
Fluorescent EVs loaded with GFP or GFP-tagged CFTR were used to help us track and quantitate the uptake of EVs by target cells. 27 We assumed that EVs isolated from A549 cells transduced by HAdV5 vectors overexpressing either the GFP or GFP-CFTR protein would incorporate relatively higher amounts of GFP or GFP-CFTR glycoproteins and their corresponding mRNAs, compared with EVs from Calu-3 cells. SDS-PAGE and Western blot analysis, using anti-CFTR and anti-GFP antibodies, were performed on A549 cell lysates and compared with Calu-3 cell lysates. As expected, the CFTR glycoprotein content of HAdV5-GFP-CFTR-transduced A549 cells could be estimated to be 10–30 times higher than the endogenous CFTR content of Calu-3 cells, after normalization to equivalent protein loads (Fig. 3a).
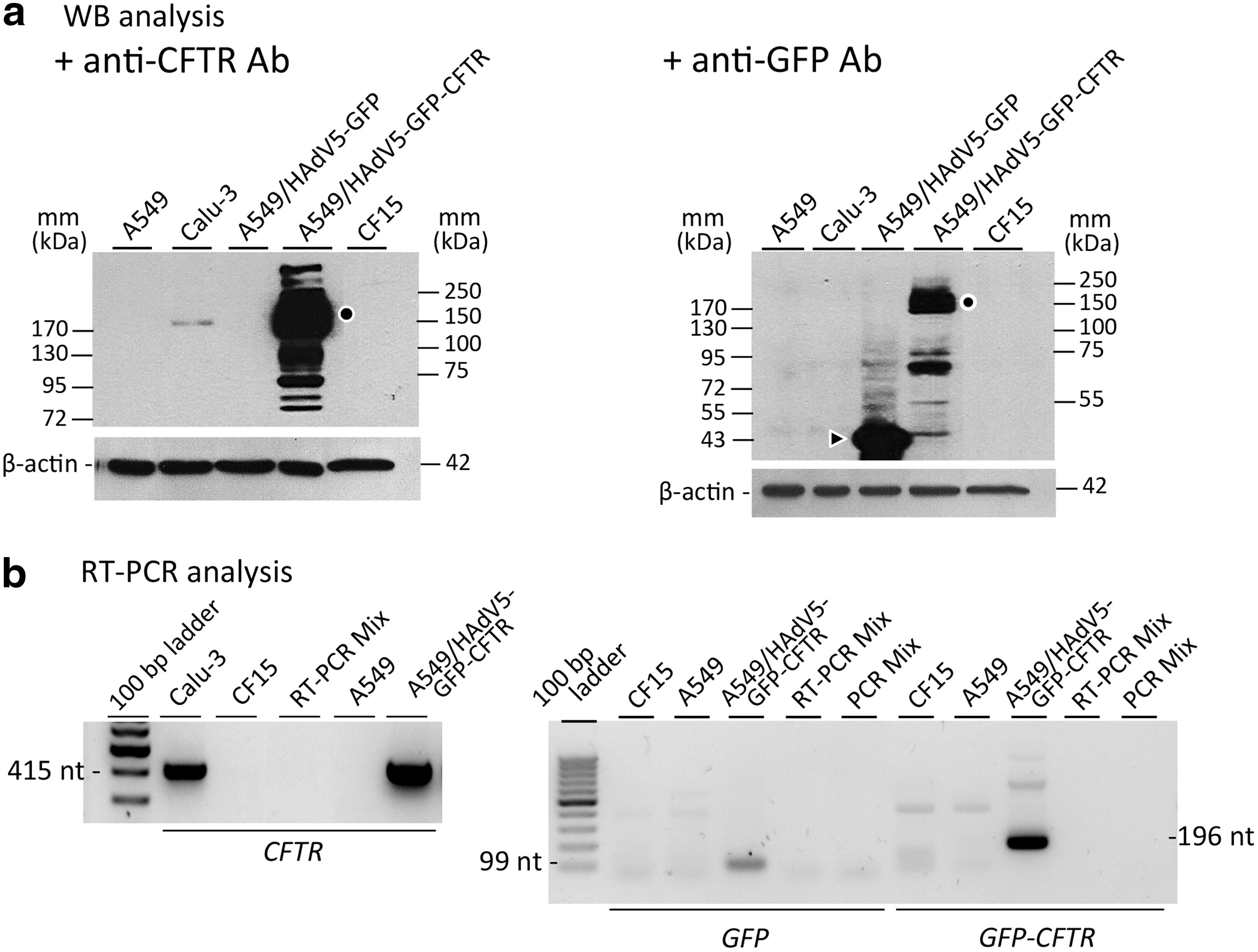
Characterization of EV-donor cells. Calu-3 and A549 cells, as indicated at the top of the panels, were probed for their CFTR or GFP-CFTR glycoprotein contents
RT-PCR amplified a 415 nt fragment from the endogenous mRNACFTR present in Calu-3 cells, and the same fragment was amplified from the mRNAGFP-CFTR transcript coding for the GFP-CFTR fusion protein in HAdV5-GFP-CFTR-transduced A549 cells. No endogenous mRNACFTR was detectable in control, nontransduced A549 cells, or in CF15 cells (Fig. 3b, left panel). A 99 nt fragment, amplified from the GFP gene, and a 196 nt fragment, specific of the GFP-CFTR junction, were found only in the RT-PCR samples of HAdV5-GFP-CFTR-transduced A549 cells, and not in the other samples (Fig. 3b, right panel). We therefore used HAdV5-GFP-CFTR-transduced A549 cells as EV-donor cells, and both A549 and CF15 as target cells for homologous and heterologous EV cell transfer, respectively.
Characterization of EVsGFP and EVsGFP-CFTR from A549 cells
MVs and EXos prepared from control, nontransduced A549 cells and from HAdV5-GFP- or HAdV5-GFP-CFTR-transduced A549 cells were analyzed for their GFP fluorescent signal by flow cytometry (Fig. 4a). Over 80% MVs and EXos from HAdV5-GFP-transduced A549 cells carried the GFP fluorescent signal (Fig. 4a). In contrast, the populations of EVs recovered from HAdV5-GFP-CFTR-transduced A549 cells contained only 10–12% fluorescent MVs, and 2–3% fluorescent EXos, namely, 4–5-fold less for the latter (Fig. 4a). These results were consistent with the fact that GFP is an abundant cytoplasmic and nuclear protein, whereas GFP-CFTR is a tightly regulated channel protein, localized in specialized microdomains of the cell plasma membrane. 24 –26
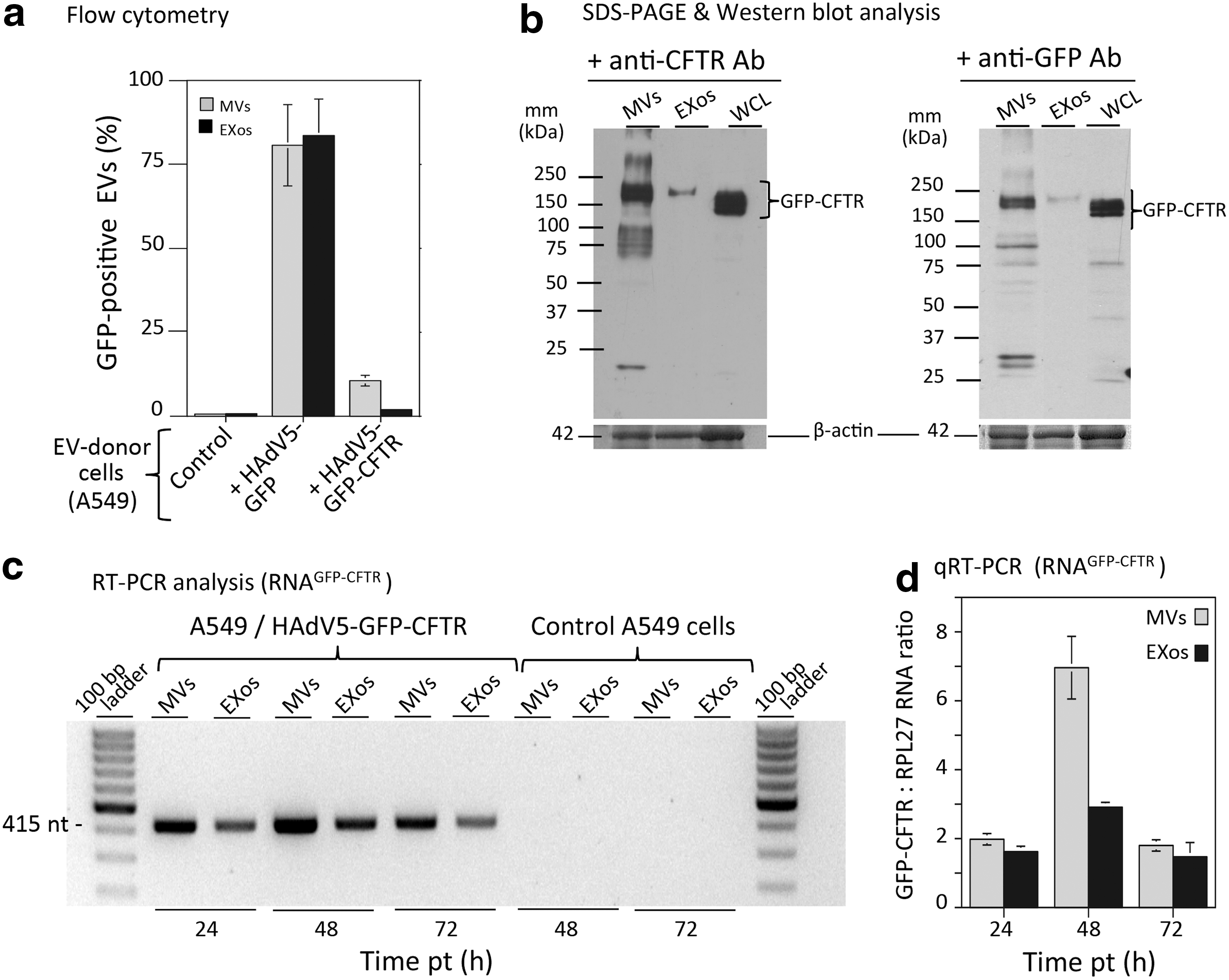
Characterization of EVs isolated from A549 cells.
Samples of MVsGFP-CFTR and EXosGFP-CFTR were then analyzed by SDS-PAGE and Western blotting, using anti-GFP or anti-CFTR antibodies to detect the GFP-CFTR fusion protein (Fig. 4b). The GFP-CFTR protein signal in MVsGFP-CFTR was estimated to be 10–20-fold higher, on a per-EV basis, compared with that of EXosGFP-CFTR (Fig. 4b). Of note, a single band of the CFTR protein migrating with an apparent molecular mass of 210–220 kDa was visible in the EXosGFP-CFTR fraction, which was compatible with that of the GFP-fused mature CFTR glycoprotein (25 + 190 kDa). In the MVsGFP-CFTR samples, however, several bands were detected, including a major doublet band at 200–220 kDa, and extra bands of immature CFTR and/or cleavage products (Fig. 4b).
MVsGFP-CFTR and EXosGFP-CFTR isolated from the A549 cell culture medium at different times after transduction with HAdV5-GFP-CFTR were then probed for their mRNAGFP-CFTR content. RT-PCR analysis, performed using primers overlapping the GFP-CFTR junction, or primers within the CFTR gene sequence, suggested that the maximum incorporation of mRNAGFP-CFTR into both MVs and EXos was attained during the first 48 hr after cell transduction with HAdV5-GFP-CFTR (Fig. 4c). This was confirmed by quantitative RT-PCR, which also showed that the mRNAGFP-CFTR content was twofold higher in MVsGFP-CFTR, compared with EXosGFP-CFTR (Fig. 4d).
Because MVsGFP-CFTR and EXosGFP-CFTR were isolated from A549 cells transduced by the HAdV5-GFP-CFTR vector, we determined whether these EVs could also contain adenoviral vector DNA or fragments thereof. Quantitative PCR using primers specific of the GFP-CFTR gene junction was performed on MVsGFP-CFTR and EXosGFP-CFTR samples harvested at 24 and 48 hr after HAdV5-GFP-CFTR transduction. Vector DNA was found in both types of EVs, and their DNA content increased with time posttransduction. There was no significant difference in vector DNA content between MVsGFP-CFTR and EXosGFP-CFTR (Supplementary Fig. S1a; Supplementary Data are available online at
Cellular uptake of MVsGFP-CFTR and EXosGFP-CFTR by target cells
EV dose dependence of the GFP-CFTR transfer
MVsGFP-CFTR and EXosGFP-CFTR from HAdV5-GFP-CFTR-transduced A549 cells were added to A549 or CF15 cell monolayers (5 × 105 cells per well) at different transducing doses, ranging from 5 × 103 to 4 × 104 EVs/cell, and incubated for 6 hr at 37°C. The cells were then harvested, and cell-associated fluorescent signal was analyzed by flow cytometry. There was a progressive increase in GFP-positive cells from 5% to 40% with increasing doses of MVsGFP-CFTR. A dose-dependent progressive augmentation of GFP-positive cells was also observed with EXosGFP-CFTR, reaching a maximum of 10% GFP-positive cells at the highest dose tested (Fig. 5a).
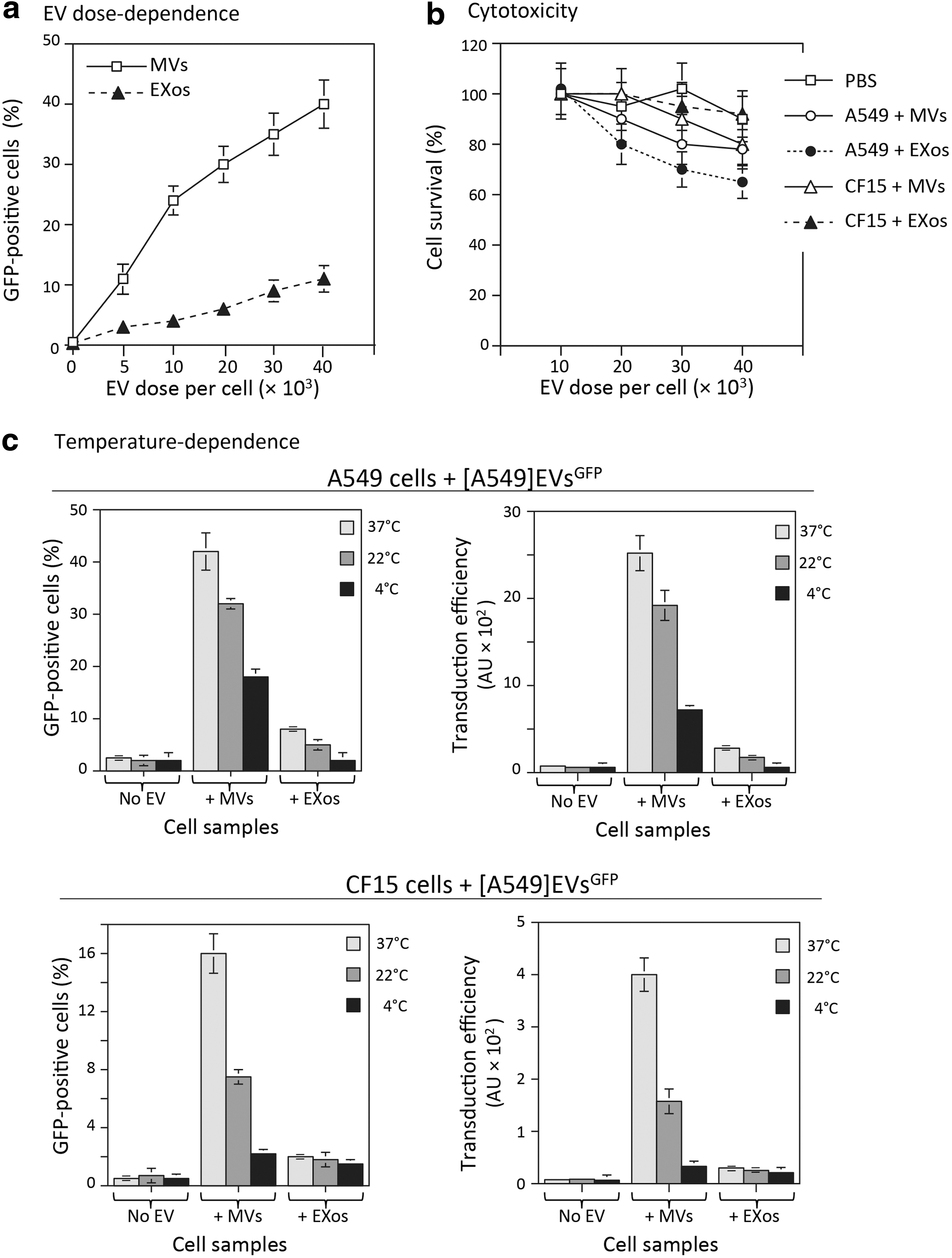
Dose dependence, cytotoxicity, and temperature dependence of EV-mediated transduction of target cells.
Cytotoxicity of MVsGFP-CFTR and EXosGFP-CFTR
The potential cytotoxicity of MVsGFP-CFTR and EXosGFP-CFTR was tested at different doses on A549 and CF15 cells, using MTT assays. The viability of EXosGFP-CFTR-transduced A549 cells decreased to 60% at the highest dose of 4 × 104 EVs/cell, but the other cell samples showed more that 80% viable cells within the range of 2 × 104 to 4 × 104 EVs/cell. Interestingly, the CF15 cells showed the highest degree of tolerance to both MVsGFP-CFTR and EXosGFP-CFTR, with no significant cytotoxity detected at the highest dose of 4 × 104 EVs/cell (Fig. 5b).
Temperature dependence
We next explored the effect of the incubation temperature on the cellular attachment and uptake of EVs in both homologous and heterologous systems. Aliquots of MVsGFP and EXosGFP from HAdV5-GFP-transduced A549 cells were added to A549 or CF15 cell monolayers (2 × 104 EVs per cell), and cells were incubated for 2 hr at 37°C, 22°C, and 4°C, respectively. At the end of this period, cells were collected, fixed, and analyzed by flow cytometry. The uptake of MVsGFP and EXosGFP was temperature dependent for both types of cell targets, with a progressive decrease of the EV-mediated transduction with the lowering of the incubation temperature (Fig. 5c). In comparison to MVsGFP-transduced A549 cells at 37°C, a reduction of about 25% was observed at 22°C, and 50% at 4°C. In MVsGFP-transduced CF15 cells, the reductions were 50% and 90% at 22°C and 4°C, respectively. Because of the relatively low values at 37°C, the variations with the temperature lowering were less pronounced for the cellular uptake of EXosGFP by both targets (Fig. 5c). In addition, the proportion of transduced cells and the transduction efficiency were significantly higher in the homologous cellular uptake compared with the heterologous uptake at all temperatures tested (Fig. 5c; compare upper and lower pairs of panels). These results suggested that the cellular uptake of A549-issued MVsGFP and EXosGFP by A549 and CF15 cells was a temperature-dependent, dynamic process, which required an optimal fluidity of the membrane lipid bilayers. However, the absence of total inhibition of cellular uptake of EVs at 4°C suggested that a temperature-independent mechanism might also be involved, such as the recognition between receptor and/or adaptor molecule(s) between the EV membrane and cell plasma membrane.
Homologous versus heterologous cellular uptake
To evaluate the efficiency of homologous and heterologous EV-to-cell transfer, GFP-tagged EVs (MVsGFP and EXosGFP) were prepared from HAdV5-GFP-transduced A549 cells. The advantage of using GFP-loaded EVs was the high proportion of fluorescent EVs for GFP (over 80%), compared with GFP-CFTR (around 10%; refer to Fig. 4a). For homologous cellular uptake of EVs, EVs isolated from A549 cells were incubated with A549 cells, whereas for heterologous cellular uptake, EVs isolated from A549 cells were incubated with CF15 cells. Incubation was carried out at 37°C for 1, 2, 3, 4, 6, 12, and 24 hr, at an input of 2 × 104 EVs/cell. At each time point, the target cells were fixed for fluorescence microscopy and assayed by flow cytometry for the proportion of GFP-positive cells and the index of transduction efficiency (TE). The TE, expressed in arbitrary units (AU), was calculated using the following formula: TE = GFP-positive cells (%) × MFI. 34
Homologous cellular uptake (Fig. 6a)
About 60% of A549 cells were GFP-positive after 3 hr incubation with MVsGFP, and 75% after 6 hr. The proportion of GFP-positive cells reached a plateau of 80% after 12 hr. The values obtained for EXosGFP-transduced cells were lower, with 5% and 10% at 3 and 6 hr, respectively, and a plateau at 25% after 12 hr. The transduction efficiency was significantly higher with MVsGFP, compared with EXosGFP, with values ranging from 4- to 8-fold higher at all time points (Fig. 6a, upper and lower leftmost panels). Fluorescence microscopy observation showed that GFP dots were more abundant in MVsGFP-transduced cells than in EXosGFP-transduced cells at all time points tested (Fig. 6a, upper and lower rightmost panels).

Homologous versus heterologous EV-mediated cell transduction.
Heterologous cellular uptake (Fig. 6b)
The uptake of heterologous EVs was slower, and its efficiency was significantly lower (about 2-fold), compared with homologous EV uptake: only 40% MVsGFP-transduced CF15 cells were found to be GFP-positive at 12 hr, and a maximum of 50% at 24 hr. This difference was also observed with EXosGFP-transduced CF15 cells, which showed no fluorescent signal over the background before the 4 hr time point, and a maximum value at 5% GFP-positive cells at 12 and 24 hr. As observed with A549 cells, the difference in transduction efficiency between MVsGFP and EXosGFP ranged between 6- and 8-fold within the late time period (Fig. 6b, upper and lower leftmost panels), which was confirmed by fluorescence microscopy (Fig. 6b, upper and lower rightmost panels). These results suggested that the cellular uptake of EVs was more efficient in homologous EV-to-cell transfer system, compared with the heterologous type, with MVs being more efficient than EXos in both cases.
Mechanisms of cellular uptake of EVs
Cell entry inhibitors
Metabolic inhibitors 48,49 were used to further explore the cell entry pathway of MVsGFP and EXosGFP in homologous and heterologous transfer systems. Only cytochalasin B, a cytoskeletal drug that acts as an endocytosis blocker, 48,49 and the macropinocytosis inhibitor amiloride 50 provoked a significant diminution (about twofold) of the GFP signal in EV-treated cells. This suggested that clathrin-dependent endocytosis and macropinocytosis were involved to some extent in the cellular uptake of EVs. The effects observed with the other drugs were moderate, and varied with the cell types and the nature of the EVs, in the negative or positive sense (data not shown). This implied that MVs and EXos used different mechanisms and/or alternative internalization pathways, of which the relative importance would depend on the nature of EVs and the type of target cells. In the presence of a given metabolic inhibitor responsible for the blockade of one pathway, the EVs might be redirected to alternative routes for cell entry, which, paradoxically, would result in an apparent higher transduction efficiency. This confirmed the diversity of mechanisms of cellular attachment and uptake of EVs previously observed. 21,51
Imaging of EV–cell interaction by electron microscopy
Monolayers of A549 cells were incubated with A549-issued MVsGFP or EXosGFP (2 × 104 EVs/cell) at 37°C for 2 hr, and the cells were fixed and processed for EM. For both MVs and EXos, EM observation showed close contacts between EVs and the cell plasma membrane (Supplementary Fig. S2a, b), and between intravesicular EVs and the vesicular membrane (Supplementary Fig. S2d, f). Electron-dense material connecting MVs and EXos with plasma and vesicular membranes was also visible. These images were reminiscent of a receptor–ligand recognition, usually followed by cell internalization via membrane fusion, or alternatively via endocytosis (Supplementary Fig. S2d, f). Images evoking macropinocytosis were also observed with both MVs and EXos (Supplementary Fig. S2c, e).
Confocal fluorescence microscopy of EV–cell interaction
The intracellular trafficking of EVs after their cellular uptake was investigated using GFP-loaded EVs, and antibodies against markers of the early, intermediate, and late vesicular compartments of the endocytic pathway. These included antibodies against Rab5, Rab4a, Rab11, and Lamp1. A549 cells were incubated with MVsGFP and EXosGFP at an input of 2 × 104 EVs/cell, harvested at 2, 6, and 24 hr posttransduction, permeabilized, and reacted with primary antibodies followed by secondary, red-fluorescent antibodies. The cell samples were then processed for confocal fluorescence microscopy. No colocalization with Rab5, Rab4a, or Rab11 was detectable with MVsGFP or EXosGFP at any of the time points considered (not shown). At 24 hr posttransduction, however, some degree of colocalization of EXosGFP and Lamp1 was observed (Supplementary Fig. S3a). Colocalization with Lamp1 was not observed with MVsGFP, even at late times posttransduction (Supplementary Fig. S3b). These observations indicated that the MVs and EXos did not accumulate in the same vesicular compartments at late times after cell entry, and suggested that MVs and EXos differed in their intracellular trafficking.
Kinetics of EV-mediated delivery of GFP-CFTR to CF target cells
MVsGFP-CFTR and EXosGFP-CFTR isolated from HAdV5-GFP-CFTR-transduced A549 cells were incubated with CF15 target cells at 2 × 104 EVs/cell for 6 hr at 37°C, and cells collected at time points 6, 12, 24, and 48 hr after transduction. At 6 hr posttransduction, the percentage of GFP-CFTR-positive cells was 10–15% for MVsGFP-CFTR-transduced cells and 5–6% for EXosGFP-CFTR-transduced cells. At 12 hr, the percentage of GFP-CFTR-positive cells decreased for both types of EVs, with only 3% fluorescent cells with MVsGFP-CFTR, and less than 2% for EXosGFP-CFTR (Fig. 7a, left panel). Interestingly, the proportion of GFP-CFTR-positive cells progressively increased at later times posttransduction with both types of EVs, with 8–9% GFP-CFTR-positive cells at 48 hr for MVsGFP-CFTR-transduced cells, and 5–6% for EXosGFP-CFTR-transduced cells (Fig. 7a, left panel). The TE values followed a similar U-shaped pattern (Fig. 7a, right panel), and the transduction efficiency was consistently 5–10-fold higher with MVsGFP-CFTR than with EXosGFP-CFTR.
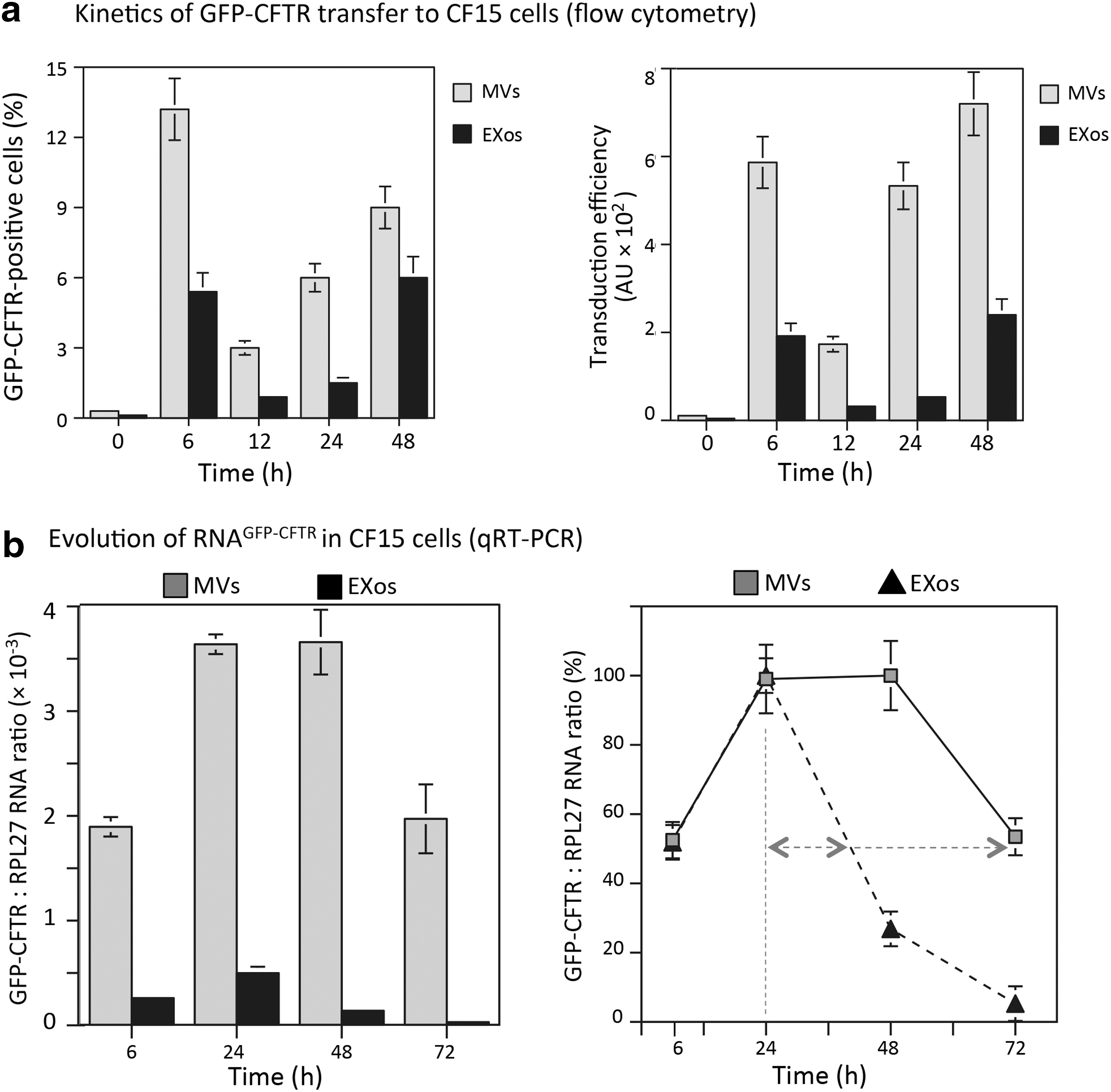
Kinetics and efficiency of EV-mediated transfer of GFP-CFTR into target cells.
The transfer of mRNAGFP-CFTR molecules to target cells was evaluated by RT-PCR and qRT-PCR analyses, using primers overlapping the GFP and CFTR sequences, and RPL27 RNA as the internal control. Reaction was performed on samples of MVsGFP-CFTR- and EXosGFP-CFTR-transduced CF15 cells, harvested at different time points after EV-transduction. Both types of samples contained mRNAGFP-CFTR as early as 6 hr posttransduction (Fig. 7b, left panel). Interestingly, in MVsGFP-CFTR-transduced cells, the number of mRNAGFP-CFTR copies increased between 6 and 24 hr posttransduction and remained stable until 48 hr, before decreasing progressively. In EXosGFP-CFTR-transduced cells, however, no plateau was observed between 24 and 48 hr, and the mRNAGFP-CFTR content rapidly decreased after 24 hr, to almost undetectable levels at 72 hr. The difference in mRNAGFP-CFTR levels between MVsGFP-CFTR- and EXosGFP-CFTR-transduced CF15 cells was estimated to be 5–7-fold between 6 and 24 hr, and 25–30-fold between 48 and 72 hr (Fig. 7b, left panel).
The half-life of intracellular mRNAGFP-CFTR transferred, deduced from the kinetic curves, was t ½ = 48 hr for MVsGFP-CFTR, and t ½ = 16 hr for EXosGFP-CFTR (Fig. 7b, right panel), that is, three times shorter in EXosGFP-CFTR-transduced cells compared with MVsGFP-CFTR-transduced cells. These results suggested that the cellular uptake of heterologous EVs by the target cells CF15 was a progressive process that prolonged until 24 hr after the initial EV-cell contact. These results also suggested that the mRNAGFP-CFTR molecules conveyed by MVsGFP-CFTR into CF15 cells did not follow the same metabolic pathway as those conveyed by EXosGFP-CFTR, and/or did not accumulate in the same cellular compartments at late times after transfer. These two possibilities, which are not mutually exclusive, were consistent with the observation by confocal fluorescence microscopy, suggesting different cell trafficking pathway for MVs and EXos (refer to Supplementary Fig. S3).
Vector DNA, carried over by EVs, was also detected in EV-transduced CF15 cells. Quantitative PCR analysis using primers specific of the GFP-CFTR junction showed that there was 50–80-fold less vector DNA in EXosGFP-CFTR-transduced cells compared with MVsGFP-CFTR-transduced cells (Supplementary Fig. S1b). There was no increase in the vector DNA content between 24 and 48 hr posttransduction. There was no detectable adenovirus hexon gene transcript in MVsGFP-CFTR- or EXosGFP-CFTR-transduced cells (not shown). These results suggested that the template of the amplified DNA fragments in both EVs and EV-transduced cells consisted of incomplete or fragmented vector DNA, naked or partially uncoated.
Restoration of chloride (Cl−) channel function in CF cells transduced by CFTR-loaded EVs
The CF15 cells, which carry the deltaF508 mutation in the CFTR gene, are defective in the CFTR Cl− channel function. The next important issue to address was whether transduction of CF15 cells by CFTR-loaded EVs was able to restore or correct the CFTR Cl− channel function in these cells. We therefore tested for the gain in Cl− channel function in CF15 cells incubated with different doses of MVsGFP-CFTR or EXosGFP-CFTR, by measuring at 24 hr postincubation the iodide efflux using an iodide-selective microelectrode. 26,42,43 As positive controls of vector-transduced CFTR activity, 28,29,35 CF15 cells were transduced by HAdV5-GFP-CFTR at several vector doses. At 50 vector particles (vp) per cell, a strong and rapid response of iodide efflux was detectable as early as 1–2 min after activation (Fig. 8a). There was no detectable Cl− channel activity at low vector doses (5 and 10 vp/cell), implying a threshold effect as previously observed in human airway primary CF cells transduced by the same HAdV5-GFP-CFTR vector. 28,29
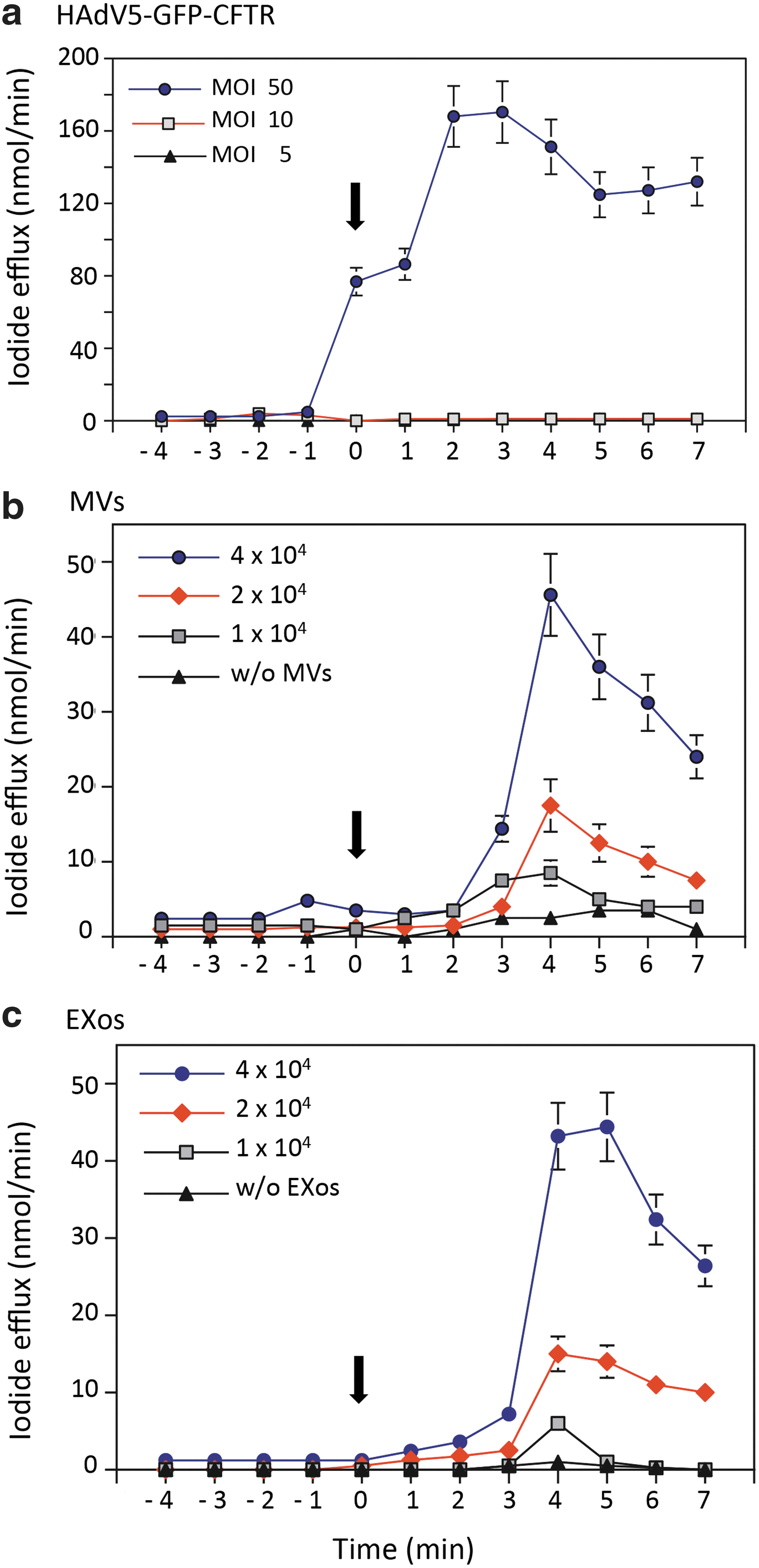
Measurement of the CFTR channel activity in EV-transduced CF cells. CF15 cells were incubated with MVsGFP-CFTR or EXosGFP-CFTR at increasing transducing doses (1 × 104, 2 × 104, or 4 × 104 EVs/cell). As positive control, CF15 cells were transduced with HAdV5-GFP-CFTR at increasing vector doses (5, 10, or 50 PFU/cell). At 24 hr posttransduction, cells were loaded with iodide, and cAMP-containing stimulation cocktail was added at time 0 (arrow), to activate the anion efflux through CFTR channels. The amount of iodide released from the cells was measured at 1 min intervals, using an iodide-selective electrode (mean ± SEM; n = 3).
A dose–response study was performed with increasing transducing doses of MVsGFP-CFTR or EXosGFP-CFTR at 1 × 104, 2 × 104, and 4 × 104 EVs/cell. For both MVsGFP-CFTR or EXosGFP-CFTR, none or low Cl− channel activity (10–20 nmol I−/min) was detected at the lowest dose of 1 × 104 EVs/cell. A modest Cl− channel activity was detected with MVsGFP-CFTR and EXosGFP-CFTR at the transducing dose of 2 × 104 EVs/cell (10–15 nmol I−/min), and more importantly, a relatively high CFTR activity (40–50 nmol I−/min) was obtained with MVsGFP-CFTR and EXosGFP-CFTR used at 4 × 104 EVs/cell (Fig. 8b, c). Compared with HAdV5-GFP-CFTR-transduced cells, the peak of CFTR activity in EV-transduced cells was about 2- to 3-fold lower (40–50 vs. 160–200 nmol I−/min), and slightly delayed (4 min after activation; Fig. 8b, c). Nevertheless, the Cl− channel activity in EV-transduced CF15 cells transduced at 2 × 104 EVs/cell was within the same range of values as those measured in Calu-3 cells, which naturally express endogenous CFTR (refer to Fig. 2b,c). These promising results demonstrated that transduction of CF cells with MVsGFP-CFTR or EXosGFP-CFTR was able to restore the CFTR Cl− channel function as early as 24 hr posttransduction. The CFTR Cl− channel activity in these EV-transduced CF15 cells was found to persist at detectable levels up to 5 days (not shown).
Discussion
In an earlier study, we demonstrated that EVs isolated from CHO cells modified to express transmembrane glycoproteins such as the Coxsackie adenovirus receptor (CAR), CD46, and the more complex dodecaspanin CFTR had the capacity to transfer the CAR-, CD46-, and CFTR-encoding mRNA molecules to naive CHO cells. This EV-to-cell transfer resulted in the gain of specific functions associated with CAR, CD46, and CFTR in the target CHO cells. 27 In the present study, we investigated the capacity of EVs in mediating the delivery of CFTR to human CF cells. Preliminary experiments using EVs from Calu-3 cells incubated with CF15 cells, which carry the CFTR ΔF508 mutation, showed a correction of the CFTR chloride channel defect in CF15 cells. However, the level of this newly acquired CFTR activity was 10–20-fold lower than that of Calu-3 cells. We therefore explored different conditions and parameters to improve the delivery of CFTR to target cells.
Important factors in CFTR delivery included the CFTR cargo in EVs, the homology between EV-donor cells and EV-recipient cells, and the EV transducing doses. To augment the CFTR cargo in EVs, and to visualize in situ the transfer of the CFTR glycoprotein, we used EVs isolated from A549 cells transduced by HAdV5 vectors expressing GFP-tagged CFTR at high levels. 28,29 Both types of EVs, MVs and EXos, were capable of incorporating and transferring GFP-CFTR to target cells in both homologous (EVs from A549 to A549 cells) and heterologous systems (EVs from A549 to CF15 cells). Not surprisingly, we found that the cellular uptake of EVs was more efficient in EV-to-cell homologous transfer. This parameter should be taken into consideration for future clinical applications of EV-based human biotherapy, and notably for the design of autologous EV-donor cells.
Upon uptake of MVsGFP-CFTR and EXosGFP-CFTR, the CFTR-deficient CF15 cells acquired the CFTR-associated Cl− channel activity as rapidly as 24 hr posttransduction. The newly acquired CFTR activity was EV-dose dependent in the range of 1 × 104 to 4 × 104 MVs or EXos per cell. However, no significant Cl− channel activity was detectable at doses inferior to 104 EVs/cell. Of note, a similar threshold effect had been observed in human airway primary CF cells transduced by adenoviral vectors. 29 Interestingly, the transduction efficiency of CF cells over time by MVsGFP-CFTR and EXosGFP-CFTR showed a curve with two maxima separated by a minimum of GFP-CFTR-positive cells at 12 hr posttransduction (Fig. 8a). This U-shaped pattern suggested that the fluorescent signal detected at early and late times posttransduction resulted from the contribution of two types of molecules, the GFP-CFTR glycoprotein and its encoding mRNA: (1) mature GFP-CFTR glycoproteins transferred by EVs to target cells would be responsible for the early GFP signal; (2) de novo synthesized GFP-CFTR, translated from the exogenous mRNAGFP-CFTR by the target cell translational machinery, would be responsible for the retarded GFP signal. These results implied that EV-packaged mRNA molecules in general, and mRNAGFP-CFTR in particular, played a major role in the gain of a function by target cells, as exemplified by the CFTR Cl− channel activity gained by CF cells. This corroborated our previous observations of the functional role of exogenous mRNA in conveying a desired biological activity in target cells. 27
However, EVs isolated from HAdV5-GFP-CFTR-transduced A549 cells were found to contain remnants of adenoviral vector DNA, which were transferred to target cells. Although no infectious viral vector could be rescued when MVsGFP-CFTR and EXosGFP-CFTR were incubated with trans-complementing HEK-293 cells, the possibility existed that the GFP-CFTR gene carried over by EVs was transcribed into mRNAGFP-CFTR, and this neosynthesized mRNA translated into the GFP-CFTR protein at late times after transduction. Such a mechanism was supported by a recent study on the intracellular fate of EV-transferred biomolecules, and on the active role played by a plasmid DNA compared with RNA molecules, considering their relative instability and short lifetime within the recipient cells. 52
In the present study, we first demonstrated that functional correction of the CFTR Cl− channel defect in CF cells could be achieved by EVs carrying mRNACFTR isolated from Calu-3 cells. In the case of CF15 cells transduced by EVs isolated from viral vector-transduced cells, such as HAdV5-GFP-CFTR/A549 cells, the mRNAGFP-CFTR responsible for the newly acquired CFTR Cl− channel activity might result from the translation of two separate pools of molecules, mRNAGFP-CFTR presynthesized in EV-donor cells (A549) and transferred by EVs to target CF cells, and mRNAGFP-CFTR neosynthesized in target CF15 cells. Whatever the respective contribution of one or the other mechanism, the neosynthesis of GFP-CFTR glycoprotein molecules would explain the persistence of the CFTR activity in CF cells for several days after the EV-mediated transfer.
Flow cytometry analysis of CF15 cells transduced with MVsGFP-CFTR and EXosGFP-CFTR at the same dose, and RT-PCR determination of the mRNAGFP-CFTR cellular content, suggested a higher transfer efficiency by MVs, compared with EXos. Likewise, mRNAGFP-CFTR had a longer half-life in MVsGFP-CFTR-transduced cells, compared with EXosGFP-CFTR-transduced cells. However, the CFTR Cl− channel activity newly acquired by EV-transduced CF15 cells and its duration over time were found to be equivalent in EXosGFP-CFTR- and MVsGFP-CFTR-transduced cells. These apparently contradictory results implied that only a fraction of the mRNAGFP-CFTR transferred by MVsGFP-CFTR was metabolically active, and that the majority remained sequestered in a dead-end cell compartment. This hypothesis was supported by fluorescence confocal microscopy, which suggested that MVsGFP-CFTR and EXosGFP-CFTR followed different trafficking and/or metabolic pathways after their cellular uptake by CF cells.
Collectively, our study confirmed the potential application of EVs for the delivery of CFTR to CF cells and the correction of the chloride channel deficiency. The present limitation to the use of CFTR-loaded EVs was the threshold observed in the acquisition of the CFTR-associated channel function, which required relatively high transducing doses of EVs, compared with conventional viral vectors. This could be solved by in vitro transcription of mRNACFTR molecules from a CFTR-encoding plasmid, and their encapsulation at high concentration into EVs, as already developed for therapeutic RNAs and liposomes. 53 Such a technique could find its application for EVs isolated from individual patients, in the context of the personalized medicine of CF pathology using autologous EVs.
Footnotes
Acknowledgments
The work in Lyon was financed by the Cystic Fibrosis French Association (VLM Contract RF20130500796 and RF20140501171). C.V. was the recipient of doctoral fellowships from the Région Rhône-Alpes (ADR Cluster-ARC 2011–2014), CMIRA Explora'doc-2012, and VLM (Vaincre la Mucoviscidose). S.S.H. is an INSERM senior scientist (Chargée de Recherche) and the recipient of a Contrat d'Interface Hospices Civils de Lyon-INSERM (2009–2013). The work in Geneva was supported by Grant #310030_134907/1 from the Swiss Science Foundation. We are grateful to Sébastien Dussurgey and Thibault Andrieu (AniRA-Cytométrie, SFR BioSciences), Bariza Blanquier (AniRA-Analyse génétique SFR BioSciences), Catherine Ott and Laurence Générénaz (Laboratoire Commun de Recherche BioMérieux-HCL), and Nathalie Calin (ENS-Lyon) for their valuable advice and technical help. We also thank Sylvie Farget for her constant secretarial aid.
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.