
Abstract
The beta-thalassemias are inherited anemias caused by mutations that severely reduce or abolish expression of the beta-globin gene. Like sickle cell disease, a related beta-globin gene disorder, they are ideal candidates for performing a genetic correction in patient hematopoietic stem cells (HSCs). The most advanced approach utilizes complex lentiviral vectors encoding the human β-globin gene, as first reported by May et al. in 2000. Considerable progress toward the clinical implementation of this approach has been made in the past five years, based on effective CD34+ cell mobilization and improved lentiviral vector manufacturing. Four trials have been initiated in the United States and Europe. Of 16 evaluable subjects, 6 have achieved transfusion independence. One of them developed a durable clonal expansion, which regressed after several years without transformation. Although globin lentiviral vectors have so far proven to be safe, this occurrence suggests that powerful insulators with robust enhancer-blocking activity will further enhance this approach. The combined discovery of Bcl11a-mediated γ-globin gene silencing and advances in gene editing are the foundations for another gene therapy approach, which aims to reactivate fetal hemoglobin (HbF) production. Its clinical translation will hinge on the safety and efficiency of gene targeting in true HSCs and the induction of sufficient levels of HbF to achieve transfusion independence. Altogether, the progress achieved over the past 15 years bodes well for finding a genetic cure for severe globin disorders in the next decade.
Introduction
T
The successful transplantation of donor HSCs in thalassemic patients is potentially curative, but this option is not available to the majority of thalassemic subjects, for whom a suitably matched related donor cannot be found. 2 Because of the greater risks associated with matched-unrelated or mismatched transplants, most thalassemia patients thus have to settle for life-long transfusion therapy and all of its consequences. Moreover, despite the considerable improvement in the life expectancy of transfusion-dependent individuals in the last decades, 3 –5 the risk of serious complications arising over the long-term from viral infections, iron toxicity, and liver cirrhosis still remains. 6 These medical risks, together with the socioeconomic cost of chronic beta-thalassemia, warrant the pursuit of curative therapies.
Rationale for Globin Gene Transfer to Cure Beta-Thalassemia
The goal of globin gene transfer is to restore the capacity of the thalassemic subject's own blood-forming stem cells to generate RBCs with a normal hemoglobin content. 7,8 Only transduced HSCs can provide long-term clinical benefits through productive erythropoiesis based on a normalized alpha:beta globin chain synthesis ratio. The goal of cell and gene therapy for thalassemia is thus to achieve transfusion independence without exposing patients to the risks of HSC transplantation from a suboptimally matched donor. For patients who lack an HLA-matched donor and thus have a higher risk of mortality after allogeneic HSC transplantation, globin gene transfer in autologous stem cells offers the prospect of a curative stem cell-based therapy. 2
Preclinical Proof-Of-Principle and Safety Studies
The implementation of globin gene transfer for the treatment of severe beta-thalassemia requires the efficient introduction of a regulated human β- (or β-like) globin gene in HSCs. The beta-globin gene must be expressed in erythroid-specific fashion and at high level, especially for the treatment of transfusion-dependent β0-thalassemias. After exhaustive and systematic testing of numerous lentiviral vector designs, we identified in the late 1990s several combinations of genomic sequences that could be stably transferred at high efficiency in murine HSCs and express in their erythroid progeny the human beta-globin gene at therapeutic levels. 9 This study opened up the field, which for well over a decade had previously failed to achieve this goal using various other vector systems. The highest performing globin lentiviral vector termed, TNS9 (Fig. 1), encodes a particularly potent combination of promoter, intron, enhancer, and locus control region elements, 10 sufficient to effectively correct the thalassemia syndrome in beta-thalassemic mice. 9 In this and subsequent studies, 9,11,12 we demonstrated that thalassemic mice engrafted with TNS9-transduced bone marrow cells corrected their anemia, extramedullary hematopoiesis, and iron accumulation in peripheral tissues and organs. 9 In a lethal model of β0-thalassemia major, wherein mice succumb within 60 days of birth to severe anemia, massive splenomegaly, extramedullary hematopoiesis, and hepatic iron overload, we showed rescue and long-term survival after TNS9 transfer in fetal liver HSCs. 12 In large cohorts of mice, we did not observe evidence of vector silencing over time in primary, secondary, or even tertiary chimeras (unpublished observations), indicating that the TNS9 vector functioned continuously over the 26-month study duration. Several groups subsequently generated variants of the TNS9 vector and also reported curative responses in similarly treated mice presenting with beta-thalassemia or sickle/beta-thalassemia (reviewed in refs. 2,7,8,13 ). Thus, multiple studies in different animal models established that correction of anemia and secondary organ damage caused by iron accumulation was feasible using lentiviral vectors encoding a regulated human beta- or beta-like globin gene (β-like chains include the gamma chain and mutant beta chains) and strongly supported the merit of transferring a human globin gene in autologous HSCs as a rational alternative to high-risk, non–matched-related donor transplantation in patients with severe beta-thalassemia. In view of the high performance of the TNS9 vector and the vast amount of data collected over several years with this vector, including extensive safety data summarized below, we selected the TNS9 transcription unit for clinical investigation.
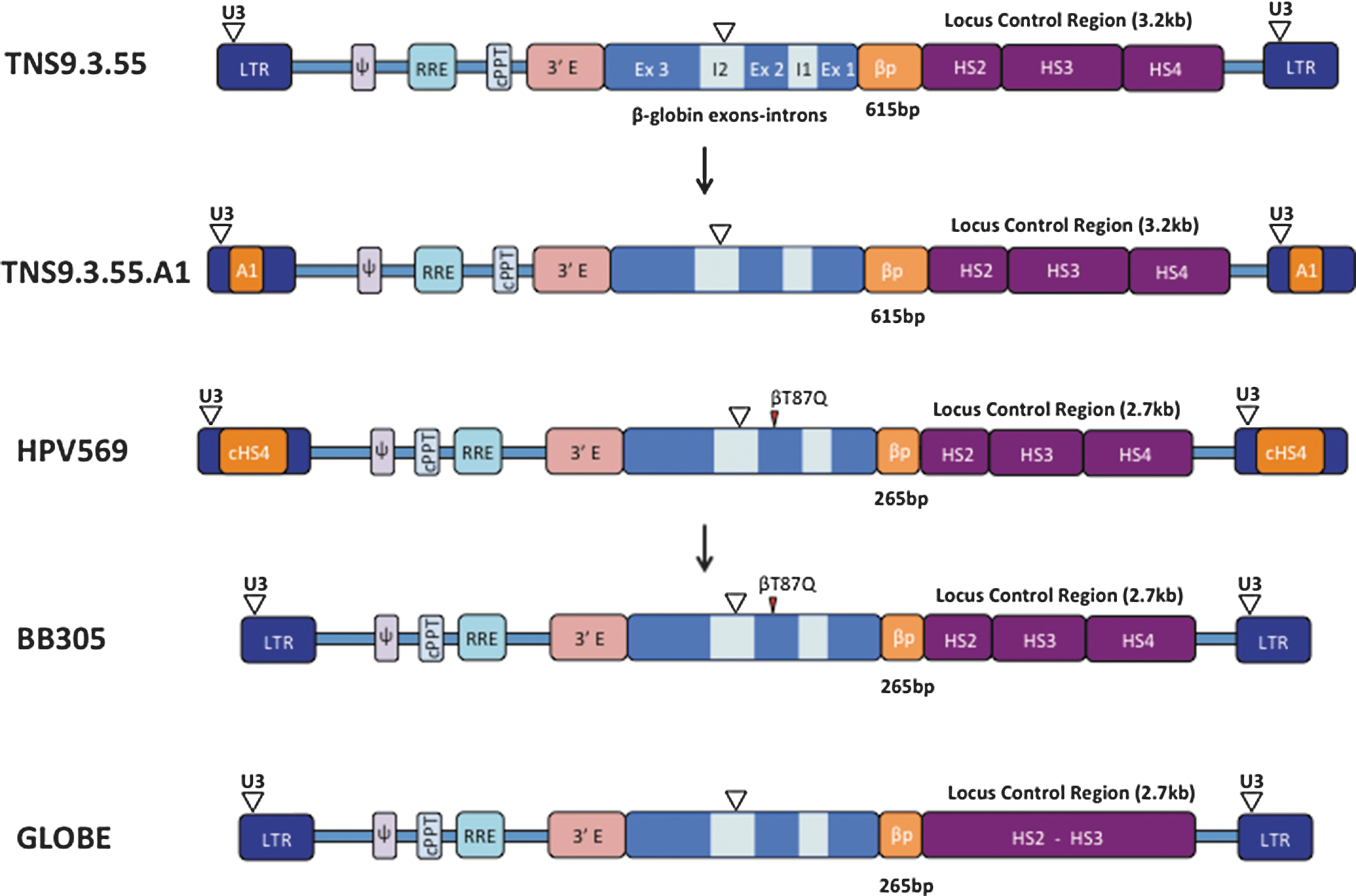
Structure of integrated self-inactivating lentiviral vectors currently approved for clinical trials in β-thalassemia. From top to bottom: TNS.9.3.55 (LTR, HIV long terminal repeat; U3, LTR U3 region, partially deleted; Ψ, HIV Psi sequence; RRE, HIV Rev responsive element; cPPT, HIV central polypurine tract; 3′E, human β-globin 3′ enhancer; Ex, exon; I, intron; βP, β-globin promoter; HS, DNAseI hypersensitive site; bp, base pair; kb, kilobase; inverted open triangle, deletion). TNS9.3.55.A1, a TNS9.3.55-derivative containing the human insulator A1. HPV569 (cHS4, chicken HS4 region insulator; βT87Q, β-globin T87Q mutation, which confers additional antisickling activity to the beta chain 80 ). BB305, a HPV569-derivative without the cHS4 insulator. GLOBE, a β-globin lentivector without the HS4 region.
A critical aspect in designing and selecting a vector for therapeutic application is its safety profile. The major concern associated with the use of retroviral vectors is that of insertional oncogenesis, which in its extreme form may lead to leukemia. Leukemia formation is caused by a combination of events involving the activation of an endogenous oncogene by an integrated retroviral vector complemented by additional mutations accumulating over time in the same clone. 14 Such serious adverse events have been observed in patients with X-linked severe combined immunodeficiency and Wiskott–Aldrich syndrome that were treated with HSCs modified with gamma-retroviral vectors comprising two complete long terminal repeats. 15 –18 Unlike these vectors, globin vectors are erythroid-specific and do not express in HSCs or in progenitor cells of any lineage, which considerably reduces the risk of transforming these cells. 19 Furthermore, globin vectors activate only in late-stage erythroid cells, shortly before enucleation, which serves as a natural safeguard against the oncogenic transformation and clonal expansion of erythroblasts. These advantageous features of globin vectors notwithstanding, we conducted, as part of our Investigational New Drug (IND) application to the U.S. Food and Drug Administration (FDA), extensive safety studies evaluating a series of TNS9-like vectors and control vectors in large cohorts of thalassemic mice (unpublished observations). In nearly 300 recipient mice, including primary and secondary recipient mice, followed for an average 12 and 20 months, respectively, we did not observe a single case of leukemia. Although such results cannot guarantee the safety of these vectors, these outcomes are superior to those obtained with vectors encoding long terminal repeats. Based on the above efficacy and safety data, the FDA granted us in 2012 the first approval in the United States for a clinical trial investigating globin gene transfer in thalassemia patients. This critical approval paved the way for other groups to subsequently obtain FDA approval for phase I/II globin gene transfer studies for the treatment of beta-thalassemia and sickle cell disease. In Europe, the team of Philippe Leboulch and Marina Cavazza-Calvo in Paris, France, proceeded to evaluate a variant vector called β87/HPV569 20 (Fig. 1).
First Clinical Steps: CD34+ Cell Mobilization Studies
Before proceeding to clinical studies, we decided to first assess the safety and feasibility of harvesting HSCs from thalassemia patients. These collected CD34+ cells from thalassemic subjects were also useful to optimize globin gene transfer under “current Good Manufacturing Practice” (cGMP) conditions. We conducted a pilot trial to investigate the safety and effectiveness of mobilizing CD34+ HSCs in adults with beta-thalassemia major.
21
A secondary objective of this clinical study was to assess whether patient CD34+ HSCs could be transduced under cGMP conditions at levels sufficient for proceeding to a therapeutic clinical trial, utilizing the TNS9.3.55 lentiviral vector. All five subjects enrolled tolerated G-CSF well with minimal side effects, confirming prior mobilization studies in pediatric
22
and adult
23
thalassemic subjects. All CD34+ cell collections achieved the minimum targeted dose of 8 × 106 CD34+ cells/kg after leukophereses on days 5 and 6. Using clinical-grade TNS9.3.55 vector stock, we demonstrated gene transfer in the range of 0.2–1.5 vector copies per cell in small-scale patient CD34+ cell transduction studies, 0.2–0.6 in scaled-up validation runs performed under cGMP conditions. The transduced CD34+ cells maintained their potential to engraft NOD/scid-γc
null mice, maintaining a stable vector copy number 6 months posttransplant.
21
This validated procedure for stem cell collection and globin gene transduction was approved by the FDA and implemented in the first U.S. trial to evaluate globin gene transfer in patients with severe inherited globin disorders (NCT01639690 at
The group of Evangelia Yannaki in Thessaloniki, Greece, subsequently conducted another mobilization study in adult thalassemic subjects, this time combining the use of a CXCR4 antagonist with G-CSF. 23 This resulted in higher CD34+ yields, without incurring additional toxicity. Interestingly, the CD34+ cells mobilized in this way, which were also transduced with the TNS9.3.55 vector, appeared less effectively transduced than CD34+ cells mobilized with G-CSF alone. This difference may, however, be offset by the larger cell yield and possibly a greater hematopoietic quality of the cell product. Both mobilization regimens appear to be well suited for collecting CD34+ cells in thalassemic subjects.
Phase I Clinical Trials
Four trials evaluating globin gene transfer in thalassemic subjects have opened and accrued patients. The first two opened in Paris and New York, evaluating the β87/HVP569 and TNS9.3.55 vectors, respectively (Fig. 1). Two other trials subsequently opened, one utilizing the vector BB305, a variant of β87/HVP569 (sponsored by bluebird bio, a biotechnology company), and the other the vector GLOBE, developed by Giuliana Ferrari in Milano, Italy. We will soon bring to the clinic the TNS9.3.55.A1 vector, a variant of TNS9.3.55 (Fig. 1).
The Paris trial opened using a low-titer vector stock and without an optimized CD34 cell collection procedure. Nonetheless, one of the four enrolled subjects showed clinical benefit, despite low engraftment of β87/HVP569-transduced HSCs, owing to the remarkable emergence of a single myeloid progenitor clone, spurred by the fortuitous trans-activation of the HMGA2 gene at the site of vector integration. 25 Remarkably, the erythroid progeny of this dominant clone expressed the vector-encoded β87 chain, which accounted for about one-third of the total hemoglobin starting one year after the cell infusion. Another third came from the subject himself, who did not have β0-thalassemia but a β+ form of the disease. The vector alone would not have resulted in a sufficient increase in total hemoglobin to achieve transfusion independence, but another remarkable event occurred in this patient, consisting of an unusually elevated and sustained expression of hemoglobin F (which results from the activation of the endogenous fetal or gamma-globin gene). The sum total of endogenous HbA, the induced HbF, and the vector-derived HbAβ87 added up to 9–10 g Hb/dl, crossing the threshold for transfusion independence. This extraordinary adverse event, owing to a single clone providing clinical benefit for 7 years, eventually regressed with the presumed exhaustion of the mutated clone. Most importantly, this clone never progressed to leukemia. The other three subjects did not achieve transfusion independence. 26 Owing to the genomic instability of the β87/HVP569 vector, 25 bluebird bio removed the duplicated cHS4 sequence that had been inserted into the vector U3 region upon taking over the therapeutic program from Genetix Pharma, thus creating the BB305 vector (Fig. 1). The use of HVP569 has now been discontinued. Seven thalassemia subjects have been infused with CD34+ cells transduced with BB305, with clinical benefit in patients with β+- or βE-thalassemia but not in patients with β0-thalassemia. 27
The New York trial, which has been conducted in close collaboration with Aurelio Maggio in Palermo, Sicily, and Paolo Moi and the late Renzo Galanello in Cagliari, Sardinia, opened in 2012, enrolling adult subjects with transfusion-dependent beta-thalassemia major who lack an HLA-matched donor. The treatment is based on the administration of autologous CD34+ hematopoietic cells transduced with the TNS9.3.55 vector, a lentiviral vector encoding the wild-type human β-globin gene. The cells are transduced ex vivo; frozen down; tested for transduction efficiency, sterility, and other release criteria; and thawed immediately before infusion after host conditioning. Most significantly, this protocol differs from the Paris and bluebird bio trials in calling for reduced intensity conditioning rather than a fully myeloablative regimen as used in the HPV569/BB305 studies. It is indeed established for other blood disorders treated with transduced autologous CD34+ cells that conditioning with busulfan at 8 mg/kg is sufficient to achieve therapeutic engraftment of modified HSCs. 28,29 Our results in four subjects infused to date have shown very stable engraftment without clonal dominance. 30 Although there is general consensus that a nonmyeloablative conditioning regimen would have many advantages, including decreased toxicity, rapid hematopoietic recovery, and shortened hospitalization, it remains to be determined whether it is sufficient to support the efficacy of this treatment.
Instead of producing another lot of the same vector, we plan to resume our trial with a variant vector that incorporates a safety element termed an insulator. Indeed, although our extensive preclinical studies did not reveal a single case of vector-induced leukemia, and though no severe adverse event has been reported to date in any of the globin trials, the occurrence of clonal expansion in a subject treated with the HPV569 vector commands caution and stands in support of incorporating additional safety features in the vector. Insulators are common genomic elements that exhibit barrier function, which marks transitions between euchromatin and heterochromatin, and/or enhancer blocking activity, which directionally controls enhancer–promoter interactions. 31,32 Within retroviral vectors, insulators can diminish position effects and vector silencing. 33 –35 George Stamatoyannopoulos and colleagues in Seattle, WA, have conducted the first systematic search for all human insulators marked by a highly occupied CTCF binding site. 36 In collaboration, we extensively characterized one of these elements and have incorporated it into the TNS9.3.55.A1 vector (Fig. 1).
The Milano trial utilizes another variant of TNS9, this time lacking the HS4 element 37 (Fig. 1). The protocol just opened and the first subject was infused in late 2015. 38 It differs from the above by the cell infusion route, which is not intravenous but medullary via the iliac crest. The conditioning used by the TIGET team includes thiotepa and treosulfan. The required age for enrollment onto this protocol will be 18 years for the first three, followed by three children age 8–17 years, before allowing the treatment of subjects aged 4–7 years.
Emergence of Gene Editing and Gene Repair Technologies
While lentiviral vectors have entered the critical stage of first-in-human clinical evaluation, new genetic technologies are emerging in the laboratory. Thus, an expanding set of genome editing tools is creating novel prospects for impacting directly on disease mutations and targeting genes or regulatory sequences. 39 These technologies exploit different physiological DNA repair pathways, depending on the desired outcome.
Homology-directed repair (HDR) is the main pathway for the repair of mutated genes. It is active in the S/G2 phases of the cell cycle and requires an exogenous DNA template containing the correct DNA sequence flanked by sequences homologous to the targeted mutation. Another pathway, termed the nonhomologous end joining (NHEJ) pathway, is co-opted to introduce mutations. It is active throughout the cell cycle and does not require homologous DNA templates during the repair process. Though NHEJ-based approaches are more efficient than HDR-based ones, NHEJ is error-prone and the outcome of DNA modifications cannot be controlled. In contrast, HDR permits the incorporations of specific, predetermined changes to the target sequence. 40,41
The laboratory of Maria Jasin demonstrated that the deliberate introduction of double-strand breaks (DSBs) at the target site stimulates HDR several hundred-fold. 42 Since then, a variety of DNA sequence-specific nucleases have been developed, including zinc-finger nucleases (ZFNs), meganucleases (MNs), transcription-activator-like effector nucleases (TALENs), and the RNA-guided nuclease system CRISPR/Cas (clustered regularly interspaced short palindromic repeats/CRISPR-associated). 43,44 To carry on their function, all of these nucleases rely on specific DNA interaction modules and a nuclease domain. Meganucleases utilize a single domain for both activities. ZFNs and TALENs share the same nuclease domain, whereas the DNA-binding modules are based on either the zinc-finger domain or TALE domain, respectively. CRISPR/Cas is a ribonucleoprotein, wherein the RNA component (gRNA) binds to the target DNA, and the Cas protein (Cas9 or Cpf1) introduces the DSB using two nuclease domains (for details see 44,45 ). DNA nickases or nuclease-null enzymes fused to chromatin-modifying modules have been recently developed for most of the nuclease platforms, thus expanding the number of tools for specific biological applications. 46
Familiar Challenges: Delivery and Specificity
A major challenge to clinical application is to efficiently introduce the required materials in human stem cells to enable efficient and dependable completion of these targeted reactions. Over the years, a number of approaches have been used to deliver these genome-editing components. DNA transfection has been the standard approach. Alternative nonintegrating viral vehicles have emerged to address the toxicity to primary cells in the instances where these (e.g., hematopoietic cells) are sensitive to either transfection or nucleic acids. These recombinant viral vectors belong to three groups: lentivirus (LV), adenovirus (AdV), and adeno-associated virus (AAV). 39 All these vectors have been used to deliver the genome-editing tools required for NHEJ-based approaches. Only AdV vectors can carry all the components necessary for HDR-based genome editing in a single vector, independent of the nuclease platform and the HDR approach. Extrachromosomal HDR DNA templates can consist of either double- or single-stranded DNA (dsDNA or ssDNA) of variable length. 47 For gene addition or incorporation of selection DNA cassettes to enrich for corrected cells, large extrachromosomal DNAs are needed. For gene mutation repair or addition of short DNA sequences, the donor template can be of smaller size, usually requiring short homology arms. Synthetic single-stranded oligodeoxynucleotides (ssODNs) have been successfully utilized, with targeting efficiencies enhanced by using nuclease-resistant modified ssODNs. 48
Another major challenge is to ensure the specificity of targeting. Unwanted cleavage/modification of potential genomic off-target sites is a known side effect. 49 Nucleases with improved DNA specificity are being developed to address this issue. Limiting nuclease expression can also provide further safety. 50 Chimeric nucleases can be delivered as RNA, which allows for high short-term expression resulting in efficient genomic target cleavage. Several current strategies using any of the above nuclease platforms to modify genomic sites in human primary hematopoietic cells take advantage of this approach. 51 –54
Genome Editing Strategies to Correct B-Globin Deficiencies
Two main research directions based on gene editing have emerged in the globin field, one aiming to repair the β-globin gene (HBB) and the other to reactivate the γ-globin gene (HBG), which is silenced after birth but is sometimes capable of compensating for a β-gene defect if it remains expressed at a sufficient level.
HBB gene repair
ZFNs, TALENs, and CRISPR/Cas9 nucleases have been designed to target the β-globin gene, which in combination with plasmid DNA, nonintegrating lentiviruses (NILV), or ssODN have enabled the correction of specific β-thalassemia mutations, the sickle cell mutation, or the incorporation of a normal β-globin cDNA in human stem cells. 52,55 –60 Most of these studies have been performed in pluripotent stem cells, whose potential clinical application is not within reach owing to the lack of an efficient differentiation process to produce adult long-term hematopoietic stem cells (LT-HSCs) and other safety concerns. On the other hand, β-globin gene repair has been obtained in human primary HSCs; however, the levels of correction in LT-HSCs is less than 1% of the total alleles, which is not clinically suitable. 52 Similar results have been obtained for other gene targets in human HSCs. 51 Evidently, improvements in HDR efficiencies in LT-HSCs need to be made before undertaking any clinical application.
HBG gene re-activation
A fraction of patients with β-thalassemia or sickle cell disease (SCD) present a condition called hereditary persistence of hemoglobin F (HPHF). 61,62 In general, HPHF patients present a mild form of β-thalassemia or SCD. Increased γ-globin expression in β-thalassemia attenuates the imbalance of α/non-α globin chains, which ameliorates erythropoiesis. In SCD-HPHF patients, increased levels of HbF delay HbS polymerization and increase Hb solubility, thus reducing RBC sickling. Pioneering genome-wide association studies (GWAS) conducted by Antonio Cao and collaborators in HPHF subjects identified the responsible cis-acting genetic variants within the β-globin gene cluster, as well as variations at two loci, Bcl11a on chromosome 2 and HBS1l-MYB on chromosome 6. 63 –65 Further investigation aiming to determine the role of Bcl11a in globin gene regulation found that Bcl11a acts as a negative regulator of γ-globin expression, promoting the switch from fetal to adult hemoglobin production (Fig. 2). 66 –68
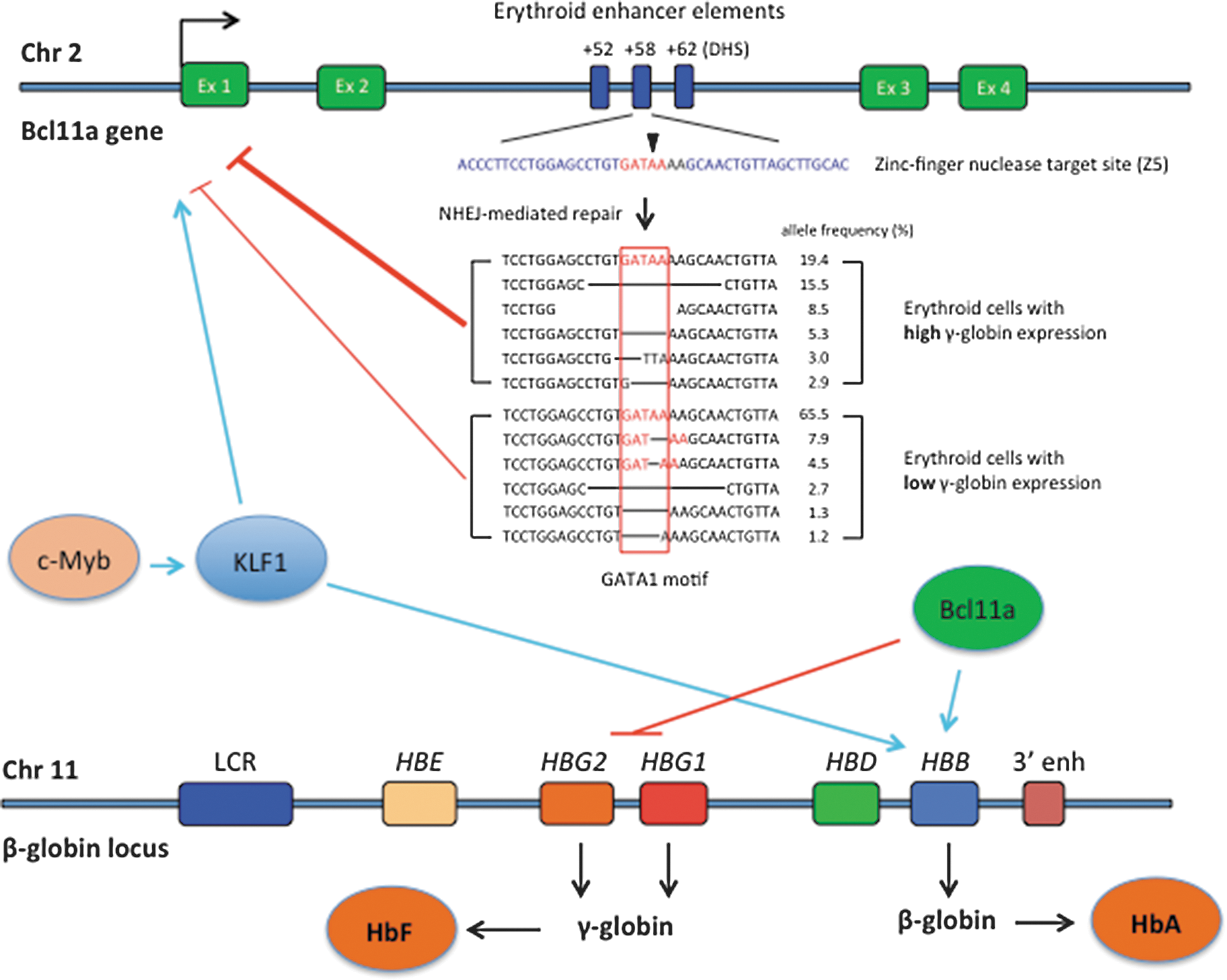
Reversal of globin switching through Bcl11a gene editing. Genes c-Myb, KLF1, and Bcl11a positively regulate β-globin gene expression (light blue arrows). Klf1 also induces Bcl11a expression (light blue arrow). Bcl11a represses the expression of γ-globin (red line). Zinc-finger nuclease-mediated cleavage at a target sequence (blue) located in the DHS (DNAse hypersensitive site) +58 region generates DNA sequences that no longer contain a putative GATA1 transcription factor binding motif (in red). High frequency of those GATA1-less sequences in cell population is associated with reduced Bcl11a expression (red lines) and consequently higher γ-globin expression (sequence data from ref. 71 ). Chr, chromosome; Ex, exon; LCR, locus-control region; HBE, ɛ-globin gene; HBG, γ-globin gene; HBD, δ-globin gene; HBB, β-globin gene; 3′ enh, 3′ enhancer; HbF, fetal hemoglobin; HbA, adult hemoglobin.
Several occurring genetic variants in the Bcl11a gene have been associated with low Bcl11a expression and concomitant increased levels of γ-globin that are associated with milder disease. 63,69 These variants are located in a region of the intron 2 of the Bcl11a gene that is associated with an erythroid-specific enhancer chromatin signature. 70 Further mapping determined that these genetic variants are located in distinct DNAse I hypersensitive sites (DHSs), designated +55 and +62 (Fig. 2), located 55 and 62 kb from the transcriptional start site of the human Bcl11a gene, respectively. 70 Interestingly, a third DHS, +58, shows no association with any genetic variant obtained through GWAS.
Using gene-editing technology, two studies have determined that the +58 region plays a major role in regulating the expression of Bcl11a. In the first study, TALENs were designed to target each of the above DHSs. TALEN-induced mutations at +58 yielded the higher levels of γ-globin expression. Further screening of transcription factor footprints in human erythroblasts revealed a 162 bp region of the +58 DHS. ZFNs were designed to specifically target 5 footprints in human CD34+ cells. Modified cells were differentiated to erythroid cells and analyzed for Bcl11a and γ-globin mRNA levels. Genetic modifications with a ZFN cleaving at a GATA1 motif resulted in marked increase of γ-globin transcripts associated with a corresponding decrease of Bcl11a mRNA levels. 71 Further analysis of erythroid cells with high and low γ-globin mRNA levels showed marked enrichment of alleles without the GATA1 motif (see Fig. 2 for details). In the second study, Canver et al. performed an in situ saturating CRISPR/Cas9-based mutagenesis of the Bcl11a enhancer region in human erythroblasts. Analysis of the high-HbF expressing cells showed an enrichment of gRNA sequences targeting mainly the +58 DHS. 72 The top gRNAs targeted either a sequence containing the predicted GATA1 motif or a sequence containing predicted motifs for RXRA, EHF, ELF1, and STAT1. Sequences contained modifications at these sites at higher frequencies in cells expressing high HbF levels. Combined, these studies showed that the GATA1 motif in the +58 DHS plays a major role in the regulation of Bcl11a expression in human erythroid cells. These studies not only have undercovered the role of Bcl11a in this complex regulation of globin gene expression but also revealed a potential new genetic approach to reverse globin switching for the treatment of hemoglobinopathies. Further clonal analyses are needed to establish the correlation between allele modification, Bcl11a expression levels, and ultimately the levels of HbF and HbA in erythroid cells. It remains to be demonstrated whether targeting Bcl11a will suffice to revert β+ or β0 thalasssemias.
Several other exciting uses of genome-editing tools have been described that could potentially result in clinical benefit. The common theme among these new strategies is the direct activation of the gamma-globin gene, through three distinct mechanisms: chimeric transcription factors, 73 chimeric histone modifiers, 74 and forced DNA looping. 75 Of note, all of the above approaches rely on the ex vivo modification of autologous HSCs, which are to be later returned to the patient. Current efforts to develop an in vivo beta-globin gene repair approach have focused in the use of biodegradable PNA/DNA nanoparticles, which can be systemically delivered. 76
Expectations for Cell and Gene Therapy of the Beta-Thalassemias
The β-thalassemias and the companion disorder sickle cell disease are entering a new era where the anticipation of a curative genetic intervention cure is increasingly plausible. Lentiviral vectors are the most advanced approach and have already entered the phase of clinical testing. Gene editing approaches are still at a preclinical stage but offer exciting prospects as well.
Early clinical data show that globin gene delivery through improved and well-manufactured self-inactivating lentiviral vectors can provide significant clinical benefits to β-thalassemia patients without, so far, any major adverse event although a case of major clonal expansion has been documented. In aggregate, 16 patients have been treated on all combined thalassemia protocols and 6 have become transfusion independent. Globin lentiviral vectors are promising, even though their efficacy may be limited by integration site-dependent variegation. Despite their tissue-specific expression, safety concerns remain owing to random gene disruption, as exemplified in one subject in the Paris trial. The impact of this single occurrence is at present difficult to gauge, as no other patient on any of the trials has so far developed a comparable clonal expansion. In this light, the inclusion of genetic elements with enhancer-blocking and chromatin barrier function into globin lentiviral vectors represents an appreciable advance that may enhance the performance and safety of integrating vectors. 77
Current technologies for nuclease-based genome editing provide very efficient cleavage of their genomic target. However, safety concerns regarding cleavage, and concomitant modification, of off-target sites remain to be addressed. Several high-throughput genome-wide analyses of off-target cleavage have been developed, and improvements in sensitivity are expected to quantify with more precision the true specificity of targeted nucleases. It should also be reminded that strategies relying on NHEJ-mediated repair suffer from uncontrolled genomic modifications at the nuclease-targeted site. Additional research to improve HDR efficiency is needed to enable more precise editing.
A comparison of addition and editing approaches is difficult at this time, given that only the lentiviral vectors are in the clinic and that overall patient data are still limited. The advantage of lentiviral vector-mediated globin gene transfer is the proven ability of LVs to efficiently target HSCs, even though this is more challenging with globin vectors than most other retroviral and lentiviral vectors, which have simpler and smaller structures. The remaining challenge of efficient gene transduction seems to be only a technical hurdle at this time, which presumably can be resolved by further optimization of vector design and vector manufacturing. Effecting efficient targeting gene delivery in authentic HSCs, on the other hand, is still constrained by the biology of DNA repair in HSCs. As this is intensely studied in academia and industry, one can anticipate rapid progress in our understanding of these barriers. In terms of therapeutic efficacy, globin vectors appear to express enough β or β-like chain to ameliorate the condition of the less severe forms of β-thalassemia, such as HbE disease and β+ thalassemias. It remains to be established whether the present vectors will be effective in the more severe β0-thalassemias, without requiring a high vector copy number. As to the derepression of γ-globin expression through Bcl11A targeting, it remains to be seen what levels of HbF will actually be induced and what clinical conditions could be alleviated in this manner.
Both approaches raise genotoxicity concerns. Those associated with semirandom retroviral and lentiviral vectors are by now well known to clinical investigators and regulators, and have the benefit of established methods for their tracking. 78,79 The undesirable mutations introduced by targeted nucleases are more difficult to assess, in part owing to their small size and easier escape from detection. Novel monitoring assays, however, hold the promise of advancing this field.
One thing is certain at this time: the prospects for finding a genetic cure for the severe thalassemias and sickle cell disease have dramatically improved over the past 15 years. One may anticipate dramatic changes in the management of these disorders in the next decade.
Footnotes
Acknowledgments
The authors thank the Stavros Niarchos Foundation, The New York Stem Cell Science (NYSTEM), and the Doris Duke Charitable Foundation for their support of their research on thalassemia and sickle cell disease.
Author Disclosure
No competing financial interests exist.