
Abstract
T cells engineered to express CD19-specific chimeric antigen receptors (CARs) have shown breakthrough clinical successes in patients with B-cell lymphoid malignancies. However, similar therapeutic efficacy of CAR T cells in solid tumors is yet to be achieved. In this study we systematically evaluated a series of CAR constructs targeting glypican-3 (GPC3), which is selectively expressed on several solid tumors. We compared GPC3-specific CARs that encoded CD3ζ (Gz) alone or with costimulatory domains derived from CD28 (G28z), 4-1BB (GBBz), or CD28 and 4-1BB (G28BBz). All GPC3-CARs rendered T cells highly cytotoxic to GPC3-positive hepatocellular carcinoma, hepatoblastoma, and malignant rhabdoid tumor cell lines in vitro. GBBz induced the preferential production of Th1 cytokines (interferon γ/granulocyte macrophage colony-stimulating factor) while G28z preferentially induced Th2 cytokines (interleukin-4/interleukin-10). Inclusion of 4-1BB in G28BBz could only partially ameliorate the Th2-polarizing effect of CD28. 4-1BB induced superior expansion of CAR T cells in vitro and in vivo. T cells expressing GPC3-CARs incorporating CD28, 4-1BB, or both induced sustained tumor regressions in two xenogeneic tumor models. Thus, GBBz CAR endows T cells with superior proliferative potential, potent antitumor activity, and a Th1-biased cytokine profile, justifying further clinical development of GBBz CAR for immunotherapy of GPC3-positive solid tumors.
Introduction
C
Glypican-3 (GPC3), a membrane-bound proteoglycan, is expressed in several solid tumors including hepatocellular carcinoma (HCC), hepatoblastoma, embryonal sarcoma, malignant rhabdoid tumor (MRT), yolk sac tumor, Wilm's tumor, squamous cell carcinoma of the lung, and liposarcoma. 19 –29 GPC3 is an attractive target for immunotherapy since it is not expressed at detectable levels in nonmalignant tissues including normal or cirrhotic liver. 19 –22 In addition, GPC3 expression in HCC is associated with significantly worse prognosis even after complete tumor removal, likely due to GPC3’s ability to enhance wnt signaling and stimulate tumor growth. 30 –37 Two recent early phase clinical trials tested a GPC3-specific GC33 mAb and demonstrated that targeting GPC3 is safe, well tolerated and, depending on the density of GPC3 expression on tumor cells, can achieve significant antitumor responses in patients with advanced HCC. 38,39
Unlike clinically tested CD19-CARs, which contained either CD28 or 4-1BB co-stimulatory endodomains, the only GPC3-CAR construct reported contained both CD28 and 4-1BB endodomains. 19,40 The inclusion of two endodomains in GPC3-CAR may be necessary, as in contrast to B-cell malignancies, solid tumors generally express little or no costimulatory ligands for T cells. 41 However, relative contribution of CD28 and 4-1BB in the effector functionality of GPC3-CAR T cells remains unknown and the clear benefit of their combined expression has not been experimentally established. 42 –45 In this study, we systematically evaluated the antitumor properties of T cells expressing GPC3-CARs encoding CD28, 4-1BB, either endodomain, or none. In addition to using a well-established HCC tumor model for evaluating GPC3-CARs, for the first time we tested the antitumor potential of GPC3-CAR T cells against MRT. We found that the costimulatory endodomain composition of the CARs has significant effect on the cytokine polarization in T cells and 4-1BB induces a Th1 polarized profile. We show that the inclusion of 4-1BB endodomain alone in GPC3-CAR is sufficient to generate T cells with enhanced proliferation, in vivo persistence, and potent therapeutic activity in xenogeneic tumor models.
Materials and Methods
Cell lines
HepG2, Hep3B, A549, G401, and HEK 293T cell lines were purchased from the American Type Culture Collection (Manassas, VA). Huh-7 was a kind gift from Dr. Xiao-Tong Song (Baylor College of Medicine, Houston, TX), and its identity was confirmed at the Characterized Cell Line Core Facility at MD Anderson Cancer Center (Houston, TX) by short-tandem repeat method. Cell lines were maintained according to the manufacturer's manual. Huh-7 and G-401 cells expressing an enhanced green fluorescent protein/firefly luciferase fusion gene (eGFP.Ffluc) 46 were generated by single cell cloning after transduction with a retrovirus encoding eGFP.Ffluc. A549 cells expressing GPC3 (A549.GPC3) were generated by transducing wild type A549 with a retrovirus encoding GPC3.
Generation of retroviral constructs
A codon-optimized gene was synthesized by GeneArt® (Thermo Fisher Scientific, Waltham, MA) encoding the GPC3-specific scFv from GC33, 47 and subcloned in frame into retroviral vectors containing expression cassettes encoding an IgG1 short hinge, a CD28 transmembrane domain, and CD3ζ, CD28ζ, 4-1BBζ, or CD28.4-1BBζ signaling domains (Fig. 1). The sequence of each cloned CAR was verified by sequencing (Seqwright, Houston, TX). In addition to nontransduced (NT) T cells, disialoganglioside (GD2)-specific CAR derived from 14g2a.scFv and CD19-specific CAR from FMC63.scFv containing CD3ζ and CD28.4-1BB costimulatory endodomains were used as negative controls. 48,49
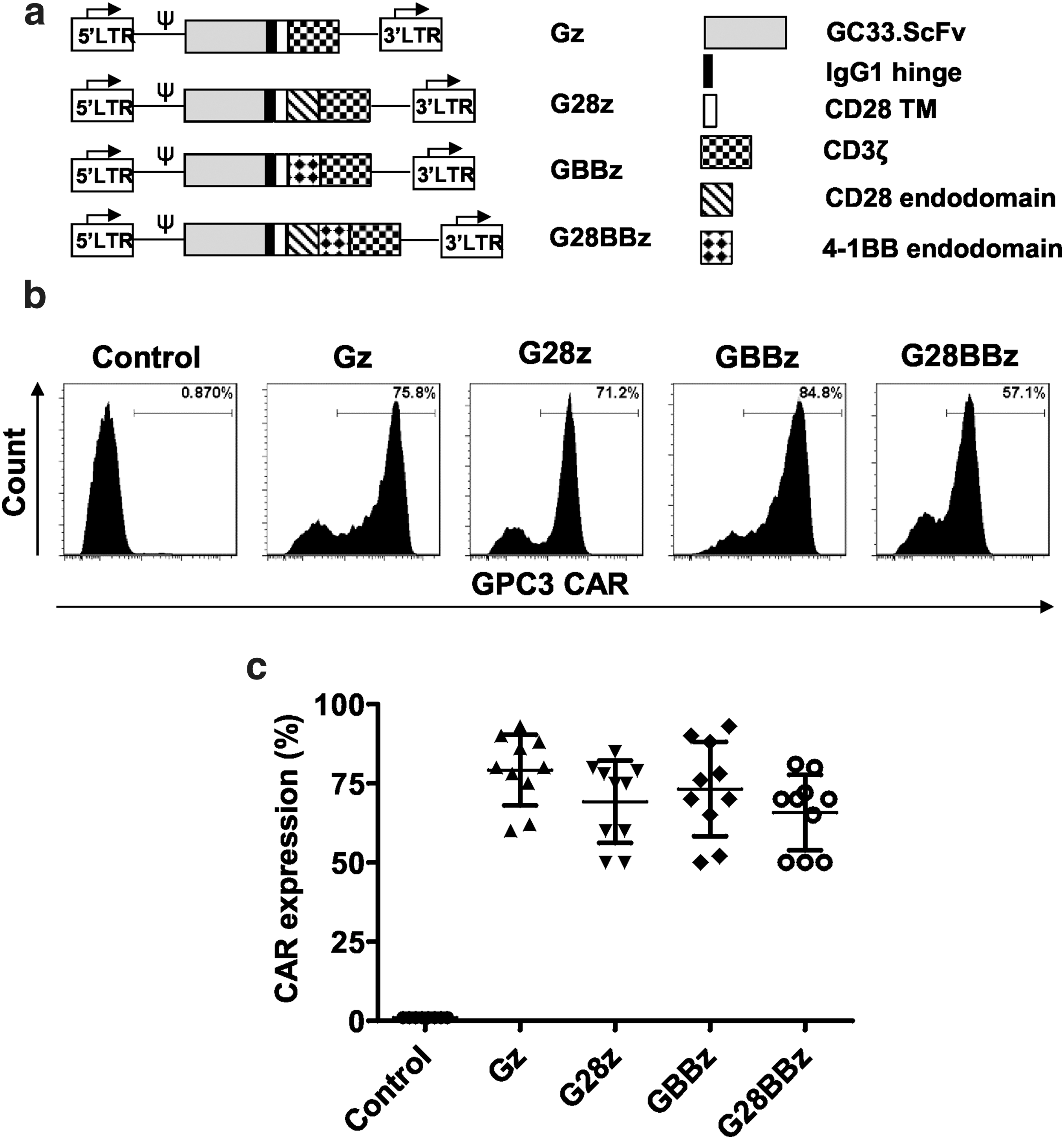
Generation of glypican-3 chimeric antigen receptor (GPC3-CAR) T cells. The GPC3-specific single chain variable fragment (scFv) derived from GC33 monoclonal antibodies was cloned in frame into retroviral vectors encoding the immunoglobin 1 (IgG1) short hinge, the CD28 transmembrane domain and a CD3ζ signaling domain with or without the costimulatory endodomains derived from CD28, 4-1BB, or their combination.
Generation of CAR T cells
Retroviral supernatants were produced by transient transfection of HEK 293T cells with plasmids containing one of GPC3-CARs, RDF plasmid encoding the RD114 envelope and PegPam3 plasmid encoding the MoMLV gag-pol as previously described. 50 Human peripheral blood mononuclear cells isolated from healthy volunteer donors (Gulf Coast Regional Blood Center, Houston, TX) were stimulated with OKT-3/CD28 mAb–coated plates for 48 h in complete RPMI medium (HyClone RPMI 1640, 10% heat inactivated fetal bovine serum and 2 mM Glutamax) with interleukin 7 (IL-7; 10 ng/mL) (PeproTech, Rocky Hill, NJ), and IL-15 (5 ng/mL) (Peprotech). IL-7 and IL-15 were used to optimize CAR T cell expansion. 51 After 48 h of stimulation, cells were transduced on Retronectin (Kusatsu, Japan)–coated and retroviral particle–loaded plates and after 48 h cells were removed, washed, and cultured in IL-7 and IL-15 containing complete RPMI media for further expansion.
Flow cytometry
For all flow cytometry analyses, FACSArray or LSR-II instruments were used (BD Biosciences, Franklin Lakes, NJ). Results were analyzed by FlowJo (FlowJo, LLC; Ashland, OR). GPC3-CAR expression was detected by anti-F(ab)2 Alexa Fluor 647–conjugated antibody (Jackson ImmunoResearch, Cat No. 115-605-006) and anti-goat IgG1 isotype control (Jackson ImmunoResearch, Cat #: 115-605-006). GPC3 expression of tumor cell lines was detected with YP7 mAb 52 provided by Mitchell Ho (National Institutes of Health, Bethesda, MD).
Cytotoxicity assay
The ability of GPC3-CAR T cells to kill GPC3-positive tumor cells in vitro was tested in a standard 4-h chromium 51 ( 51 Cr) release assay as previously described. 53 In brief, target cells were loaded with 51 Cr for 1 h, washed three times, and mixed with effector cells in 96-well plates at various effector to target ratios. Supernatant was collected after 4 h of incubation and radioactivity was measured to determine specific cytotoxicity.
Multiplex cytokine quantification
Resting GPC3-CAR T cells were co-cultured at a 1:1 ratio with target cells (Supplementary Fig. S1; Supplementary Data are available online at
In vitro serial killing and CAR T cell proliferation assay
Tumor cells and effector cells were cocultured at a 1:1 ratio in complete RPMI without cytokines. Effector cells were counted and re-plated with fresh tumor cells every 3 days in fresh media. Carboxyfluorescein succinimidyl ester (Invitrogen, Carlsbad CA) dilution was measured on day 3 after first stimulation, as described by Hawkins et al. 54 , and 7-amino-actynomycin (BD Biosciences) staining was performed on day 7 according to the manufacturer's manual.
Animal models
NOD/SCID/IL2γnull (NSG mice, The Jackson Laboratory) were maintained at the Small Animal Core Facility of Texas Children's Hospital and were treated according to the protocols approved by Baylor College of Medicine's Institutional Biosafety Committee and Institutional Animal Care and Use Committee. Twelve-week-old, gender-matched NSG mice were injected with different tumor cell lines intraperitoneally (IP) respectively to generate the tumor-bearing model. To determine the in vivo proliferation of GPC3-CAR T cells and controls, T cells were cotransduced with CAR and eGFP.Ffluc 46 constructs and intravenously (IV) injected into Huh-7 tumor–bearing mice followed by bioluminescence imaging every other day. To determine the antitumor potential of GPC3-CAR T cells, mice were injected with eGFP.Ffluc expressing Huh-7 (2 × 106 per mouse), G401 (5 × 106 per mouse), A54955 (2 × 106 per mouse) tumor cells followed by the IV injection of T cells 5 days (A549), 2 weeks (Huh-7) or 3 weeks (G401) later, respectively. Tumor bioluminescence was measured by weekly imaging and survival was documented for each group.
Statistical analyses
Descriptive analysis was performed to summarize cytokine production and CAR expression on T cells. Plots of growth curves were generated to visually illustrate patterns of survival and expansion of CAR T cells. Area under the curve (AUC) values for CAR T-cell frequencies were calculated using trapezoidal rule for each treatment group. The normality assumption was examined, and if the assumption was in doubt, the data were transformed when necessary. Statistical comparisons were analyzed using ANOVA and t-test with Bonferroni adjustment as appropriate. Antitumor efficacy was analyzed using the Kaplan–Meier method and compared using the Gehan–Breslow—Wilcoxon test. Statistical analyses were carried out using GraphPad Prism 5.0 (GraphPad Software) and SAS 9.4. A p value <0.05 was considered statistically significant.
Results
GPC3-CARs are stably expressed on T cells and costimulatory endodomains do not alter expression levels
The scFv of GC33 mAb was cloned in frame into SFG gamma-retroviral vectors containing CAR expression cassettes with CD3ζ (Gz), CD28.CD3ζ (G28z), 4-1BB (GBBz), or CD28.4-1BB.CD3ζ (G28BBz) endodomains (Fig. 1a). CD3/CD28-activated T cells were transduced with RD114-pseudotyped retroviral vectors encoding the indicated constructs. Cell surface expression of CARs was measured by flow cytometry (Fig. 1b, c). All 4 CARs were stably expressed on the cell surface of T cells with no significant difference between constructs. The median CAR expression was 79.2% (range 60–93%) for Gz, 80% (range 50–85%) for G28z, 80% (range 52–93%) for GBBz and 70% (range 50–81%) for G28BBz (Fig. 1c). The generated CAR T-cell lines contained >95% CD3-positive T cells, which were comprised of CD4- and CD8-positive T-cell subsets with the same ratio as in NT T cells (Supplementary Fig. S1).
GPC3-CAR T cells recognize and kill GPC3-positive tumor cells
To test whether transgenic expression of GPC3-CARs renders T cells cytotoxic toward GPC3-positive targets, we performed standard 51 Cr-release cytotoxicity assays at the indicated effector to target ratios using GPC3-positive [HepG2 (hepatoblastoma), Huh-7 (HCC), Hep3B (HCC), G401 (MRT)], GPC3-negative, unmodified A549 (lung cancer) cells, and A549 that were genetically modified to express GPC3 (A549.GPC3, Supplementary Fig. S2, Supplementary Table S1). NT- and GD2-specific CAR-expressing (CAR-GD2) T cells served as negative effector controls. GPC3-CAR T cells specifically killed A549.GPC3 but not unmodified A549 cells, confirming the specificity of the generated CARs. Furthermore, GPC3-CAR T cells effectively killed all other tumor cells (HepG2, Huh-7, Hep3B, G401) that were GPC3 positive (Fig. 2). CAR-GD2 and NT T cells did not kill any of the target cells, demonstrating that the cytolytic activity of T cells depends on the expression of GPC3 on target cells and GPC3-CARs on T cells.
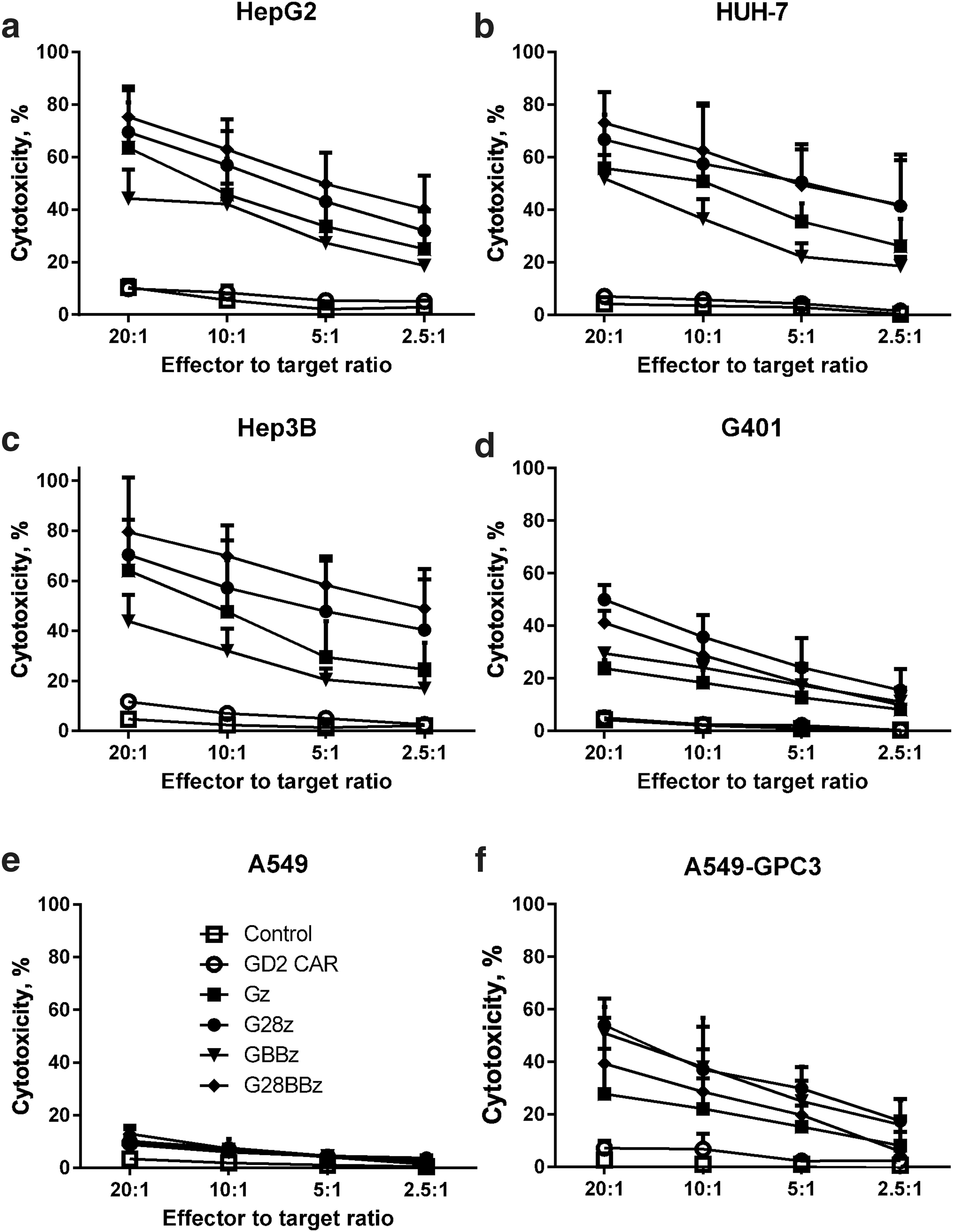
GPC3-CAR T cells recognize and kill GPC3-positive tumor cells. Tumor cell lysis was measured with standard 4 h chromium 51 (
51
Cr) release assay at indicated effector to target ratios against GPC3-positive solid tumor cell lines HepG2, Huh-7, Hep3B, and G401. GPC3-negative A549 cell line served as negative control, and A549, genetically modified to express GPC3 (A549.GPC3), as positive control.
Costimulation by 4-1BB and CD28 have opposing effects on Th1 and Th2 cytokine production by GPC3-CAR T cells
Having estiablished that GPC3-CAR T cells recognize GPC3-positive target cells in an antigen-specific manner, we next determined in coculture assays the ability of GPC3-CAR T cells to secrete Th1 (interferon gamma [IFNγ], granulocyte macrophage colony-stimulating factor [GM-CSF], IL-2) and Th2 (IL-4, IL-10) cytokines. We used the same panel of GPC3-positive and GPC3-negative tumor cells, and effector controls as for the cytotoxicity assays. GPC3-positive target cells induced cytokine production of GPC3-CAR T cells and not of NT or CAR-GD2 T cells confirming specificity (Supplementary Fig. S3a, b), and striking differences were noted in cytokine release pattern dependent on the costimulatory endodomains of GPC3-CARs. After controlling for donor-to-donor variation of absolute amount of cytokine release we found that 4-1BB costimulation induced higher levels of IFN-γ and GM-CSF production compared with CD28 costimulation compared to Gz (p < 0.05 in 3 of 4 and 2 of 4 tested cell lines, respectively, Fig. 3). In regard to Th2 cytokines, CD28 costimulation induced significantly more IL-4 and IL-10 production than 4-1BB costimulation (p < 0.05 in 3 of 4 tested cell lines for both cytokines, Fig. 3). Thus, CD28 costimulation skews cytokine productions of T cells toward a Th2 pattern, whereas 4-1BB costimulation enhances Th1 cytokine production. Introducing 4-1BB costimulation into G28z T cells could not overcome the Th2 bias of CD28 costimulation, and G28BBz T cells produced more IL-10 than Gz T cells for all cell lines tested (Fig. 3).
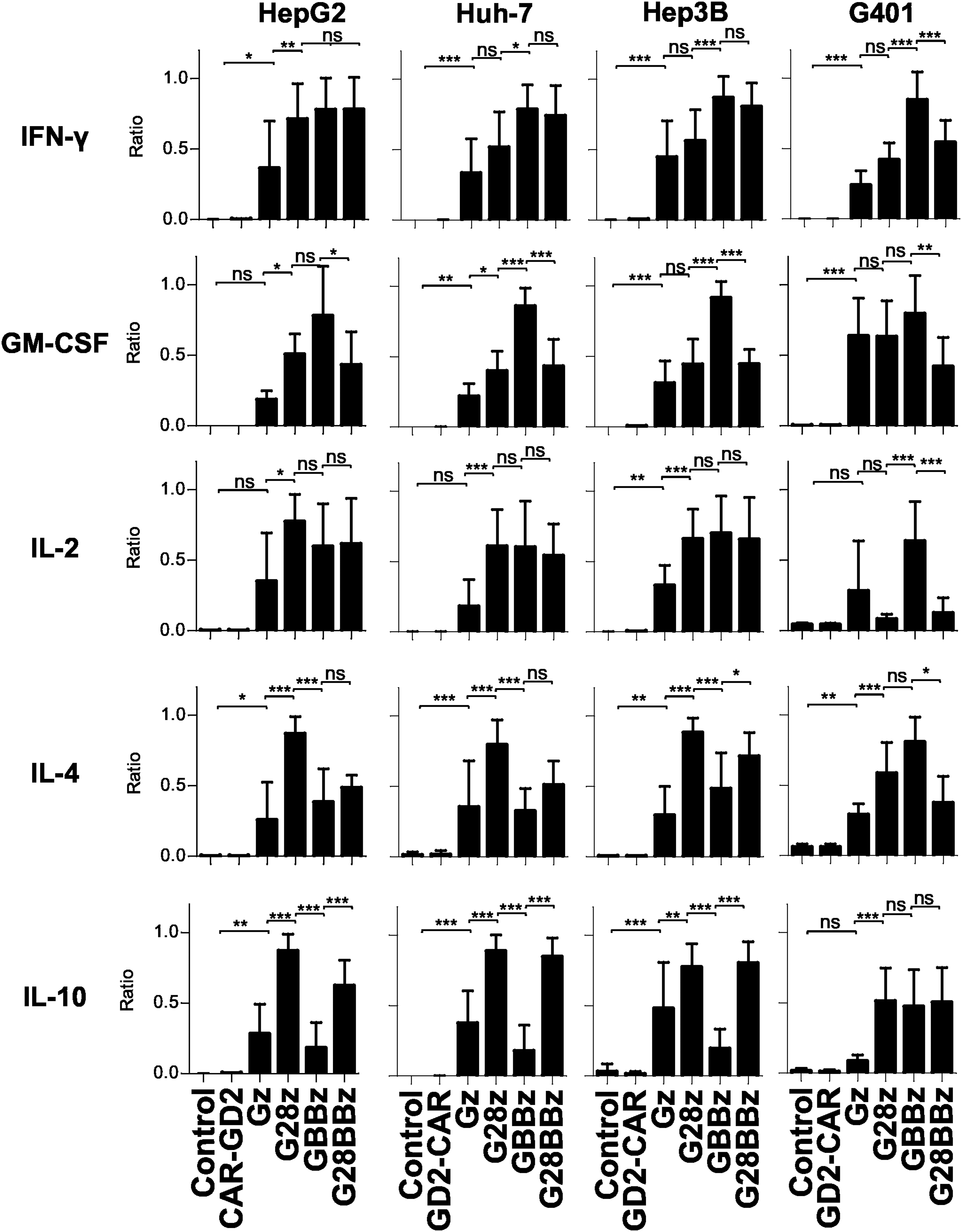
Differential cytokine production of GPC3-CAR T cells dependent on costimulatory endodomains. GPC3-positive HepG2, Huh-7, Hep3B, and G401 cells were cocultured with GPC3-CAR T cells for 24 h at 1:1 ratio and indicated cytokine levels in tissue culture supernatant were measured using Luminex. To correct for donor to donor variability of absolute cytokine concentrations, the data were normalized as a ratio relative to the maximum in each experiment, with the maximal cytokine level in each experiment being 1 (formula: sample value / maximum value of the experiment for the tested cytokine; 3–5 independent donors for all GPC3 CAR constructs). Mean and SD are shown. *p < 0.05; **p < 0.01; ***p < 0.001, one-way ANOVA with Bonferoni post-test analysis. ns, not significant (p ≥ 0.05).
Costimulation by 4-1BB results in improved GPC3-CAR T cell survival during serial killing of tumor cells in vitro
To determine the effect of costimulatory endodomains on GPC3-CAR T cell expansion in vitro, we performed a 7-day proliferation assay using carboxyfluorescein succinimidyl ester–labeled CAR T cells that were stimulated with GPC3-positive tumor cells. NT and CAR-GD2 T cells were used as controls. G28z, GBBz, and G28BBz induced greater T cell proliferation than did Gz (Fig. 4a) and increased their viability as judged by 7-amino-actynomycin staining (Fig. 4b). Next, we performed co-cultures in which GPC3-CAR T cells were restimulated every 3 days with fresh GPC3-positive tumor cells in the absence of cytokines. G28z, GBBz, and G28BBz outperformed Gz as judged by fold-increase of absolute number of CAR T cells at the indicated timepoints (Fig. 4c, d; AUC: Gz vs. controls p < 0.001, Gz vs. G28z or GBBz or G28BBz p < 0.001). GBBz T cells expanded faster and to a greater extent than G28z T cells (day 7: G28z mean 3.2, SD 0.021, GBBz mean 6.0, SD 0.25, G28z vs. GBBz p < 0.001; day 10: G28z mean 4.9, SD 0.19, GBBz mean 7.2, SD 0.54, G28z vs. GBBz p < 0.001). No difference was observed between GBBz and G28BBz T cells. Thus, inclusion of the 4-1BB endodomain in GPC3-CARs results in enhanced T-cell proliferation and improved survival of T cells after serial killing of GPC3-positive tumor cells in vitro.
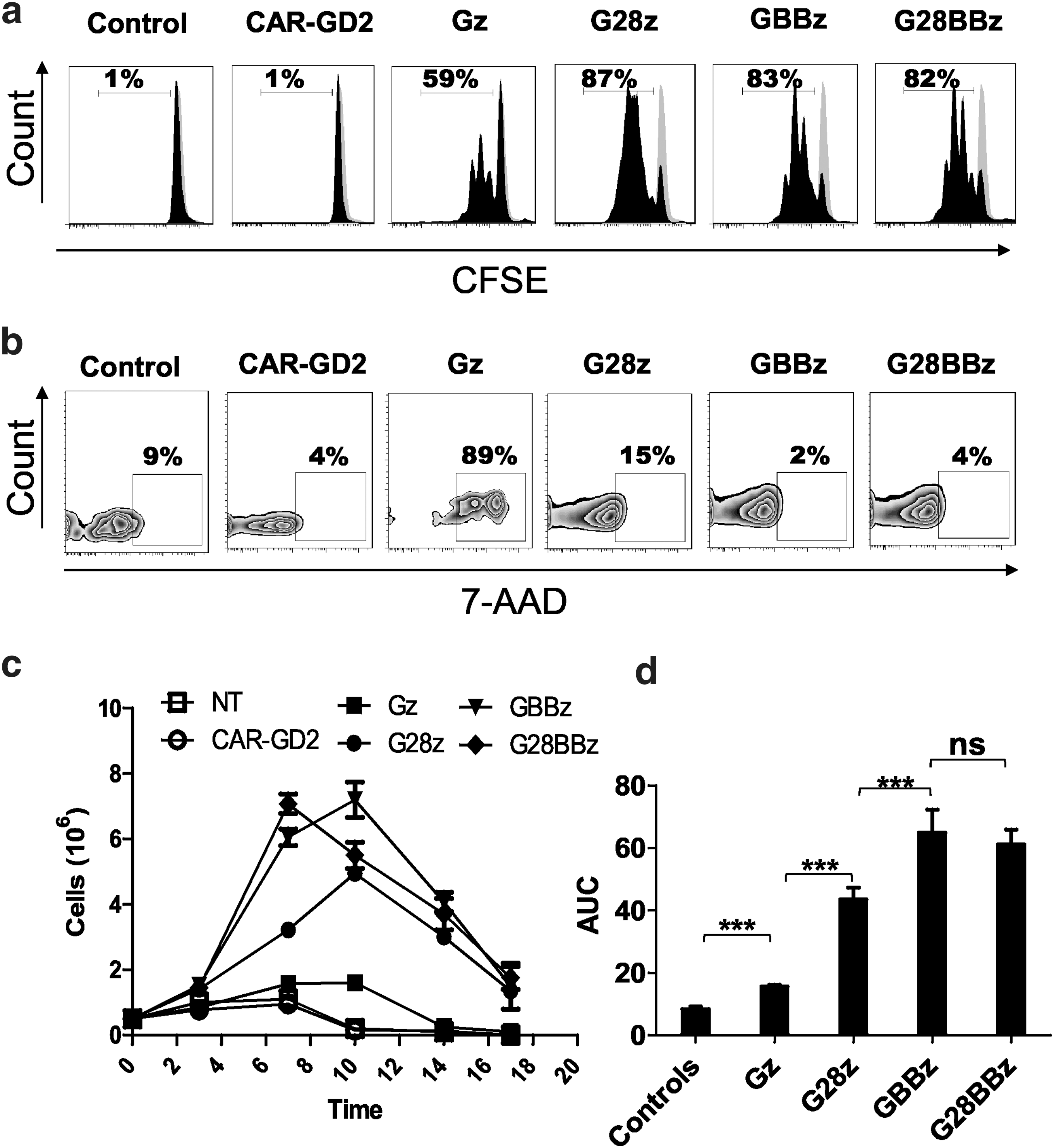
4-1BB improves survival of GPC3-CAR T cells after repeated stimulation by GPC3-positive target in vitro. GPC3-positive Huh-7 was cocultured at 1:1 ratio with GPC3-CAR T cells and cells were replated every 3 days with fresh tumor cells without the addition of cytokines. (
Costimulation by 4-1BB results in enhanced antigen-specific GPC3-CAR T-cell expansion in vivo
Having established that 4-1BB costimulation results in superior T-cell proliferation in vitro after repeated stimulation, we next determined the ability of GPC3-CAR T cells to expand in vivo. NSG mice were IP injected with 2 × 106 Huh-7 cells followed by the IV injection on day 14 of 5 × 106 GPC3-CAR T cells that were genetically modified to coexpress eGFP/Ffluc. The eGFP.Ffluc construct encodes an enhanced Ffluc, which is optimal to track small number of engineered cells in vivo. 46 Ffluc-expressing T cells and CD19-CAR T cells served as controls. Gz, G28z, GBBz, and G28BBz T cells expanded in comparison to control T cells within 3 days post T-cell injection (p < 0.001; Fig. 5a, b). After day 3, Gz and G28z T-cell populations contracted, as judged by a decrease in bioluminescence signal, whereas GBBz and G28BBz T cells continued to expand peaking on day 6 (GBBz vs. G28z: p < 0.001; G28BBz vs. G28z: p < 0.001) (Fig. 5c). These results demonstrate that 4-1BB costimulation provides superior GPC3-CAR T-cell in vivo expansion.
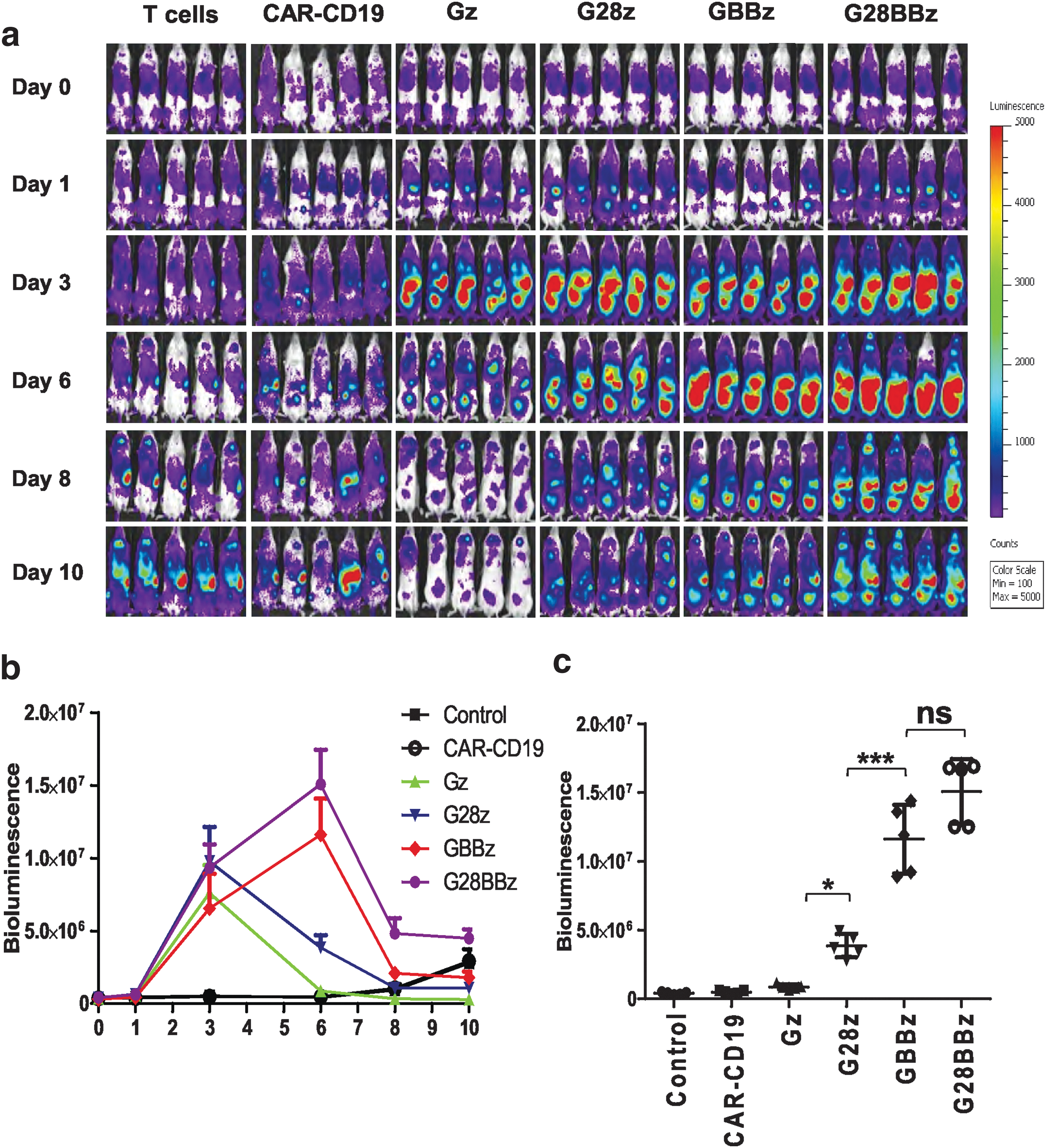
4-1BB improves the expansion of GPC3-CAR T cells induced by GPC3-positive target in vivo. NOD/SCID/IL2γnull (NSG) mice were injected with 2 × 106 GPC3-positive Huh-7 tumor cells intraperitoneally (IP) followed by injection of 5 × 107 green fluorescent protein/firefly luciferase fusion gene (eGFP.Ffluc)–expressing GPC3-CAR T cells intravenously (IV) on day 21. Parental T cells and CAR-CD19 T cells coexpressing eGFP.Ffluc served as controls.
GPC3-positive tumor xenografts can be completely eliminated by GPC3-CAR T cells in vivo
To evaluate the in vivo therapeutic potential of GPC3-CAR T cells, NSG mice were injected with 2 × 106 Huh-7.Ffluc cells IP followed by the IV injection of 1 × 107 Gz, G28z, GBBz, or G28BBz T cells on day 14. Mice injected with phosphate-buffered saline, NT or CAR-GD2 T cells served as controls. GPC3-CAR T cells produced sustained tumor regression as judged by bioluminescence imaging resulting in a survival advantage in comparison to control groups (controls vs. GPC3-CAR T cells p < 0.001; Fig. 6a–c). Since we could not find significant differences between GPC3-CAR constructs, we decreased the dose of GPC3-CAR T cells 10-fold. Mice that had received T cells expressing GPC3-CARs with costimulatory endodomains had significant survival advantage compared with control groups or Gz (p < 0.001, p < 0.001, respectively, Supplementary Fig. S4). However, there were no significant differences among treatment groups of G28z, GBBz or G28BBz T cells judged by tumor bioluminescence or survival of tumor-bearing mice. To confirm that the observed antitumor activity depends on the expression of GPC3 on tumor cells, NSG mice were injected with 2 × 106 A549.Ffluc cells IV followed by the IV injection of 1 × 107 GBBz, or NT T cells on day 5. After injection of GPC3-CAR or NT T cells, tumors continued to grow as judged by bioluminescence imaging, with no survival difference between both groups (Supplementary Fig. S5).
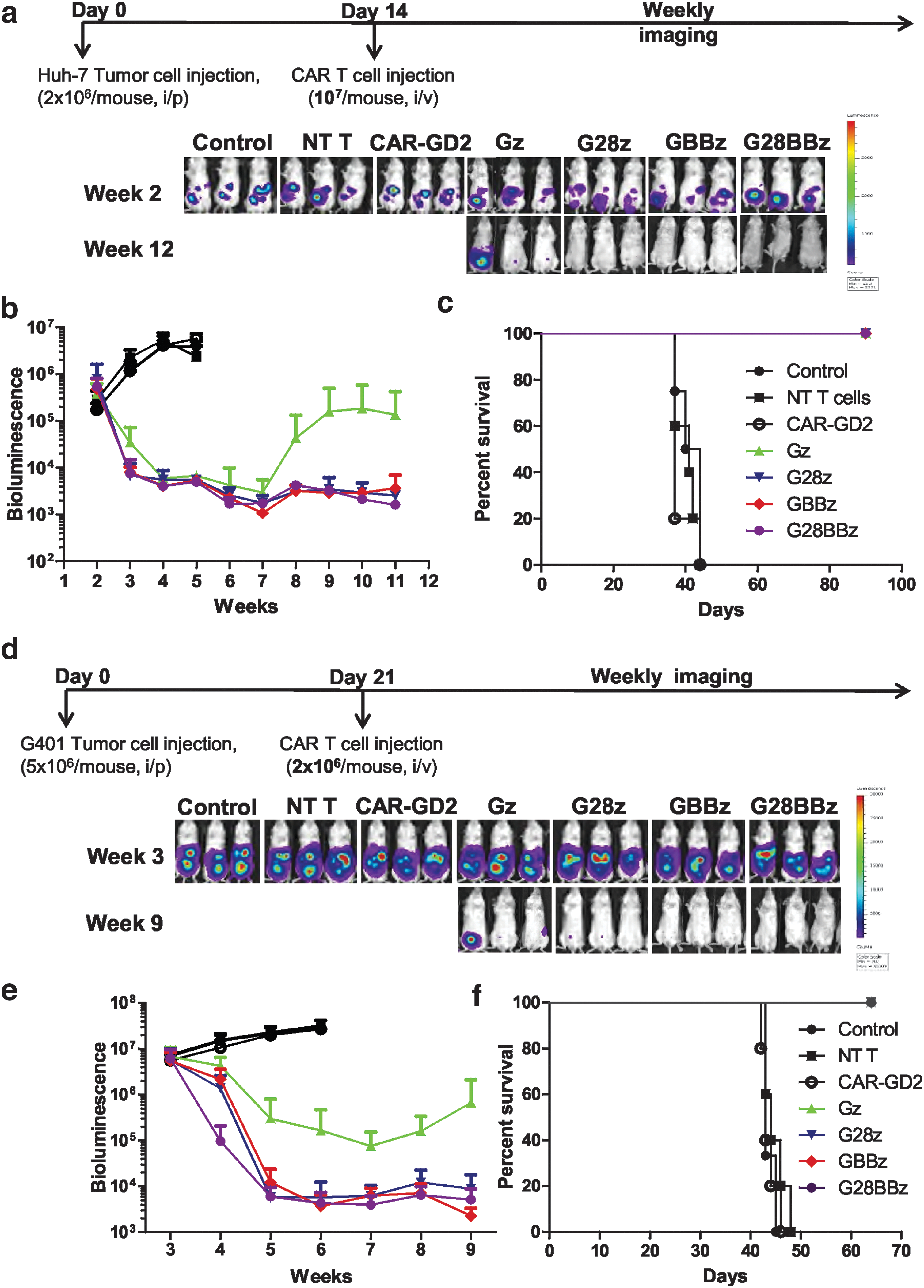
GPC3-CAR T cells eliminate HCC and MRT xenografts in vivo.
To test the ability of GPC3-CAR T cells to eliminate another GPC3-positive tumor we focused on G-401, since compared with other tested tumor cells, it was the least sensitive to cell-mediated cytotoxicity and induced the lowest amount of cytokine production in GPC3-CAR T cells in our in vitro experiments (Figs. 2, 3). On day 21, 5 × 106 G-401.Ffluc cells were injected IP, followed by the IV injection of 2 × 106 Gz, G28z, GBBz, or G28BBz T cells (Fig. 6d). Mice injected with phosphate-buffered saline, NT, or CAR-GD2 T cells served as controls. All GPC3-CAR T cell treated mice had a survival advantage compared to controls (p < 0.001) (Fig. 6f). Injection of G28z, GBBz, and G28BBz T cells resulted in significantly greater reduction in tumor burden compared with Gz T cells and as judged by tumor bioluminescence over time (AUC p = 0.0083) (Fig. 6e). Similar to the Huh-7 xenograft model, we did not find significant differences between G28z, 4-1BBz, and G28BBz T cells in regards to antitumor activity and overall survival (Fig. 6f).
Discussion
The early phase testing of CAR T cells in patients with solid tumors have generated modest results compared to results obtained testing CD19-specific CAR T cells in patients with B-cell leukemias and lymphomas, highlighting the need for systematic testing and optimization of this technology for solid tumors. We generated a set of GPC3-CARs and compared their antitumor potential against GPC3-positive solid tumors (hepatoblastoma, HCC, MRT) in vitro and in vivo with the goal of defining the CARs with the most promising antitumor properties for further clinical development. We found that the inclusion of 4-1BB in GPC3-CARs induced Th1-like polarization of T-cell cytokine production and stimulated higher rates of in vitro proliferation and in vivo expansion compared with CARs with CD28 endodomain or without costimulation. In contrast, the inclusion of CD28 induced high levels of IL-10. We show that GPC3-CAR T cells have robust antitumor activity in two xenogeneic tumor models. In addition to hepatoblastoma and HCC, we demonstrate for the first time that malignant rhabdoid tumors can be effectively targeted with CAR T cells.
Since GPC3 is expressed on several solid tumors but not expressed on mature healthy tissues, it is an attractive immunotherapeutic target. 19 –25,36,37,56 We tested the specificity and antitumor potential of T cells expressing GPC3-CARs with or without CD28 and/or 4-1BB costimulatory endodomains. The generated CAR T cells specifically killed GPC3-positive tumor cells.
Studies have shown that the inclusion of costimulatory endodomains in CARs can enhance T-cell IFN-γ and IL-2 production.
44,49,50,57,58
We noticed that antigen dependent stimulation through GPC3-CARs with costimulatory endodomains did not always result in higher production of cytokines compared with the first generation GPC3-CAR and that cytokine production was differentially affected by CD28 and 4-1BB costimulatory endodomains within GPC3-CARs. Indeed, the inclusion of 4-1BB induced significantly higher levels of Th1 cytokines IFN-γ and GM-CSF, whereas the inclusion of CD28 resulted in higher levels of IL-10 and IL-4. Our group has recently described a similar pattern of Th1/Th2 polarization of natural killer T (NKT) cells expressing CAR-GD2s with 4-1BB- or CD28 endodomains.
48
However, the preferential induction of Th2 cytokines by CD28
Consistent with other published studies, 63,64 we found improved proliferation and expansion of T cells with the inclusion of costimulatory endodomains in GPC3-CARs. We observed that GBBz and G28BBz induced superior T-cell expansion both in vitro and in vivo compared to Gz and G28z. Clinical testing of CAR T cells have shown that longer T-cell persistence is associated with better antitumor effect and overall outcome. 6,13 However, in our xenograft models the improved T-cell expansion and/or Th1 cytokine profile endowed by GBBz and G28BBz did not translate into a survival benefit compared with G28z in treated mice. This is most likely due to the limitations of NSG mouse models—foremost, the induction of xenogeneic GvHD by mature human T cells and the lack of an immunosuppressive tumor microenvironment. 63,65 Thus, novel models, preferentially immune competent animal models, are needed to study CAR T-cell/tumor cell interactions in vivo. However, even immune competent animal models do not adequately recapitulate “human tumors.” Thus, the definitive answer for choosing GBBz or G28BBz for later stage clinical testing may have to come from a clinical trial in which both CARs are compared within individual patients. 66
In conclusion, GPC3-CARs with 4-1BB endodomains endow T cells with a Th1-biased cytokine profile, superior proliferative potential, and potent therapeutic activity in vivo, justifying further clinical development.
Footnotes
Acknowledgments
The authors would like to thank Dr. Malcolm Brenner (Center for Cell and Gene Therapy, Baylor College of Medicine, Houston, TX) and Dr. Milton Finegold (Department of Pathology, Baylor College of Medicine, Houston, TX) for constructive discussions. We also thank Dr. Mitchell Ho (National Institutes of Health, Bethesda, MD) for providing YP7 mAb. This work was supported by National Institutes of Health/National Cancer Institute grants (K12 CA090433 to A.H.; RO1 CA116548 to L.M.) Pablove Foundation Research Award (A.H.), and the Pediatric Pilot Award of Baylor College of Medicine (A.H.).
Author Disclosure
The Center for Cell and Gene Therapy has a research collaboration with Cell Medica and Bluebird Bio. S.G., L.M., G.D., W.L., and A.H. have patent applications in the field of T-cell and gene-modified T-cell therapy for cancer.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.