
Abstract
GM2 gangliosidosis is a family of three genetic neurodegenerative disorders caused by the accumulation of GM2 ganglioside (GM2) in neuronal tissue. Two of these are due to the deficiency of the heterodimeric (α–β), “A” isoenzyme of lysosomal β-hexosaminidase (HexA). Mutations in the α-subunit (encoded by HEXA) lead to Tay-Sachs disease (TSD), whereas mutations in the β-subunit (encoded by HEXB) lead to Sandhoff disease (SD). The third form results from a deficiency of the GM2 activator protein (GM2AP), a substrate-specific cofactor for HexA. In their infantile, acute forms, these diseases rapidly progress with mental and psychomotor deterioration resulting in death by approximately 4 years of age. After gene transfer that overexpresses one of the deficient subunits, the amount of HexA heterodimer formed would empirically be limited by the availability of the other endogenous Hex subunit. The present study used a new variant of the human HexA α-subunit, μ, incorporating critical sequences from the β-subunit that produce a stable homodimer (HexM) and promote functional interactions with the GM2AP– GM2 complex. We report the design of a compact adeno-associated viral (AAV) genome using a synthetic promoter–intron combination to allow self-complementary (sc) packaging of the HEXM gene. Also, a previously published capsid mutant, AAV9.47, was used to deliver the gene to brain and spinal cord while having restricted biodistribution to the liver. The novel capsid and cassette design combination was characterized in vivo in TSD mice for its ability to efficiently transduce cells in the central nervous system when delivered intravenously in both adult and neonatal mice. This study demonstrates that the modified HexM is capable of degrading long-standing GM2 storage in mice, and it further demonstrates the potential of this novel scAAV vector design to facilitate widespread distribution of the HEXM gene or potentially other similar-sized genes to the nervous system.
Introduction
T
To study the disease mechanisms and to test various therapeutic approaches, mouse models have been developed for both these diseases. The mouse model for TSD, developed by the targeted disruption of the hexa gene encoding the α-subunit, 3 –5 has no or a mild behavioral phenotype but displays many of the neuropathological features characteristic of TSD, in particular GM2 ganglioside accumulation. The absence of clinical signs in TSD mice is due to the presence of a lysosomal mouse sialidase capable of hydrolyzing GM2 to its neutral asialo derivative, GA2, which then interacts with HexB to be further degraded to glucosylceramide. 5,6 This alternative metabolic pathway helps TSD mice to keep GM2 accumulation below an acutely toxic level and thereby escape clinical symptoms. The hexb –/– mouse model for Sandhoff disease exhibits a severe disease phenotype as it is deficient in both HexA and HexB and has extensive GM2 accumulation throughout the brain and spinal cord with a humane end point of approximately 16 weeks of age. 4,5
Several gene replacement therapies have been tested in the mouse models for these diseases. The heterodimeric nature of HexA, which is the only isoenzyme capable of degrading GM2, makes the treatment for TSD and SD more challenging. Adenoviral gene therapy of TSD mice, using a vector encoding HEXA alone, when injected intravenously produced only a slight increase in HexA (or possibly HexS mistaken for HexA) activity in the serum, whereas simultaneous injection of vectors encoding both HEXA and HEXB increased the serum HexA activity to 42% of wild-type levels in these mice. 7 SD mice injected intracranially with a recombinant adeno-associated viral (rAAV) vector expressing the β-subunit, or a combination of vectors with α- and β-subunits, showed reduction in GM2 levels and increased survival. 8 However, the effect of the single-subunit vector in this study could be attributed to higher levels of HexB and the mouse sialidase driving the alternative metabolic pathway for hydrolyzing GM2. A single striatal injection of a mixture of vectors expressing the β-hexosaminidase α- and β-subunits in SD mice prevented the neuronal loss in the brain. 9 Cachon-Gonzalez and colleagues reported that intracranial coinjection of rAAV vectors expressing the α- and β-subunits into 1-month-old SD mice prevented the disease pathology throughout the brain and spinal cord, which helped the mice survive up to 2 years. 10 In that study, an infusion of an rAAV vector encoding the β-subunit alone resulted in detectable expression of only the HexB isozyme. AAVrh8 vectors encoding feline α- and β-subunits were injected intracranially into SD cats at a 1:1 ratio, and this resulted in a significant reduction in cerebrospinal fluid (CSF) biomarkers of the disease. It ultimately resulted in extension of the life span for the treated cats. 11 Intracranial stereotaxic infusion of a mixture of rAAV2/1α and rAAV2/1β into 4-week-old SD mice extended the median survival from 131 to 615 days. 12
The gene replacement therapies so far discussed all used direct injections to the brain, using viral vectors. The possibility of translating this highly invasive delivery approach into humans is problematic simply given the size differential between the brains of animal models and humans, along with the limited spread of AAV vectors from the injection site. The emergence of AAV9 vectors for global gene transfer into the brain and spinal cord, due to its ability to penetrate the blood–brain barrier, has resulted in therapeutic approaches for multiple CNS diseases via intravascular gene delivery.
13
–17
Intravenous administration of self-complementary AAV9 vectors has been demonstrated as a viable and translatable approach to target the CNS in mice, cats, and nonhuman primates,
18
–21
and this approach is being applied in a human trial for spinal muscular atrophy (
An ideal vector capable of treating TSD and SD would express both HEXA and HEXB genes simultaneously, but this would be beyond the packaging capacity of scAAV, which is needed to achieve efficient global CNS gene transfer. Therefore, there is a conceptual advantage to creating an engineered subunit that could be delivered, using a single scAAV vector, and that could form a stable and functional homodimer when delivered in vivo and degrade GM2 in a GM2AP-dependent manner. The crystallographic structures of human HexA and HexB have been elucidated in detail. 24,25 Matsuoka and colleagues attempted to design and develop a modified HexB reported to have GM2-degrading activity and GM2AP-binding ability, based on the amino acid homology between the Hex α- and β-subunits. 26 They purified the modified HexB recombinant enzyme with the altered substrate specificity, that is, the ability to efficiently bind negatively charged substrates, and an α-loop sequence for enhancing its interaction with the GM2AP–GM2 complex. They administered the modified HexB by intracerebroventricular injection directly to the CNS of 10-week-old SD mice and observed a remarkable reduction in GM2 storage in the thalamus, cerebral cortex, and also the liver. Their results indicated that the modified HexB enzyme can function in vivo if used in enzyme replacement therapy (ERT). However, Sinici and colleagues 27 tested this construct and another more extensive hybrid β-subunit construct in cellulo, using Tay-Sachs cells preloaded with a fluorescent GM2 derivative, and found that neither modified β-subunit was capable of significant human GM2AP-dependent hydrolysis of GM2. Likely, the encouraging results reported by Matsuoka and colleagues are attributable to restoration of HexB-like activity that takes advantage of the alternative sialidase pathway in mice. As an alternative approach to develop a hybrid subunit, Tropak and colleagues 28 designed a modified α-subunit incorporating both the unique β-subunit sequences required to form a stable, HexB-like, homodimer and those sequences needed for the homodimer to interact with the GM2AP–GM2 complex. The α-active site was retained in order to facilitate the hydrolysis of negatively charged substrates, for example, GM2. This modified subunit (μ) was confirmed to form a stable dimeric enzyme, HexM, which could bind to the GM2AP–GM2 complex, and degrade GM2 in live human embryonic kidney (HEK) cells rendered HEXA–/– and HEXB–/– by CRISPR (clustered regularly interspaced short palindromic repeats) gene editing. In vivo experiments in this study further demonstrated the ability of HexM to reduce accumulated GM2 in Tay-Sachs and Sandhoff mice. Therefore, this homodimer has HexA-like activity and can be expressed from a single gene packageable within an scAAV vector. This construct is discussed further in the present study.
It is important to note that systemic delivery of AAV9 vectors results in substantially higher biodistribution to the liver compared with the CNS, which could lead to transgene-specific liver toxicity. 15,29 Furthermore, some studies have reported incidence of liver tumors with intravenous delivery of AAV9 into neonates. 14,30,31 A liver-detargeted version of AAV9 with comparable levels of brain and spinal cord transduction could be a safer reagent to treat neurological disorders. Pulicherla and colleagues 32 reported the generation of AAV9-derived vectors displaying selective loss of liver tropism. One AAV9 variant, denoted as AAV9.47 with four point mutations (S414N, G453D, K557E, and T582I), showed a significant 110-fold decrease in the liver transduction in mice injected via the tail vein and analyzed 4 weeks postinjection by vector DNA biodistribution and transgene (luciferase) expression. The transduction pattern in the brain did not show any significant drop, which suggests AAV9.47 as a good candidate for global gene delivery to the CNS via systemic injection.
In the present study, we discuss the functional ability of the new hexosaminidase variant HexM and the design of a novel expression cassette to package it into an scAAV vector for in vivo applications. We show that the novel cassette has the ability to provide ubiquitous transduction throughout the CNS. We provide further characterization of the liver-detargeted AAV9.47 variant to demonstrate its ability to transduce neurons and glia throughout the CNS after intravenous injection into neonatal and adult mice. We demonstrate that the HexM homodimer is capable of degrading GM2 in a localized manner when injected intracranially into adult TSD mice, and that the scAAV9.47/HEXM vector can mediate long-term GM2 reduction when injected intravenously into neonatal TSD mice.
Materials and Methods
Plasmid constructs
The plasmids carrying the transgene cassette required for packaging into scAAV vectors were designed for GFP, HEXA, and HEXM. This novel design includes the combination of a short synthetic and ubiquitous promoter derived from the JeT promoter
33
and a synthetic intron (UsP, also referred to as the JeTI promoter) along with the simian virus 40 polyadenylation signal [SV40 poly(A)] to drive expression of the encoded gene (Fig. 1). A Kozak consensus sequence was incorporated at the start codon. Pilot in vitro studies showed that the UsP expression cassette had approximately 15% of the activity when using a CMV promoter (Supplementary Fig. S1; supplementary methods and data are available online at
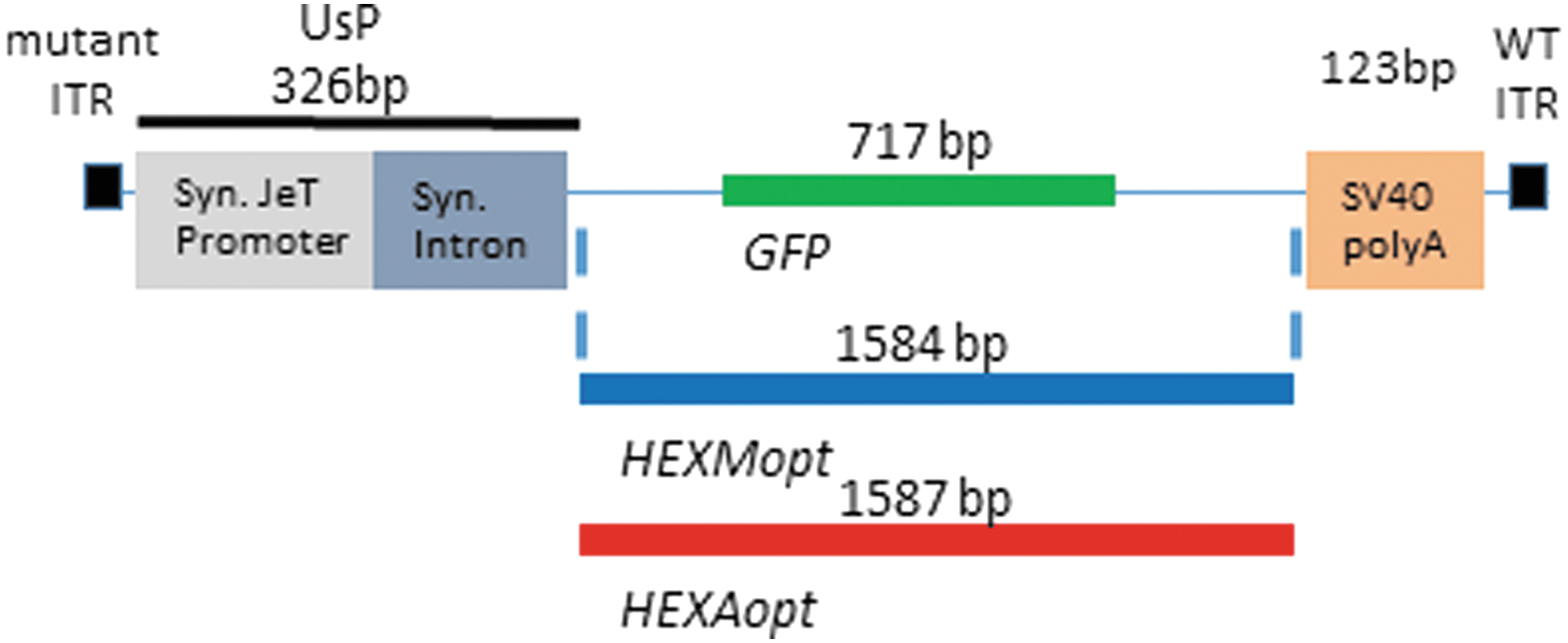
Adeno-associated viral (AAV) construct diagrams. The expression cassette design for packaging GFP and codon-optimized versions of human HEXA and HEXM included a novel synthetic promoter–intron combination (UsP) along with the simian virus 40 polyadenylation signal (SV40 polyA) to drive expression. These elements are flanked by the mutant inverted terminal repeat (ITR) on the 5′ end and the wild-type (WT) AAV2 ITR on the 3′ end to package into a self-complementary AAV vector. Color images available online at
The human HEXA and HEXM genes were codon-optimized by DNA2.0 (Menlo Park, CA). The construct expressing GFP from the CBh promoter 22 with the bovine growth hormone (BGH) poly(A) was used as a reference standard for comparative gene expression for the newly designed constructs.
Vector preparation
Recombinant AAV vectors were generated as described,
34
using proprietary methods developed at the University of North Carolina Gene Therapy Center Vector Core facility (Chapel Hill, NC). The following AAV9 or AAV9.47 vectors were produced and used for this study: AAV9/CBh-GFP, AAV9.47/CBh-GFP, AAV9/UsP-GFP, AAV9/UsP-HEXA, AAV9/UsP-HEXM, AAV9.47/UsP-GFP, and AAV9.47/UsP-HEXM. All of these were scAAV vectors. Peak fractions were dialyzed in phosphate-buffered saline (PBS) containing 5%
Mouse studies
All investigations were approved by the University of North Carolina–Chapel Hill Institutional Animal Care and Use Committee. The Tay-Sachs disease mouse model [strain B6;129S-hexa –/–] 3 and wild-type C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and bred at the University of North Carolina.
For intravenous delivery into wild-type adult female C57BL/6 mice, 200 μl of scAAV9/GFP (2 × 1011 VG) or AAV9.47/GFP (2 × 1011 VG) was injected and expression/biodistribution was analyzed 3–4 weeks postinjection. Adult TSD knockout mice were injected via the tail vein with scAAV9.47/GFP (5 × 1011 VG) in a volume of 200 μl when 4–5 months old and were analyzed 6 months postinjection.
Neonatal facial vein injections into TSD mice were performed on postnatal days 0–2 (P0–P2), using scAAV9.47/UsP-GFP (5 × 1010 VG) or scAAV9.47/UsP-HEXM (5 × 1010 VG) in a volume of 20 μl. The injections were carried out before the genotypes of the mice were determined and as such included both heterozygous and knockout mice. They were euthanized when 15 months old.
Intracranial injections were performed as described 35 into 15-month-old TSD knockout mice, using the following stereotaxic coordinates, relative to bregma: rostral/caudal, +0.5 mm (toward rostral); left/right: left, +3 mm; up/down, −4 mm. These mice received a mixture of AAV9/GFP (1 × 109 VG) and either AAV9/HEXM (1 × 109 VG) or AAV9/HEXA (1 × 109 VG) in a total volume of 1 μl delivered over 10 min. Two mice were injected per vector mixture. They were analyzed 4 weeks postinjection. At sacrifice, the mice were transcardially perfused with PBS containing heparin at 1 U/ml (Abraxis, Schaumburg, IL) and samples were collected for downstream analysis.
Immunohistochemistry
After 48 hr of fixation in freshly made PBS containing 4% paraformaldehyde, the entire brain and portions of the cervical and lumbar spinal cords were sectioned at 40 μm, using a Leica vibrating microtome. Every fifth section was processed for immunohistochemistry (IHC). Sections were treated with hydrogen peroxide to remove endogenous peroxidase activity before blocking. Samples were incubated for 1 hr at room temperature in blocking solution (10% goat serum, 0.1% Triton X-100, 1 × PBS) and then incubated for 48–72 hr at 4°C in primary antibody solution (3% goat serum, 0.1% Triton X-100, 1 × PBS, and primary antibody). Primary antibodies were as follows: rabbit anti-GFP (diluted 1:1000; #AB3080, Millipore, Bedford, MA) or the human anti-GM2 ganglioside antibody (diluted 1:1000; KM966, gift from Kyowa Hakko Kirin, Tokyo, Japan). After incubating with the appropriate secondary antibody for 1 hr at room temperature, color development was performed with a VECTASTAIN Elite ABC kit (#PK-6100; Vector Laboratories, Burlingame, CA) with 3,3′-diaminobenzidine tetrachloride (DAB) (#04008; Polysciences, Warrington, PA) substrate and nickel–cobalt intensification of the reaction product.
DAB-processed brain sections were digitized with a ScanScope slide scanner (Aperio Technologies, Vista, CA). Virtual slides were viewed with the ImageScope software package (version 10.0; Aperio Technologies).
For the data provided in Supplementary Fig. S3B, a region of interest was created in three representative images of an identical cortex area of the brain per vector treatment, and the number of astrocytes and neurons (identified by morphology) were manually counted. The results from two independent counters were averaged. Data are expressed as number of cells per millimeter squared.
Quantitative PCR analysis of vector genomes
Quantitative PCR (qPCR) was used for vector biodistribution studies. Tissue DNA was purified and quantified as described. 18 Data are reported as the number of double-stranded GFP or HEXM DNA molecules per two double-stranded copies of the murine LaminB2 locus, or in other words, the number of vector DNA copies per diploid mouse genome.
Biochemical detection of GM2 gangliosides
Gangliosides were extracted by a modification of a method described earlier. 36 Briefly, previously frozen murine mid-brain sections were weighed and sonicated in 0.5 ml of methanol at amplitude 15–20% for 3 × 10 sec pulses with cooling on ice between pulses (Sonic Dismembrator model 500; Fisher Scientific, Waltham, MA). Samples were centrifuged for 15 min at 10,000 rpm at 4°C, and 0.4 ml of supernatant was removed for enzyme activity studies; the remainder was suspended (including the pellet) up to a final volume of 1.5 ml of methanol added to 2.5 ml of chloroform. Acidic gangliosides were isolated using C18-E columns (Phenomenex, Torrance, CA) and eluted sequentially in 2 ml of methanol, and then in 2 ml of a 1:1 methanol–chloroform mixture. The eluate was then evaporated under a stream of nitrogen, and the residue was resuspended in 1:1 methanol–chloroform and then applied to an HPTLC silica gel 60 plate (Merck, Darmstadt, Germany) by applying 10 μl at a time and drying between applications. Plates were developed with running buffer (chloroform–methanol–CaCl2, 0.22% [55:45:10]). After staining with orcinol (Sigma-Aldrich, St. Louis, MO) and baking at 105°C for 10 min, plates were immediately scanned for subsequent spot densitometric analysis (ImageJ software), with a mix of monosialoganglioside standards (Matreya, Pleasant Gap, PA) alongside. The ganglioside ratios are presented as GM2:total gangliosides.
Results
Liver-detargeted AAV9.47 capsid transduces neural cells after intravenous administration in adult mice
AAV9.47 capsid was engineered as described. 32 It was categorized as a liver-detargeted version of the AAV9 capsid, with a significant 110-fold decrease in liver transduction relative to the naturally occurring AAV9 capsid. The level of overall brain transduction was comparable to that of the original AAV9 capsid, making it a good candidate for targeted delivery to brain and other CNS organs via the intravenous route while avoiding the high biodistribution of AAV9 to the liver. The liver-detargeted characteristics of the AAV9.47 vector reported previously were confirmed in our study by a biodistribution study (Supplementary Fig. S3A). However, previous studies did not identify which cell types were transduced by AAV9.47 within the CNS. In our study, we observed that the capsid is able to transduce neural cells, predominantly neurons, when delivered intravenously into adult wild-type mice (Fig. 2B and Supplementary Fig. S4). GFP expression was visible throughout various regions of the brain and spinal cord. When compared with the transduction profile of AAV9 capsid packaged with the same transgene cassette containing GFP under the control of the CBh promoter and injected into wild-type mice (Fig. 2A), there was an approximately 4-fold reduction in the number of astrocytes transduced by the AAV9.47 capsid and an approximately 50% increase in the number of transduced neurons in the cortex (Supplementary Fig. S3B). Overall, AAV9.47 transduced approximately 20% less cells than AAV9 in the cortex.

CNS transduction pattern of AAV9/CBh-GFP, AAV9.47/CBh-GFP, and AAV9/UsP-GFP after intravenous administration in wild-type mice. Adult wild-type mice were injected intravenously with 2 × 1011 vector genomes (VG) of scAAV9/CBh-GFP
Synthetic UsP promoter–intron drives ubiquitous expression in the brain
Previously, our laboratory described the use of a modified β-actin promoter with a shortened MVM (minute virus of mice) intron to drive sustained high levels of transgene expression in both neurons and glia throughout the CNS. 22 Combined with the BGH poly(A) signal, this scAAV construct can package a transgene of up to approximately 1.2 kb in length. The HEXA and HEXM transgenes are approximately 1.6 kb in length, requiring a novel expression cassette to provide efficient and ubiquitous expression within the contexts of an scAAV genome. The newly designed expression cassette (Fig. 1), using the synthetic UsP promoter–intron combination, paired with the SV40 poly(A) signal, is capable of widespread expression in the CNS (Fig. 2C and Supplementary Fig. S4) after intravenous delivery in wild-type mice. The intensity of GFP expression in astrocytes is noticeably decreased for the UsP-GFP construct, compared with the CBh-GFP construct, when both were packaged in the AAV9 capsid. However, the novel design has the advantage of being capable of packaging a transgene up to approximately 1.7 kb within an scAAV vector, which will accommodate the HEXA or HEXM transgene.
AAV9.47 capsid is capable of widespread gene transfer to the brain after intravenous administration in both adult and neonatal Tay-Sachs mice
To assess the ability of the UsP construct to mediate long-term sustained expression in the Tay-Sachs mouse model, we packaged the newly designed UsP-GFP construct within the AAV9.47 capsid and injected it intravenously into adult TSD mice at 4–5 months of age and into neonatal mice, consisting of both TSD and their heterozygous littermates, at age P0–P2. These mice were euthanized 6 and 15 months postinjection, respectively, and were analyzed for the biodistribution of the AAV vector by qPCR and for CNS transduction by IHC against GFP. As previously reported, the vector was significantly detargeted from the liver compared with the biodistribution pattern of AAV9, 18,32 while retaining biodistribution to the brain and spinal cord (Figs. 3A and 4A). IHC images of the brain clearly showed that the AAV9.47 capsid can deliver the gene to all regions of brain and facilitate expression in neurons and glia within the CNS of hexa –/– and hexa +/– mice, and that this expression persisted for 15 months (Figs. 3B and 4B).

Biodistribution and brain transduction after intravenous administration of AAV9.47/UsP-GFP in adult Tay-Sachs mice. Adult Tay-Sachs knockout mice were injected intravenously when 4–5 months old with 5 × 1011 VG of scAAV9.47/UsP-GFP and analyzed 6 months postinjection.

Vector biodistribution and immunohistochemical analysis after intravenous administration of scAAV9.47/UsP-GFP in neonatal Tay-Sachs mice. Tay-Sachs mice, both knockout (KO) and heterozygous (het), were injected via the facial vein on postnatal days 0–2 (P0–P2) with 5 × 1010 VG of scAAV9.47/UsP-GFP and analyzed 15 months postinjection.
The hexosaminidase variant, HexM, can clear GM2 storage similar to HexA after intracranial administration into aged Tay-Sachs mice
To directly test the expression of the new hexosaminidase gene variant, HEXM, to reduce long-standing accumulated GM2 in the TSD mouse brain, compared with that of the unmodified HEXA gene, these were packaged into scAAV9 vectors and injected stereotaxically into 15-month-old TSD mice along with an identical titer of scAAV9/UsP-GFP vector to track vector spread. Each experimental group consisted of two animals receiving the same vector combination. The mice were euthanized 4 weeks postinjection and the brain sections were subjected to IHC analysis against GFP and GM2 simultaneously. Several paired images of similar brain regions were analyzed. The HexA-like activity was assessed by the clearance of GM2 within the injected region, compared with the contralateral brain hemisphere (Fig. 5E–G). The area of AAV transduction was visualized by staining for GFP (Fig. 5A–C). Qualitatively, a marked reduction of GM2 was apparent in the areas that overlapped with strong GFP expression. Both β-hexosaminidase vectors were able to degrade GM2 in the regions that were injected, but complete clearance of accumulated GM2 was not obtained by either vector. IHC analysis was done on brain sections from age-matched untreated heterozygous mice (Fig. 5D and H) for comparison with the GM2 levels in an unaffected brain. From these experiments, we conclude that delivery of scAAV9/UsP-HEXM results in clearance of long-standing GM2 accumulations, at least as well as that after delivery of scAAV9/UsP-HEXA.
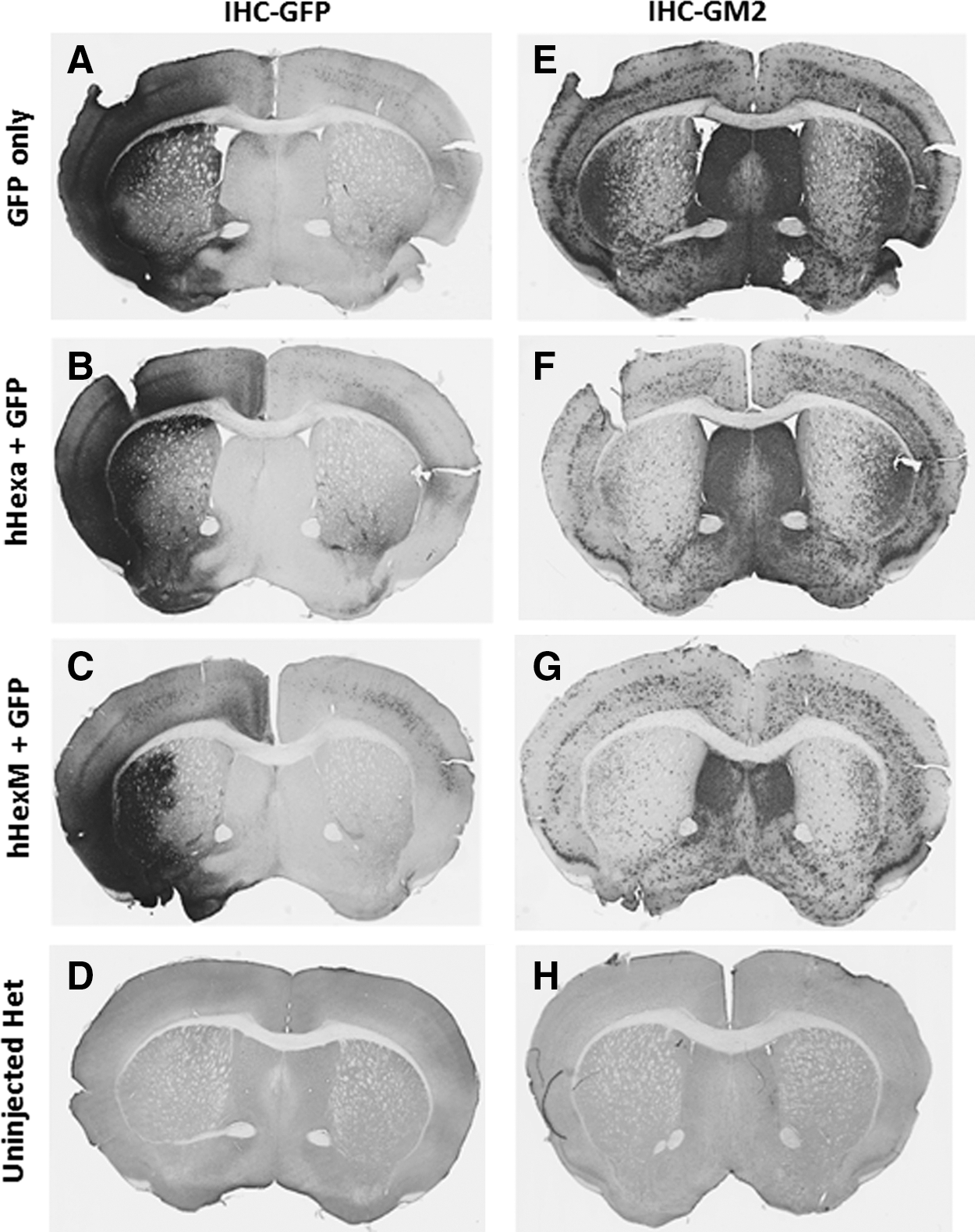
HexM is capable of reducing long-standing GM2 in aged Tay-Sachs mice. Fifteen-month-old TS knockout mice received a single unilateral intracranial injection of a two-vector mixture containing equal amounts of scAAV9/UsP-GFP and scAAV/UsP-HEXA or scAAV9/UsP-HEXM. They were killed 4 weeks postinjection and coronal sections of the brain were analyzed by IHC for GFP expression to indicate the spread of vector
Intravenous administration of AAV9.47/HEXM vector into neonatal Tay-Sachs mice results in long-term CNS reduction in GM2 gangliosides
Injections of AAV9.47/UsP-HEXM (at a dose of 5 × 1010 VG, through the facial vein) into neonatal TSD mice on P0–P2 were done; the mice included both knockout and heterozygous mice and were euthanized when 15 months old. The pattern of vector genome biodistribution when HEXM was the transgene was variable between individual mice but overall similar to the biodistribution pattern when GFP was the transgene (Fig. 6A; compare with Fig. 4A). Biochemical analysis of both rostral and caudal parts of the brain from treated mice showed a reduction in the accumulation of GM2 gangliosides compared with that of control uninjected TSD mice (Fig. 6B). The caudal (hind) brain had a more marked quantitative reduction in GM2 storage than the rostral brain (data not shown). This demonstrates the ability of the scAAV9.47/UsP-HEXM vector to mediate sustained reduction in CNS GM2 gangliosides, the hallmark of TSD pathology.

Intravenous administration of AAV9.47/UsP-HEXM in neonatal Tay-Sachs mice results in long-term reduction in GM2 ganglioside levels. Tay-Sachs mice, both knockout and heterozygous, were injected via the facial vein on P0–P2 with 5 × 1010 VG of scAAV9.47/UsP-HEXM and analyzed 15 months postinjection.
Discussion
This paper addresses several challenges in creating an ideal gene replacement therapy for the related Tay-Sachs and Sandhoff diseases. Past studies have relied on single-stranded (ss)AAV, herpes simplex virus, or adenoviral vectors administered intracranially, to deliver a single Hex subunit with one vector or both subunits by a dual-vector approach. 7 –10,12,14,37 –41 One challenge is that the functional HexA enzyme consists of the α- and β-subunits, which from a gene transfer perspective complicates a simple gene replacement strategy because overexpression of one subunit will inevitably lead to the other endogenous subunit becoming rate-limiting. The HEXM gene, which encodes the modified Hex α-subunit variant, μ, overcomes this challenge because the Hex μ-subunit can form a stable homodimer, HexM, which functionally replaces the HexA heterodimer. 28 Overexpression of HexM has been shown to result in its enhanced secretion, and secreted HexM is taken up by other deficient cells through plasma membrane mannose 6-phosphate receptors. In addition, unlike HexA, both active sites in the HexM homodimer can potentially bind the GM2AP–GM2 complex and hydrolyze GM2. Thus, overexpression of HexM should exert a greater therapeutic benefit through cross-correction than would be possible through overexpression of either the α- or β-subunit alone, which would result predominantly in the secretion of either HexS or HexB, respectively. Furthermore, the HEXM gene can be packaged within a single scAAV vector, eliminating the need for a relatively inefficient dual-vector approach.
The HEXA and HEXB genes are each approximately 1.6 kb in length. Packaging them both within a single AAV vector is theoretically possible using traditional ssAAV vectors, but impossible within an scAAV vector. Strategies to achieve widespread CNS gene transfer in rodents, pigs, cats, dogs, and nonhuman primates have been described using AAV9, AAVrh10, and AAVrh8 vectors administered either intravenously or into the CSF, but these strategies rely on the more efficient scAAV. 16 –19,21,34,42 –49 The 1.6-kb size of the HEXM gene could potentially allow the HEXM gene to be packaged within an scAAV vector, making such a vector more amenable to these approaches for widespread CNS gene transfer. The selection of small promoters and poly(A) signals to package a 1.6-kb gene within the approximately 2.2-kb packaging constraints of scAAV is quite limited, so we employed a novel synthetic promoter–intron combination (UsP) based on the previously described synthetic JeT promoter, 33 to provide a moderate level of expression when combined with the SV40 poly(A) signal. When driving GFP, the UsP construct displays similar ubiquitous expression throughout the CNS compared with the previously described CBh promoter, 22 although expression in astrocytes appears diminished somewhat. We conclude that the UsP promoter–intron, combined with the SV40 poly(A) signal, provides an ideal expression construct for HEXM within an scAAV vector.
A potential challenge related to intravenous administration of AAV vectors (AAV9, AAVrh10, or AAVrh8) targeting the CNS is that the high vector doses required also result in extraordinarily high loads of gene transfer to the liver. This creates the potential for deleterious liver-specific T cell responses, 50,51 possible toxicity related to transgene overexpression, and hepatocellular carcinoma when administered to neonates. 30,31 By using the AAV9.47 capsid, biodistribution to the liver was highly reduced relative to AAV9, while preserving the ability of AAV9 to cross the blood–brain barrier. An apparent reduction in astrocyte transduction with AAV9.47 was noted relative to AAV9, which may necessitate a slightly higher overall dose. However, in diseases that benefit from cross-correction, primary expression in neurons should be sufficient. Regarding the potential for hepatocellular carcinoma, our use of an enhancer-less expression construct would be expected to reduce the potential for oncogenesis. Whether as a result of the reduced liver biodistribution or the lack of an enhancer, our cohorts injected intravenously as neonates did not develop tumors up to their planned end point of 15 months old. Although this observation should be examined more carefully, it is encouraging. The time course of infantile TSD and SD in humans is rapid, and the earliest possible age of intervention would be ideal. Although it is unclear whether the risk of hepatocellular carcinoma with AAV vector observed in mice could translate to humans, our vector design potentially reduces that hypothetical risk.
The efficacy of gene therapy in mouse and cat models of GM2 gangliosidosis is susceptible to misinterpretation because of the sialidase metabolic by-pass pathway, which could significantly reduce GM2 storage levels with any increase in hexosaminidase activity (i.e., HexA, HexB, HexS, or HexM). This by-pass pathway does not exist in humans. The studies reported here were conducted in the TSD model, which is not ideal to evaluate treatment efficacy because of the mild phenotype of the mice. This paper provides information on the development of the reagents, along with evidence supporting the hypothesis that this has a likelihood of providing a therapeutic benefit. A companion article by Osmon and colleagues, 52 in this issue of Human Gene Therapy (see p. 497), describes the application of this approach and vector design with a mouse model of SD, which does display a severe disease phenotype, and is more appropriate for evaluating treatment efficacy.
Several technological advances were combined to generate a reagent to treat Tay-Sachs and Sandhoff diseases. This single scAAV vector design enables delivery approaches for widespread CNS gene transfer. Key to making use of scAAV are the novel hexosaminidase variant, HEXM, and the short synthetic promoter–intron UsP. Finally, by using an enhancerless expression construct and a liver-detargeted capsid, we suggest that the possible risk of hepatocellular carcinoma with this vector design would be greatly diminished. In conclusion, the collective body of work presented herein describes a novel vector design that greatly enhances the potential success of a gene transfer strategy to treat both Tay-Sachs and Sandhoff diseases. This vector design may also be useful to deliver other genes of similar size to treat CNS disorders.
Footnotes
Acknowledgments
This research was funded by the New Hope Research Foundation (
Author Disclosure
S.G. has received patent royalties from Asklepios Biopharma that are not directly related to these studies. D.M. and B.M. are inventors on a pending patent for the sequence and potential uses of HexM. Credit is given to S.G., J.K., and M.K. for the design and initial characterization of the UsP construct. J.K. and M.K. are inventors on a pending patent for UsP.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.