
Abstract
GM2 gangliosidosis is a group of neurodegenerative diseases caused by β-hexosaminidase A (HexA) enzyme deficiency. There is currently no cure. HexA is composed of two similar, nonidentical subunits, α and β, which must interact with the GM2 activator protein (GM2AP), a substrate-specific cofactor, to hydrolyze GM2 ganglioside. Mutations in either subunit or the activator can result in the accumulation of GM2 ganglioside within neurons throughout the central nervous system. The resulting neuronal cell death induces the primary symptoms of the disease: motor impairment, seizures, and sensory impairments. This study assesses the long-term effects of gene transfer in a Sandhoff (β-subunit knockout) mouse model. The study utilized a modified human β-hexosaminidase α-subunit (μ-subunit) that contains critical sequences from the β-subunit that enables formation of a stable homodimer (HexM) and interaction with GM2AP to hydrolyze GM2 ganglioside. We investigated a self-complementary adeno-associated viral (scAAV) vector expressing HexM, through intravenous injections of the neonatal mice. We monitored one cohort for 8 weeks and another cohort long-term for survival benefit, behavioral, biochemical, and molecular analyses. Untreated Sandhoff disease (SD) control mice reached a humane endpoint at approximately 15 weeks, whereas treated mice had a median survival age of 40 weeks, an approximate 2.5-fold survival advantage. On behavioral tests, the treated mice outperformed their knockout age-matched controls and perform similarly to the heterozygous controls. Through the enzymatic and GM2 ganglioside analyses, we observed a significant decrease in the GM2 ganglioside level, even though the enzyme levels were not significantly increased. Molecular analyses revealed a global distribution of the vector between brain and spinal cord regions. In conclusion, the neonatal delivery of a novel viral vector expressing the human HexM enzyme is effective in ameliorating the SD mouse phenotype for long-term. Our data could have implications not only for treatment of SD but also for Tay–Sachs disease (α-subunit deficiency) and similar brain disorders.
Introduction
G
The incidence of TSD and SD is rare but is considerably higher in some populations, such as those of Ashkenazi Jewish or French Canadian decent. 3 –5 Clinically, TSD and SD are subdivided into three categories based on the severity of the resulting phenotype. The severity is inversely correlated with the age of onset of symptoms and the residual HexA activity. 6,7 A mutation resulting in a complete enzyme deficiency produces a severe, acute, infantile form of the disease. Infants progress normally through the developmental stages until approximately six months of age, at which point development regresses and the main symptoms of neurodegeneration appear: motor spasms, seizures, loss of hearing, loss of sight, and, in severe cases, paralysis. 1,2,8 These infants do not typically survive past 4 years of age. 8 Individuals with a mutation resulting in diminished HexA activity (2–10% of normal) will develop the juvenile or adult forms of GM2 gangliosidosis, which have a delayed onset of symptoms. 6 It has been proposed that raising the level of functioning enzyme above a “critical threshold of activity” will prevent and possibly even reverse substrate storage, thus preventing further neurodegeneration. 9 Residual HexA activity between 10% and 15% of normal levels is estimated to correspond to this critical threshold. 7
The SD mouse model (β-subunit knockout [KO]) is similar in many ways to the human infantile phenotype of SD and TSD, including motor spasticity, tremors, and occasionally muscular rigidity. 10 –12 The TSD mouse model (α-subunit knockout) manifests a very mild phenotype because of an alternative GM2 ganglioside degradation pathway that is available in mice. This pathway, which utilizes sialidase and the HexB isoenzyme in mice, is not functional in humans. 10,12 GM2 gangliosides begin accumulating in the brain of the SD KO mice from birth. Symptoms of the disease begin manifesting around 12 weeks of age and progress until the mice reach their humane endpoint between 14 and 16 weeks, because of the severity of their motor impairments. Overall, the behavioral tests comparing SD mice to their heterozygous or wild-type controls show that the SD mice lose their ability to coordinate and perform movements at a relatively steady pace over the progression of the disease. 12 On the rotarod apparatus, symptomatic SD mice significantly underperform compared with heterozygous and wild-type controls. 12,13 In a measure of general locomotor activity, the SD mice travel significantly less distance in an open field test (OFT) than their normal littermates. 14
Viral gene therapy stands as a promising therapeutic tool for TSD and SD. There have been numerous attempts to identify a suitable gene vector and delivery method capable of transducing cells throughout the CNS. Vectors delivered by direct brain intraparenchymal injection have been shown to be highly efficient at transducing cells local to the site of injection, but the number of injection sites required for uniform distribution throughout the CNS limits the practicality of this method in human applications. Recent studies have demonstrated that intravenously delivered self-complementary AAV serotype 9 (scAAV9) is effective at broadly transducing cells within the CNS as it can cross the blood–brain barrier (BBB) and transduce both neurons and glia. 15 –19
A potential drawback to intravenous AAV delivery is the large uptake of the vectors by the liver. 15,20 –23 Associated with a high liver uptake is an increased probability of hepatocellular nodule/tumor formation when administered to neonatal mice. 14,23 The recent advances in AAV technology have enabled the creation and testing of chimeric AAV capsids combining aspects from multiple serotypes in order to achieve greater target specificity. 15,16,24,25 A variant of AAV serotype 9, the AAV9.47 capsid, which de-targets the liver, 21 can potentially make more vector available for CNS uptake.
A challenge in creating a gene vector using scAAV9 is in overcoming its genome size limitations. Although the overall genome size limitation of AAV is approximately 4.7 kb, the transgene and regulatory sequences must be less than approximately 2.2 kb to allow self-complementary packaging. The self-complementary packaging of both the HEXA and HEXB genes (total ∼3.2 kb) in a single vector is therefore not possible. 26,27 To resolve the packaging capacity dilemma, a single subunit was recently designed to combine the key elements of both the human α and β subunits to create a single hybrid subunit, μ, which can homodimerize and form a new stable isoenzyme, HexM. This HexM enzyme has been shown to be capable of catabolizing the GM2 ganglioside substrate in association with the human GM2AP. 28 The gene for this new μ-subunit, HEXM, is ∼1.6 kb, and, combined with short regulatory sequences, is able to fit within the scAAV packaging constraints. 29
The current study evaluates the therapeutic effects of systemic delivery of scAAV9.47/HEXM vector in the neonatal SD mouse model. We sought to examine the phenotypic and biochemical effects of a single vector injection. The primary aim of this study was to determine whether the newly developed HexM enzyme, which was derived from the human β-hexosaminidase α-subunit, can function with the murine GM2AP and provide behavioral and survival benefit. We found that the single intravenous injection of scAAV9.47/HEXM in neonatal SD mice delayed the phenotype onset and significantly extended survival to a median age of 40 weeks.
Methods
Experimental animals
The SD mouse model was purchased from Jackson Laboratories and a colony was established at Queen's University through heterozygous breeding. Genotypes were determined via PCR amplification of DNA extracted from ear punches. Experimental animals were obtained through mutant crosses. Animals were housed on a 12-hour light cycle. All experimental protocols and procedures were performed in accordance with and were approved by the Queen's University Animal Care Committee. Table 1 shows the cohorts, number of animals, treatment, dose of AAV, and age at sacrifice.
Design of the study
rAAV vector, GFP, and HEXM plasmid
The vectors containing the green fluorescent protein (GFP) and HEXM transgenes in self-complementary rAAV vectors were produced using the previously described methods 30 by Dr. Steven Gray's laboratory at the Gene Therapy Center, University of North Carolina at Chapel Hill. The expression cassette utilized a synthetic UsP promoter with an SV40 polyadenylation signal. 29 These constructs were packaged within an AAV9.47 capsid. 21
Injections
Neonatal mice were intravenously injected through the superficial temporal vein on day 0–1 as previously described. 14 The dames were removed from the pups and placed into a new clean cage during the injections. The cage of pups was placed on a heating pad to keep them warm. The pups each received a single injection of 5.0 × 1010 vector genomes (vg) (approximately 5 × 1013 vg/kg) of either scAAV9.47/HEXM or scAAV9.47/GFP (suspended in 350 mM NaCl phosphate buffer +5% sorbitol) in a total volume of 50 μl or a single injection of 50 μl of 1 × PBS. After the injections, the dame was returned to her home cage with the pups, and all the pups were monitored closely for several days postinjection to ensure survival.
Behavioral testing
Mice underwent a series of behavioral tests monthly from 12 weeks until their humane endpoint. Behavioral testing was performed in the morning during their light cycle. To assess motor coordination, mice were tested on a Rotarod (RR) apparatus (IITC Life Sciences) using an acceleration protocol. The rod accelerated from 4 to 40 rotation per minute (RPM) over the course of 5 min, and the latency to fall, end RPM, and distance traveled were recorded as measures of motor coordination. Each mouse was given three trials on the RR, spaced out by 10 min rest intervals, and the best of the trials was recorded each month for data analysis. If a mouse happened to complete the full 5 min on the RR, it received a score of the full 5 min and did not require subsequent trials. To evaluate motor strength, the mice participated in the mesh test. The mice were placed on a mesh they could grasp, and were flipped to hang upside down over an empty box approximately 20 cm high for up to a minute. Each mouse was given three trials, each spaced out by about 5 min, of which the best trial was recorded for data analysis. If a mouse made it to a minute, they required no further trials and their best time of a minute was recorded for that month. Finally, the mice were also observed in the OFT (ActiMot; TSE Systems) for an overall measure of locomotor activity, measured through time moving. Each mouse was observed in the OFT for one 5 min trial.
Tissue and serum collection and processing
Tissues were collected at 8 weeks for the short-term cohorts and at the long-term humane endpoint. The humane endpoint for the animals was determined one of two ways: (1) the loss of >15% of their body weight, or (2) the appearance of symptoms that would impede and deteriorate the quality of life of the animal (i.e., severe tremors/shakiness, inability to right itself). For comparison of the terminal long-term endpoints, untreated 12-, 14-, and 16-week SD KO mice tissues were collected. The mice were euthanized via CO2 asphyxiation followed by cardiac puncture, and then perfused with 1× PBS. For histological analysis, a section of the midbrain was taken before separating the rest of the brain into its respective sections. Midbrain sections were fixed in paraformaldehyde overnight, followed by immersion in 100% ethanol for 24 hr before being paraffin embedded. The brain and spinal cord regions were then divided based on rostral, middle, and caudal brain, and cervical and lumbar spinal cord visualizations. Visceral organs collected included the heart, lungs, liver, spleen, kidney, gonads, as well as a sample of arm muscle. All tissues collected were frozen at −20°C for further processing, including copy number analysis. Midbrain samples were used for all biochemical analysis of hexosaminidase activity and ganglioside storage. Half of the frozen midbrains were sonicated with 700 μl of 1× PBS and spun down at 4°C on maximum speed for 20 min and split in half for biochemical analysis.
Ganglioside storage analysis
Ganglioside extraction and visualization was performed as previously described. 31 –34 Briefly, frozen midbrain sections were sonicated in 3 × 10 sec bursts and spun down at 4°C on maximum speed for 20 min, after which 300 μl of the supernatant was mixed with the pellet before beginning the extraction. Gangliosides were extracted by a series of dilutions and nitrogen evaporations of the midbrain samples in methanol and chloroform solvents. These mixed ganglioside samples were then separated on a thin-layer chromatography plate using a 55:45:10 chloroform:methanol:0.2% calcium chloride mobile phase. Bands were visualized using orcinol and plates were dried at 120°C for 10 min. Densitometry analysis was performed comparing the intensity of the GM2 to the GD1A bands using ImageJ software. A mix of manufactured gangliosides is run as the standard control for each TLC plate. This assay was performed for brains of all the experimental mice as well as additionally on control untreated SD mice (−/−) with ages 12, 14, and 16 weeks (n = 3 each) for comparison of GM2 levels.
Hexosaminidase activity assay
Hexosaminidase activity was measured as previously described. 35,36 Briefly, samples of hexosaminidase enzymes were obtained from 400 μl of sonicated midbrain homogenates and the collected serum samples. Total hexosaminidase activity was determined through the use of the 4-methylumbelliferyl-2-acetamido-2-deoxy-β-D-glucopyranoside (4-MUG) assay, whereas the hexosaminidase activity (HexA, HexS, and HexM) toward negatively charged substrate was determined through the use of the 4-Methylumbelliferyl-7-6-sulfo-2-acetamido-2-deoxy-β-D-glucopyranoside (4-MUGS) assay, and comparing each of these assays to the 4-methylumbelliferone (4-MU) standard curve. The total protein content in the brain homogenates was determined through Bradford assay before the MUG and MUGS analysis. The midbrain samples were diluted to 1:10 in 1× PBS to complete the MUG and MUGS enzyme assays. Briefly, the samples were incubated with the fluorogenic substrate at 37°C for 1 hr, and then read on the plate reader with excitation wavelength of 365 nm and an emission wavelength of 450 nm.
Histology
Histological procedures were performed similarly to those previously described. 14 Paraffin-embedded midbrain samples were sectioned on a microtome to a thickness of 4–6 μm. Briefly, sections were deparaffinized and then were followed as per citrate buffer antigen retrieval protocol. Slides were then blocked for both nonspecific binding and endogenous peroxidase activity, followed by incubations in primary then secondary antibody. DAB stain was then applied and the slides were then dehydrated through a series of alcohol baths increasing in concentration. Immunohistological detection for GM2 ganglioside storage was accomplished with a 1:1000 dilution of the chimeric murine–human IgG1 anti-GM2 ganglioside antibody, 37 KM966 (Kyowa Hakko Kirin Co., Ltd.). The primary antibody was detected with an antihuman biotinylated secondary antibody at a 1:1000 dilution.
Biodistribution analysis by qPCR
Copy number analysis for our vectors was carried out as previously reported. 25 Values are reported as double-stranded copies of the HEXM vg per two double-stranded copies of the mouse LaminB2 locus. For diploid mononuclear cells, this will approximate the copies of vg per cell.
Statistics
One-way ANOVAs with Bonferroni post-hoc tests were performed on the results from the ganglioside assay, the hexosaminidase assays, as well as the 12-week behavioral time point. The remainder of the behavioral data were analyzed through two-way repeated-measures ANOVAs with the Bonferroni post-hoc tests, carrying data forward and backward for any missing samples at time points. A Kaplan–Meyer curve was used to assess the survival of the SD mice, followed by a log-rank (Mantel–Cox) test. All statistical analyses were performed in GraphPad Prism 5.
Results
Survival advantage to AAV9.47/HEXM-treated SD mice
To evaluate the effectiveness of the AAV9.47/HEXM, a single dose of the virus was administered systemically to neonatal SD mice through temporal vein injections. Separate SD cohorts were injected with AAV9.47/GFP or PBS as controls. Heterozygous mice were used in order to approximate the lifespan and biochemistry of a positive control (normal) mouse. The untreated KO SD mice typically reach a humane endpoint of approximately 14–16 weeks of age, at which point the severity of their symptoms requires them to be humanely euthanized. 10,12 In the long-term-monitored cohorts, the AAV9.47/HEXM-injected animals survived for a mean of 41.77 weeks of age, a median of 40 weeks, and an age range of 36–49 weeks (n = 6). This extension in survival is significant compared with the average 15-week humane endpoint (p < 0.001). 10,12 Figure 1 shows the comparison of the long-term AAV9.47/HEXM cohort survival against that of heterozygous mice. The historical average survival 10,12 of untreated animals is also shown.

Kaplan–Meyer survival curve depicting the HexM-treated group that outlived knockout (KO) age-matched controls. HexM-treated animals survived to a median age of 40 weeks, ranging between 36 and 49 weeks (n = 6). This extension in survival is significantly different compared with the historical 15-week humane endpoint (**p < 0.001).
Behavioral amelioration of AAV9.47/HEXM-treated KO SD mice
Phenotypically, the AAV9.47/HEXM-injected mice were visibly moving and acting similar to the heterozygous controls until the last 6–8 weeks before their humane endpoint when they began exhibiting tremors and slow, rigid movements. We objectively measured the changes in phenotype between the experimental groups through a battery of behavioral testing. The OFT measures the general locomotion of the mice through measuring the time moving. There was a significant effect of the treatment group on the time spent moving in the OFT (F 2, 16=10.83, p = 0.001) at 12 weeks (Fig. 2A). The AAV9.47/HEXM-injected mice significantly outperformed the untreated SD mice (p < 0.05), and performed similarly to the heterozygote controls (Fig. 2A). Over the course of the 4 weekly behavioral testing, the AAV9.47/HEXM-injected mice showed no significant deficits in locomotor activity compared with the heterozygous controls (F 1 = 1.42, p = 0.26) for the OFT time moving (Fig. 2B).

HexM-treated KOs show significant and long-lasting behavioral improvements.
The RR assessment on an accelerating protocol tests an animal's coordination. Once the mouse falls from the rotating rod, the end RPM was recorded. Upon examining the end RPM of the mice at 12 weeks, the effect of treatment appeared modest, and the end RPM of the scAAV9.47/HEXM-injected SD mice was not significantly above that of untreated SD mice (F 2, 13=3.25, p = 0.072) (Fig. 2C). However, further monthly testing of the scAAV9.47/HEXM-injected SD mice showed RR performance similar to heterozygous controls up to 36 weeks old (F 1 = 0.57, p = 0.47). By 40 weeks, a declining RR performance was observed (p > 0.05) and only two mice survived beyond 40 weeks (Fig. 2D).
The strength of the mice was measured through the use of the mesh test. At the 12-week behavioral assessment, there was a significant effect of scAAV9.47/HEXM injection on the latency to fall in the mesh test (F 1,7 = 11.76, p = 0.001), where the untreated SD mice were significantly outperformed by their age-matched heterozygous controls and the scAAV9.47/HEXM-injected SD mice (p < 0.05) (Fig. 2E). Over the time course of behavioral testing from 12 to 44 weeks, the scAAV9.47/HEXM-injected SD mice performed similarly to the heterozygous controls (F 1 = 1.71, p = 0.22) (Fig. 2F).
Biodistribution of scAAV9.47/HEXM in the brain and liver
The increase in length of survival and the amelioration of phenotypic symptoms suggested that the treatment of scAAV9.47 crossed into the CNS (Fig. 3). We examined the biodistribution of the intravenously injected virus through quantitative PCR (qPCR) analysis. Both HEXM and GFP vectors were found in all organs assayed in vector-injected mice. The HEXM was found to be expressed at lower level (∼5 × in general) in the long-term scAAV9.47/HEXM-injected SD mice compared with the 8-week scAAV9.47/HEXM-injected SD mice cohort especially in lumbar and cervical spinal cord region where it was almost 100-fold lower. We confirmed the previously reported biodistribution of the vector following the pattern of liver:brain ratio of approximately 1:1 using the AAV9.47 capsid, 21 compared with AAV9, which showed a liver:brain ratio of >50:1 in mice studies, indicating AAV9.47 detargeting of the liver. 25,38
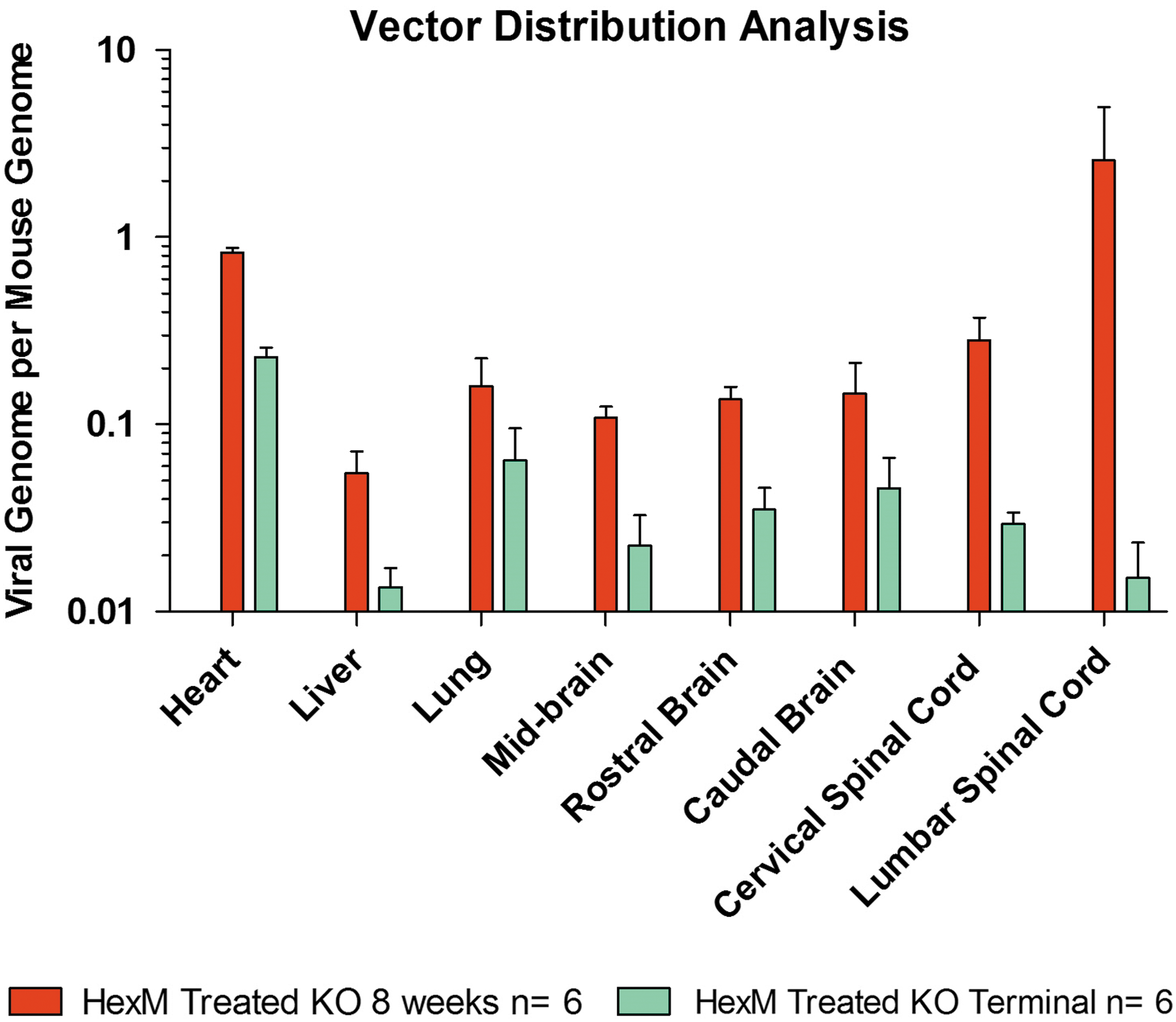
Vector biodistribution of the HexM vector. Widespread delivery achieved to the brain and spinal cord regions, as well as a relative detargeting of the liver at both the 8-week and long-term terminal humane endpoints. HexM was not detectable above 0.001 copies per mouse genome in mice dosed with vehicle or AAV9.47/GFP, and is not shown on the graph. Data are presented as the copies of vector DNA per diploid mouse genome found in each assessed organ. Color images available online at
Enzymatic changes in the terminal midbrain sections
In the MUGS assays of the midbrain samples at 8 weeks, there appeared to be an increase in activity in the AAV9.47/HEXM-injected group compared with the KO groups injected with PBS or AAV9.47/GFP, but this difference was not significant. All treatment groups differed significantly from the heterozygous controls (p < 0.001) (Fig. 4A). At the long-term terminal endpoint, there was a similar finding that the AAV9.47/HEXM vector treatment did not increase Hex activity significantly above that of untreated control mice (Fig. 4B). Any possible change in MUGS activity in the brain of the AAV9.47/HEXM-injected mice was below the level of sensitivity for this assay.

Enzymatic and chromatographic analysis of the midbrain samples.
Decrease in GM2 ganglioside accumulation in the AAV9.47/HEXM-treated SD mice
As part of the initial characterization of HexM, Tropak et al. 28 who used the same mice for short-term analysis of GM2 ganglioside, reported that these neonatal mice injected with scAAV9.47/HEXM vector had a significant reduction in the storage of the GM2 ganglioside compared with the scAAV9.47/GFP and PBS control mice, but there was still a significant amount of GM2 ganglioside stored in the scAAV9.47/HEXM-injected mice compared with the heterozygous control mice. However, at the long-term terminal endpoint, the GM2 ganglioside levels in scAAV9.47/HEXM-injected mice at 36–49 weeks of age were not different from those in untreated mice at 12–16 weeks of age (Fig. 4C). Taken together, the results indicate that injections with scAAV9.47/HEXM delay the accumulation of GM2 gangliosides but ultimately the gangliosides accumulate to fatal levels.
Histological decrease in GM2 ganglioside accumulation in the AAV9.47/HEXM-treated SD mice
A comparison of GM2 ganglioside staining patterns of PBS-injected mice to both heterozygous control mice and scAAV9.47/HEXM-injected mice was carried out with tissue samples from 8-week-old mice. Abundant GM2 staining across multiple anatomical areas of the brain (thalamus, hypothalamus, and hippocampus) was observed in the PBS-injected group that is absent in the heterozygous group, and nearly absent in the scAAV9.47/HEXM-injected group (see Fig. 5).

Histological ganglioside storage in the neurons of the murine midbrain at 8 weeks. Sections of the murine thalamus
Discussion
This study demonstrates the long-term therapeutic efficacy of the vector expressing a novel hexosaminidase μ-subunit for the treatment of GM2 gangliosidosis in SD mice. A single intravenous dose of the scAAV9.47/HEXM improved both the lifespan and the delayed onset of behavioral symptoms of the SD mice. HexM had previously been shown to catabolize GM2 ganglioside in association with the human rGM2AP in human cells. 28 Our study confirms that SD mice injected with scAAV9.47/HEXM are able to catabolize and slow the accumulation of GM2 ganglioside in an SD mouse model.
Although the activity of HexM and its ability to catabolize GM2 ganglioside in cellulo and in vivo (short-term) were established recently by Tropak et al. 28 its ability to delay disease symptoms and improve survival in a mouse model had not yet been established before this current study. A one-time scAAV9.47/HEXM injection in neonatal SD mice showed a marked delay in the presentation of symptoms and an increase in median survival to 40 weeks, increasing the lifespan of the mice by 2.5-fold (see Fig. 1). A direct comparison of the viral dose used in our study to many of the dosages previously published is difficult because of the differences in vector types 13,39 –43 and routes of administration. 43 –45 A previous study utilizing intravenous neonatal injections with a single-stranded AAV9/mhexb vector used a dose approximately 5 × higher than that used in the present study and resulted in an increase in survival of the SD mice to ∼43 weeks of age. 14 It is difficult to make direct comparisons between this study 14 and the current study, which used a human hybrid gene, an scAAV vector, a synthetic promoter, and an AAV9.47 capsid.
Previously published studies have shown the benefits of gene transfer for GM2 gangliosidosis using one or both hexosaminidase α-subunit and β-subunit genes. 13,14,39 –47 It has been hypothesized that the formation of HexA could be rate limited by the amount of the endogenous subunit if the gene being transferred is only for the deficient subunit (i.e., α-subunit gene for TSD or β-subunit gene for SD). However, the use of multiple vectors to deliver both transgenes can be problematic. A high vector dose or an integrating-type vector could lead to oncogenesis in a variety of organs in the body 13,14,48 –51 As previously mentioned, the Hex μ-subunit homodimerizes to form a fully functioning isoenzyme, HexM, which has been shown to replace or substitute the function of the HexA heterodimer. 28 Furthermore, it is derived from a single hybrid gene of ∼1.6 kb, thus complying with the packaging size requirement of scAAV vectors.
Behaviorally, previous literature indicated that SD mice traveled smaller distances in the OFT 14 and had a shorter latency to fall on the RR 12,13 compared with heterozygous or wild-type mice. Our data collected at 12 weeks support these studies. In the series of behavioral tests, at 12 weeks of age, the scAAV9.47/HEXM-injected SD mice significantly outperformed the KO age-matched controls and performed at roughly the same level as the heterozygous mice (see Fig. 2). The performance of the scAAV9.47/HEXM-injected mice on these tests was similar to the heterozygote control group, indicating that the scAAV9.47/HEXM treatment delayed phenotype onset of the SD mice, bringing it closer to that of a heterozygous mouse for approximately 40 weeks.
The hexosaminidase activity analysis of the midbrain at both 8-week and the terminal organ harvest showed slight, nonsignificant increase in the amount of hexosaminidase activity in the scAAV9.47/HEXM-injected group compared with that for the PBS or scAAV9.47/GFP-injected SD animals. It is reasonable that only a slight increase in hexosaminidase activity in the brain could be responsible for the reduced GM2 ganglioside level and for the marked increase in lifespan and quality of life of the scAAV9.47/HEXM-injected SD mice. As has been proposed for lysosomal storage diseases, a critical threshold of only 10–15% of normal enzyme activity level is required to avoid the disease symptoms. 13,43
Speculating that the effects of the treatment should be discernable biochemically before the time that symptoms would first appear, we euthanized the short-term cohorts at 8 weeks, and left the long-term cohorts for survival assessment. As previously stated, there was a striking decrease in the storage of GM2 gangliosides between the scAAV9.47/GFP- or PBS-injected SD mice and the scAAV9.47/HEXM-injected SD mice. Histologically, at 8 weeks we observed a reduction in the GM2 ganglioside level in the scAAV9.47/HEXM-injected animals compared with the GFP- or PBS-treated groups. At 8 weeks, there was approximately a 66% decrease in the amount of GM2 gangliosides stored in the midbrains of the HexM-treated group compared with the PBS-treated group, as assessed through the GM2 ganglioside storage assay. 28 A similar decrease in the storage of GM2 gangliosides was also seen at the terminal examination of the midbrains of the long-term cohort. The apparent slight increase in the amount of HexM activity enabled the scAAV9.47/HEXM-injected mice to slow the accumulation of the GM2 ganglioside, delaying the lethality of the disease until a median of 40 weeks of age. Upon performing the GM2 ganglioside storage analysis, it was observed that the long-term terminal mice had similar storage amounts as 12–14-week SD animals, presumably allowing a delay in disease progression.
The vector genome biodistribution analysis illustrated the effectiveness of the scAAV9.47/HEXM vector in that it was not only efficient at crossing through the immature blood–brain interface of neonate mice and then transducing the brain and spinal cord, but also effectively detargeted from the liver. The copy number analysis showed that the vector DNA was reduced at the long-term endpoint, especially in cervical and lumbar spinal cord region. It should be noted that the study was not designed to assess the differences in the AAV9.47 systemic distribution. The exact reason for the observation is not clear, but it is speculated that this reduction could be because of the death of transduced brain cells as widespread neurodegeneration ensues. However, neuroinflammation is a known co-morbid factor in SD, and therefore the decreased ratio at the terminal time point may also be a false reflection because of an increase in the presence of inflammatory cells (contributing to more mouse genomes), thus reducing the ratio of vector to mouse genomes (Fig. 3).
Previous studies using AAV vectors in a systemic neonatal treatments have noted the formation of liver and lung tumors in their AAV-treated animals. 14,22,50,51 In this study, we did not encounter any tumors in any of the organs harvested from any of the study mice. The mechanism underlying the tumorigenicity of AAVs is still unclear; however, the relatively low dose of the scAAV9.47/HEXM treatment and the liver-detargeting abilities of AAV9.47 are plausible explanations for the lack of tumor genesis. Synthetic promoters have been shown to be less oncogenic because we used a synthetic promoter, UsP, that may have contributed to lack of tumor development in our study. 23
The current study shows that intravenous/systemic AAV gene therapy with HEXM is a viable minimally invasive option for the therapeutic treatment of GM2 gangliosidosis. This approach has resulted in pronounced improvements in survival and quality of life of the SD mice, and it has demonstrated its ability to clear GM2 ganglioside within the CNS of mice. Our collaborators Karumuthil-Melethil et al. 29 conducted a study investigating the effects of the AAV9.47/HEXM treatment on TSD mice (complementary article in this issue of Human Gene Therapy). However, because of the unique metabolic bypass that exists in mice, TSD mice express only a mild chronic phenotype. Our work demonstrates that this therapeutic approach lays the foundation for a translatable global treatment for both TSD and SD using a single delivery of one hybrid gene in an scAAV vector. Future studies will be required looking at the effect of increasing the dose of the scAAV9/HEXM vector in the SD neonatal mice, as well as investigating the effectiveness of this vector in crossing the BBB after injection in adult SD mice. Our work also contributes to the growing literature supporting the use of AAV vectors to broadly deliver a therapeutic gene to the CNS as a treatment strategy for neurodegenerative diseases.
Footnotes
Acknowledgments
Funding for this research was provided by the New Hope Research Foundation (to J.W., S.G., and D.M.) and through donations from Uger Estate (to D.M.). Research to Prevent Blindness of the UNC Department of Ophthalmology provided secondary administrative support to S.G. A Manitoba Research Chair (Research Manitoba) provides support for B.M. Clifford Heindel and Violeta Zaric at UNC Chapel Hill are acknowledged for technical assistance with the biodistribution studies. We thank Kyowa Hakko Kirin Co. for graciously providing the KM966 antibody.
Author Disclosure
S. Gray has received patent royalties from Asklepios Biopharma for technologies not directly linked to this study. The sequence and potential uses of HexM are patent pending by the inventors D. Mahuran and B. Mark. The UsP promoter-intron is patent pending by inventor J. Keimel. The rest of the authors have no competing financial interests.