
Abstract

A
He was clearly referring to the use of viruses as viral vectors, and those words proved to be prophetic. In the first adeno-associated virus serotype 2 (AAV2) vector-mediated, liver-directed gene therapy trial for hemophilia B reported in 2006, 1 a significant immune response to AAV capsid proteins led to a complete loss of therapeutic levels of factor IX (F.IX) in one subject.
Preamble to the Problems
There is little doubt that viruses did not evolve for the purposes of delivery of therapeutic genes. Thus viral vectors, although used successfully to achieve the main objective, are unlikely to reach their full potential because of the host immune response. The use of AAV, in particular, is complicated by the following two facts: (1) the naked icosahedral capsid, which is subject to ready modification(s) by host cell enzymes; and (2) the single-stranded nature of the viral DNA, which is transcriptionally inactive.
Solution to the Capsid Puzzle
Having worked with stalwarts such as Kenneth I. Berns, MD, PhD, Nicholas Muzyczka, PhD, William W. Hauswirth, PhD, and R. Jude Samulski, PhD, on AAV2 as a virus since 1980, and subsequently as vectors, it eventually became obvious to me that the naturally occurring AAV as a virus could not be used optimally as a vector. In other words, the virus had to be modified in some way, shape, or form, such that the delivered vector was not seen by the host immune system as a virus. But how, or in what way?
Fortunately for me, this question was soon answered when my long-time colleague, Li Zhong, MD, and I, as well as others, reported in 2007 2 that after infection, the AAV2 capsid protein becomes phosphorylated at surface-exposed tyrosine residues by the cellular epidermal growth factor receptor protein tyrosine kinase. Tyrosine phosphorylation, which leads to ubiquitination, followed by proteasomal degradation of the vectors in the cytoplasm (elegantly demonstrated by Duan and colleagues), 3 provided one possible clue to circumvent this barrier. Another long-term collaborator, Mavis Agbandje-McKenna, PhD, the “structural guru” of AAV, and colleagues quickly identified the seven surface-exposed tyrosine residues on the AAV2 capsid, which were mutagenized to develop tyrosine-mutant, next-generation (“NextGen”) AAV2 vectors in 2008, 4 which circumvented the problems associated with the first-generation AAV2 vectors. Later, several of these mutations were combined to generate a “tyrosine triple-mutant,” which was not only significantly more efficient 5 but also less immunogenic. 6 A herculean effort by yet another long-term colleague, George Aslanidi, PhD, and colleagues, who mutagenized all 17 surface-exposed serines, all 15 surface-exposed threonines, and various permutations and combinations thereof, led to the identification of the most efficient AAV2 vectors to date. 7 In addition, because ubiquitination occurs on lysines, all seven surface-exposed lysine residues on the AAV2 capsid were also mutagenized. 8
Remarkably, most, if not all, of the surface-exposed tyrosine, serine, threonine, and lysine residues are highly conserved among all 10 commonly used AAV serotype vectors. However, it has proven difficult to predict which specific combination of mutations for a given AAV serotype vector would be optimal, depending on the cell type and the host species to be transduced. It has, nonetheless, become abundantly clear, as schematically illustrated in Fig. 1, that the use of capsid-modified NextGen AAV vectors is likely to overcome some of the limitations associated with the conventional AAV vectors. Indeed, three phase I clinical trials with the NextGen AAV2 vectors are currently underway 9 –11 and, it is hoped, more will follow in the not-too-distant a future.

Development of NextGen AAV vectors. Surface-exposed, specific tyrosine, serine, and threonine residues on AAV capsids can be phosphorylated, which is a signal for ubiquitination. Similarly, surface-exposed, specific lysine residues on AAV capsids can be ubiquitinated, and subsequently degraded by the host cell proteasome machinery. Site-directed mutagenesis of these residues led to the generation of NextGen AAV vectors that circumvent these problems, and are more efficient at reduced vector doses, thus minimizing host immune responses.
Solution to the Genome Puzzle
The AAV genome is composed of single-stranded DNA, and although both [+] and [–] polarity strands are encapsidated in separate mature virions with equal frequency, it is now a well-established fact that the viral second-strand DNA synthesis, rather than DNA strand-annealing, is the predominant mechanism that mediates transgene expression. 12 –15 In retrospect, it is remarkable that a therapeutic level of F.IX expression, albeit transiently, was achieved at all in the first hemophilia B trial, 1 given the well-known propensity of a vast percentage of hepatocytes to prevent viral second-strand DNA synthesis. 16 –18 Double-stranded, self-complementary AAV (scAAV) vectors, generated by the Samulski group, 19 were easily able to overcome this limitation. Indeed, the subsequent landmark successful clinical trial for hemophilia B by Nathwani and colleagues 20,21 with scAAV8 vectors is a clear testament to the fact that the use of scAAV vectors, rather than the AAV8 serotype, was largely responsible for the successful outcome, because compared with mouse hepatocytes, AAV8 vectors transduce human hepatocytes much less efficiently. 22 –24
With the exception of the second hemophilia B trial with scAAV8 vectors,
20,21
all clinical trials reported thus far with AAV vectors have been carried out with single-stranded AAV (ssAAV) vectors. In spite of that, clinical efficacy has been achieved in the potential gene therapy of four additional human diseases, including Leber's congenital amaurosis,
25
–28
lipoprotein lipase deficiency,
29
aromatic
Because not all therapeutic genes can be encapsidated in scAAV vectors, additional strategies are needed to modify the single-stranded DNA genome in some way, shape, or form, such that the delivered therapeutic gene is transcriptionally active. But how, or in what way?
Once again, fortunately for me, this question was answered when yet another long-time colleague, Keyun Qing, MD, and I, as well as others, reported in 1997 32 that a host cell protein, FKBP52, in its phosphorylated form, binds specifically to the single-stranded D sequence within the AAV inverted terminal repeat (ITR) at the 3′ end of the viral genome and strongly inhibits viral second-strand DNA synthesis, and consequently, negatively impacts transgene expression. Over the ensuing years, a number of strategies were developed that circumvented this obstacle. 33 –38 Another long-term colleague, Xu-Shan Wang, MD, and others attempted to simply delete the D sequences from the viral genome, but quickly learned that the D-sequence deletion was incompatible with genome encapsidation, thereby identifying the D sequence as the “packaging signal” for the AAV genome. 39 –41 Another colleague, Giridhara Rao Jayandharan, PhD, and others identified the presence of a putative negative regulatory element in the D sequence in the 5′ ITR, to which the NF-κB-repressing factor binds and inhibits transcription. 42 Yet another long-term colleague, Chen Ling, PhD, and others eventually developed one D sequence-deleted genome that led to the generation of the generation X (“GenX”) AAV2 vectors in 2015, 43 and documented significantly enhanced transgene expression in human cell lines in vitro as well as in murine hepatocytes in vivo.
Thus, it is clear, as schematically illustrated in Fig. 2, that the use of genome-modified GenX AAV vectors is likely to overcome the limitations associated with the conventional ssAAV vectors, once again, it is hoped, in the not-too-distant future.

Development of GenX AAV vectors. The D sequence at the 3′ end in the viral inverted terminal repeat (ITR) contains the binding site for a cellular protein, FKBP52, phosphorylated forms of which strongly inhibit viral second-strand DNA synthesis. The D sequence at the 5′ end in the ITR contains the binding site for NF-κB-repressing factor, a negative regulator of transcription. Removal of these sequences led to the generation of GenX AAV vectors that circumvent these problems, and led to more efficient transgene expression.
Future Prospects
Although it appears that both major problems—the naked capsid, subject to enzymatic modifications and degradation, and the transcriptionally inactive, single-stranded DNA genome—associated with the conventional AAV vectors have been resolved for the most part, there is still room for further improvements. The reason for this optimism is twofold. First, it is relatively straightforward to conceive of a scenario where a GenX genome is encapsidated into the NextGen capsid to generate the optimized (“Opt”) AAV vector. Such an OptAAV vector is schematically depicted in Fig. 3. Indeed, in our more recent studies, we have observed a significant increase in the transduction efficiency of both NextGen AAV2 and NextGen AAV3 vectors containing GenX AAV2 genomes both in human cell lines in vitro and in a murine xenograft model in vivo (our unpublished results).
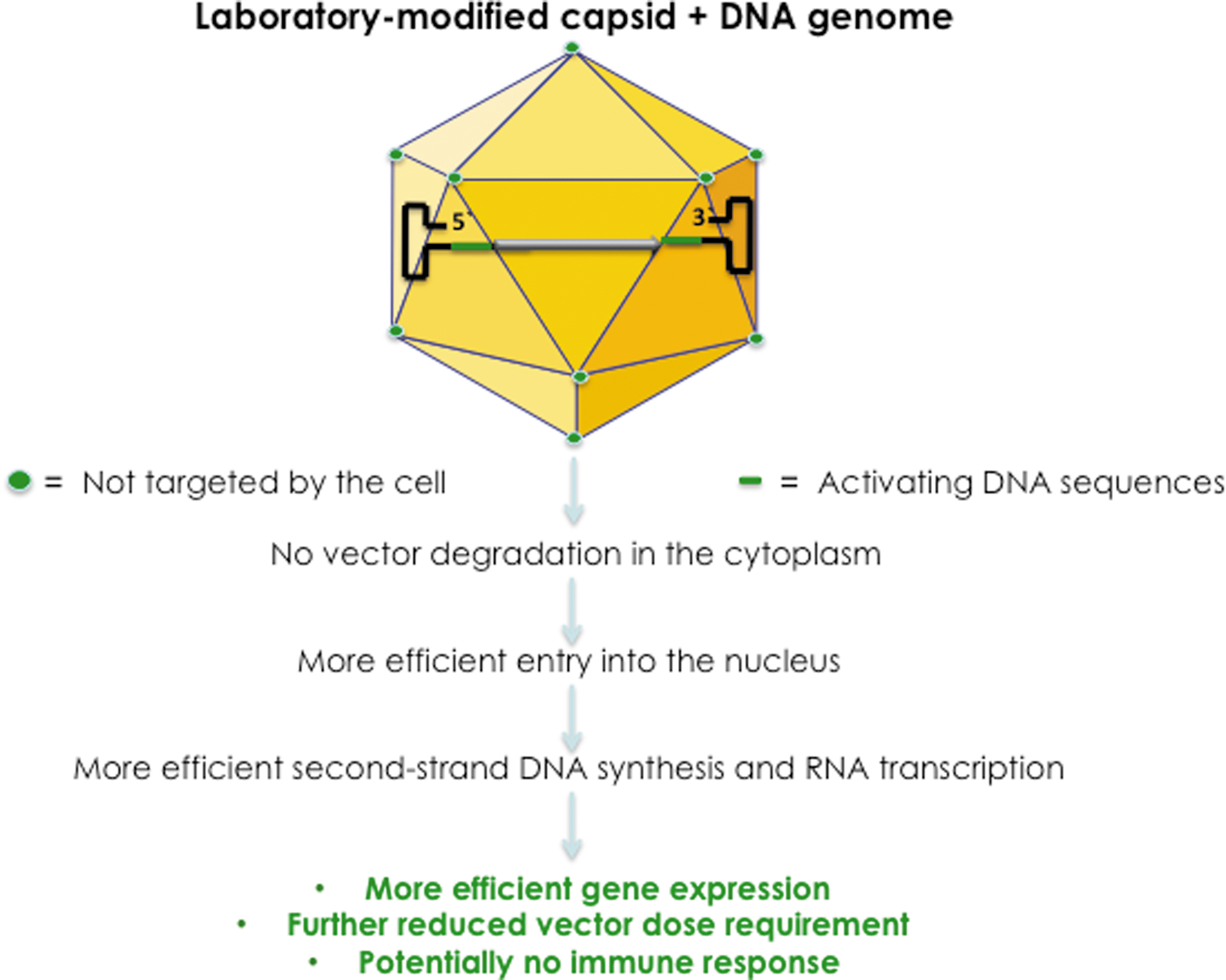
Development of OptAAV vectors. Encapsidation of GenX viral genomes into NextGen capsids led to the generation of OptAAV vectors that circumvent the problems associated with the first-generation AAV vectors, and are more efficient at further reduced vector doses, thus potentially further minimizing host immune responses.
Additional future possibilities to further increase the overall safety and efficacy of NextGen, GenX, and OptAAV vectors include the following: • Development of vectors that can overcome the preexisting immunity • Development of vectors with increased bioavailability in vivo
• Development of tissue-specific vectors • Development of vectors capable of site-specific integration
Overall Summary
As stated previously, viruses did not evolve for the purposes of delivery of therapeutic genes. Thus, gene therapy with viral vectors should be pursued only after they have been modified such that they are not targeted by, or are capable of evading, the host immune system. One review article supports the “revitalization of gene therapy,” 44 but I would suggest that the full potential of this approach is unlikely to be achieved by the first generation of viral vectors in general, and by AAV vectors in particular. Because capsid-modified NextGen AAV serotype vectors transduce cells and tissues more efficiently at lower doses, and genome-modified GenX vectors transduce cells and tissues more efficiently, combining the two to generate the OptAAV serotype vectors offers the potential advantages of being more efficient, less immunogenic, and more cost-effective. Thus, all future clinical trials in humans should be performed with NextGen, GenX, or OptAAV vectors.
To summarize, viruses did not evolve for the purposes of delivery of therapeutic genes. Thus, gene therapy with viral vectors should be pursued only after they have been modified such that they are not targeted by, or are capable of evading, the host immune system. I concur with Dr. Bluestone that we should never attempt to outsmart the immune system when it comes to viruses, but with utmost humility, I submit that we are smart enough to find ways to transiently evade the immune system when it comes to viral vectors in general, and AAV vectors in particular.
Note Added in Proof
While this article was being reviewed, the initial results of the efficacy of NextGen AAV2 vectors in the first clinical trial (Ref. 9) were reported in an article entitled: “Gene Therapy for Leber Hereditary Optic Neuropathy: Initial Results”, by Feuer and colleagues, which was published online in Opthalmology (DOI:
Footnotes
Author Disclosure
No competing financial interests exist.