
Abstract
In spite of their successes against hematologic malignancies, immunotherapeutic interventions for the treatment of patients with glioblastoma (GBM) have thus far been unsuccessful. This is in part due to the presence of a tumor microenvironment that fosters neoplastic growth and protects the tumor from destruction by the immune system. A novel genetically engineered macrophage-based platform has been developed with the potential to minimize the effects of the suppressive tumor microenvironment and improve innate and adaptive antitumor immune responses. A newly described lentiviral expression system was validated for the generation of transduced monocytes and monocyte-derived macrophages, and transgene expression was shown to be stable over the course of weeks to months, both in vitro and in a mouse xenograft model of GBM. Furthermore, the genetically engineered macrophages (GEMs) neither caused morbidity in animals nor contributed to accelerated tumor growth. The versatility of GEMs is also highlighted by showing that they can be engineered to secrete proteins that either reduce immune suppression, such as the soluble transforming growth factor beta receptor II, or promote immune cell activation, by expressing interleukin 21. There is also the potential to prevent GEM-mediated immune suppression by using the CRISPR system to knock out genes responsible for dysfunction of cytotoxic cells, including interleukin 10 and programmed death-ligand 1. Together, these results suggest that GEMs are an ideal cell type for transforming the tumor microenvironment and enhancing antitumor immunity. Importantly, it is anticipated that these findings will have broad applicability to other types of tumors with microenvironments that currently preclude successful immunotherapeutic approaches.
Introduction
G
Macrophages make an ideal therapeutic cell type for restructuring the suppressive TME because they play a central role in the crosstalk between the adaptive and innate immune systems, are efficiently recruited to and retained within the tumor, 9 and survive in the TME even after their polarization toward a pro-inflammatory phenotype. 10 –12 Furthermore, engineered macrophages may be generated from a patient's monocyte population that is discarded during the preparation of therapeutic T cell receptor (TCR) or chimeric antigen receptor (CAR) T cells. We are the first to propose the use of engineered primary macrophages for therapeutic purposes, partly due to the difficulty in genetically manipulating these cells with clinically approved vectors such as human immunodeficiency virus-1 (HIV1)-based lentivirus. Macrophages are refractory to lentiviral transduction because of their expression of a restriction factor, SAMHD1, that depletes the pool of nucleotide triphosphates available for reverse transcription. 13 Recent development of a lentiviral packaging system that generates virions containing viral protein X (Vpx), a simian immunodeficiency virus (SIV)- and HIV2-associated protein that induces the degradation of SAMHD1, has made it possible to deliver genes stably to primary human myeloid cells. 14
A platform is described that takes advantage of this method to evaluate the use of genetically engineered macrophages (GEMs) as a potential therapeutic. The study demonstrates that GEMs: (1) survive in a xenograft model of GBM without impacting animal survival, (2) can be made resistant to reprogramming by tumor-secreted signals, and (3) stably express factors that will promote persistence and activation of endogenous or adoptively transferred NK or T cells. Using this approach, we are poised to investigate the utility of GEMs as a cellular delivery vehicle that can express a multitude of factors that can overturn an immunosuppressive TME and support existing or novel immunotherapies.
Materials and Methods
Isolation of CD14+ monocytes from peripheral blood mononuclear cells
Healthy donor whole blood and apheresis product was acquired from Bloodworks Northwest (Seattle Children's Research Institute IRB #14412). Peripheral blood mononuclear cells (PBMCs) were isolated with a Ficoll-Paque density gradient (GE Healthcare) using standard protocols. 15 CD14+ monocytes were purified from PBMCs with the EasySep Human CD14 Positive Selection Kit (Stemcell Technologies) according to the manufacturer's protocols.
Macrophage differentiation
Isolated CD14+ monocytes were plated in macrophage media (RPMI-1640; Gibco) supplemented with 10% fetal bovine serum (FBS; Hyclone) on tissue culture–treated plastic dishes (Corning) at a density not exceeding 250,000 cells/cm2 in the presence of either 25 ng/mL of macrophage colony-stimulating factor (M-CSF) or 10 ng/mL of granulocyte macrophage colony-stimulating factor (GM-CSF; R&D Systems) at 37°C, 5% CO2. At 72 h after plating, half the medium was replaced with fresh macrophage media supplemented with M-CSF or GM-CSF at final concentrations of 25 and 10 ng/mL, respectively. Monocytes were considered to be fully differentiated to macrophages 6 days after isolation, and could be re-plated as necessary following detachment with 0.25% trypsin (Gibco) and gentle scraping.
Dendritic cell differentiation
Isolated CD14+ monocytes were differentiated to dendritic cells (DCs) following the protocol of Spadaro et al. 16 Briefly, CD14+ cells were plated in RPMI-1640 supplemented with 10% FBS, 100 ng/mL of GM-CSF, and 50 ng/mL of interleukin 4 (IL-4; R&D Systems). Media was replaced and fresh cytokines added 72 h later.
Lentiviral vectors
Unless otherwise stated, all constructs utilized the epHIV7 or epHIV7.2 lentiviral backbones, gifts from Dr. Michael Jensen (Seattle Children's Research Institute). In epHIV7, the CMV promoter of pHIV7 17 was replaced with the EF1α promoter. In epHIV7.2, the EF1α promoter was replaced with a minimal EF1α (lacking the HTLV-1 domain), and the gene for ampicillin resistance was exchanged for kanamycin resistance. The green fluorescent protein-firefly luciferase (GFP-ffluc) fusion protein has been previously described. 18 GFP-ffluc in the epHIV7 backbone was a gift from Dr. Michael Jensen. The mCherry construct was created by Gibson cloning into NheI/NotI of HIV7.2. To generate the soluble transforming growth factor β receptor II (sTβRII) construct described previously, 19 the extracellular portion of human TGF-βRII isoform A (aa 1–159; NCBI accession #NP_001020018.1) was cloned into the NheI/NotI sites of epHIV7.2. The truncated CD19 (CD19t) 20 was inserted by Gibson cloning upstream of the sTβRII gene, separated by a T2A sequence. The IL-21 plasmid was purchased from Origene (plasmid RC215235, containing the coding sequence of NCBI RefSeq NM_021803) and cloned into NheI/NotI of epHIV7.2. CD19t 20 was inserted by Gibson cloning downstream of the IL-21 gene, separated by a T2A sequence. The LentiCRISPRv2 vector was a gift from Dr. Feng Zhang (Addgene plasmid #52961), and guide sequences for IL-10 (GGCTGGCCCTCACCCCAGT) and programmed death-ligand 1 (PD-L1; TCCAGATGACTTCGGCCTTG) were inserted into the BsmB1 site using standard protocols. 21,22 The truncated epidermal growth factor receptor (EGFRt) 23 epitope tag was inserted to replace the puromycin resistance sequence.
Lentivirus production
All viral preparation was conducted in a BSL-2 laminar flow hood. pcVpx and pMDL-X were kind gifts of the Landau lab. 14 RSV-Rev was purchased from Addgene (plasmid #12253). pCMV-G 24 was a gift from Dr. Michael Jensen. 293T cells were purchased from ATCC (CRL-3216), and used below passage 20. 293T cells were plated at 107 cells/15 cm dish in 293T media (Dulbecco's modified Eagle's medium [DMEM] supplemented with 10 mM of HEPES and 1% Glutamax [Gibco] and 10% FBS). After overnight growth, each plate was transfected with pcVpx (4.6 μg), pMDL-X (13.5 μg), RSV-Rev (6.8 μg), pCMV-G (9.5 μg), and the transgene vector (37.8 μg) using the CalPhos Mammalian Transfection Kit (Clontech). After 16 h, media was changed to fresh 293T media. Virus-containing supernatant was collected at 48 h, followed by filtration with a 0.45 μm filter to remove debris and ultracentrifugation at 108,000 g in a Beckman Coulter Optima L-90K ultracentrifuge using the SW-28 rotor for 90 min at 4°C. The viral pellet was resuspended in serum-free DMEM, and the concentration of intact lentiviral particles (LP) was determined by the Quick Titer Lentivirus titer kit, an enzyme-linked immunosorbent assay (ELISA) for lentivirus associated p24 (Cell Biolabs).
High multiplicity of infection transduction of partially differentiated macrophages and DCs
To determine the titering units (TU) per milliliter of lentivirus packaged with or without Vpx, H9 cells were transduced with a range of concentrations of virus encoding GFP-ffluc and analyzed by flow cytometry, where TU/mL = (# of H9 cells at time of transduction) × (proportion of GFP positive cells)/(volume of virus added [mL]). This value was used to determine the multiplicity of infection (MOI) for infection of DCs or macrophages at day 3 of differentiation. 25 Virus was added with fresh media and cytokines as above. Protamine sulfate (100 μg/mL) was added to enhance transduction by lentivirus packaged without Vpx.
Lentiviral infection of monocytes or macrophages
Freshly isolated monocytes or differentiated macrophages were infected with 20–1,000 LP/cell in macrophage media (supplemented with M- or GM-CSF as appropriate for concurrent differentiation of monocytes). Media was refreshed every 3 days.
U87 cell culture
The wild-type U87 glioma cell line was purchased from ATCC (HTB-14). U87 cells stably transduced with epHIV7 encoding GFP-ffluc were a gift from Dr. Michael Jensen's lab. All U87s were cultured in DMEM supplemented with 1% Glutamax and 10% FBS at 37°C, 5% CO2, and passaged prior to the formation of neurospheres. GFP-ffluc-expressing U87s were not used for more than five passages, and GFP expression was verified using fluorescent microscopy before use.
Lentiviral copy number assay
To determine the number of lentiviral integrations per cell, CD14+ cells were transduced on day 0 of differentiation, or after 7 days of differentiation in 10 ng/mL of GM-CSF, with 100 or 250 LP/cell encoding GFP-ffluc. On day 10 of differentiation, genomic DNA was isolated using the Qiagen DNA Mini Kit qPCR for WPRE and the albumin gene on the BioRad CFX96 Real Time System using 2 × Power SYBR Green Master Mix (Thermo Fisher). Picograms of lentiviral (WPRE) and genomic DNA (albumin) was determined by comparing Cq values to those obtained from respective standard curves of known concentrations of plasmid DNA. Viral integrations per cell = 2 × (pg WPRE)/(pg ALB). WPRE-QF: ACTGTGTTTGCTGACGCAACCC; WPRE-QR: CAACACCACGGAATTGTCAGTGCC; ALB-QF: TGAAACATACGTTCCCAAAGAGTTT; ALB-QR: CTCTCCTTCTCAGAAAGTGTGCATAT.
Flow cytometry
GEMs were harvested for flow cytometry by treatment with Versene (Gibco) followed by gentle scraping. Detached cells were treated with Fc Block (BD Biosciences) to eliminate non-specific antibody binding, stained with fluorophore-conjugated antibodies, fixed with 2% paraformaldehyde, and run on a BD LSR Fortessa flow cytometer with FACS DIVA software. Analysis was performed with FlowJo for Mac, v10 (Treestar). Antihuman antibodies included CD16-V500, CD163-Alexa647, and CD80-BV786 (BD Biosciences), and CD11b-APC-Cy7, CD19-APC, HLA-DR-BV605 or HLA-DR-APC, CD45-BV785, and PD-L1-PECy7 (Biolegend), as well as the Live/Dead fixable blue stain (Thermo Fisher). Erbitux (Bristol-Myers Squibb) was biotinylated using the EZ-link biotinylation kit (Thermo Scientific) and used in conjunction with streptavidin-FITC (Biolegend).
Viral-induced cytokine production
CD14+ monocytes were isolated from healthy donor PBMCs and transduced with 0 or 250 LP/cell mCherry Vpx+ lentivirus and differentiated in GM-CSF for 7 days. Following 24 h of conditioning, media was collected at a ratio of 1 mL/500,000 cells at 24 h and 7 days post transduction. Media samples were then used undiluted on a Luminex kit that included IL-12 (p40/p70), tumor necrosis factor (TNF), and interferon alpha (IFN-α; Life Technologies).
Surveyor assay
GM-CSF-differentiated macrophages were infected with 1,000 LP/cell Vpx+ LentiCRISPRv2 virus containing control (no gRNA), IL-10 gRNA, or PD-L1 gRNA. After 5 days in culture, genomic DNA was isolated using the DNeasy Kit (Qiagen) following the manufacturer's protocols. Genomic regions surrounding the Cas9-cleaved sites were PCR amplified with the following primer sets, and the surveyor assay (Integrated DNA Technologies) carried out according to the manufacturer's protocol. The IL-10 PCR primers were F: 5′-AGAGAGGTAGCCCATCCTAAAAATAGCTG and R: 5′-GCAGGTTTCCTGCACATTTACTGTATCA. The PD-L1 PCR primers were F: 5′-TTGAATTGAATTGAGGCAGAGCTAGCAG and R: 5′-ATATGGTTTGGATGAATGGAGGTGAGGA.
Validation of PD-L1 expression downregulation
GM-CSF-differentiated macrophages were infected with 1,000 LP/cell Vpx+ LentiCRISPRv2-EGFRt virus containing control (no gRNA) or PD-L1 gRNA. After 6 days in culture, GEMs were stimulated with 100 ng/mL of lipopolysaccharide (LPS; Sigma) and 20 ng/mL IFN-γ (R&D Systems) for 24 h, and stained for flow cytometry with anti-PD-L1 and Erbitux to determine transduction efficiency based on expression of the epitope tag EGFRt.
Validation of IL-10 expression downregulation
GM-CSF-differentiated macrophages were infected with 1,000 LP/cell Vpx+ LentiCRISPRv2:EGFRt virus containing control (no gRNA) or IL-10 gRNA. After 6 days in culture, GEMs were stimulated with 100 ng/mL of LPS and 20 ng/mL of IFN-γ for 24 h at 1 mL/500,000 cells. Conditioned media was collected and used undiluted for Bio-Rad's IL-10 Bio-Plex Pro Assay on a Bio-Plex 200 instrument. Cells were stained for flow cytometry with Erbitux to determine transduction efficiency based on expression of the epitope tag EGFRt.
Validation of sTβRII expression
To verify the expression of sTβRII, GM-CSF-differentiated macrophages were transduced on day 7 with 250 LP/cell of lentivirus encoding CD19t-T2A-sTβRII or CD19t vector control. Media was collected following 24 h of conditioning in 1 mL/500,000 cells on days 5, 6, and 7 post transduction. Secreted protein was detected by the human TGF-βRII DuoSet ELISA (R&D Systems).
Validation of IL-21 expression
To verify the expression of IL-21, GM-CSF-differentiated macrophages were transduced on day 7 with 250 LP/cell of lentivirus encoding IL-21-T2A-CD19t or CD19t vector control. Media was collected 7 days post transduction (day 14 of differentiation) following 24 h of conditioning in 1 mL/500,000 cells. IL-21 secretion was detected using the Bio-Plex Pro assay (Bio-Rad).
Co-infections with epHIV7.2 and LentiCRISPRv2 Vpx+ lentivirus
GM-CSF differentiated macrophages were infected with 250 LP/cell epHIV7.2:CD19t vector control, CD19t-T2A-sTGF-βRII, or IL-21-T2A-CD19t virus concurrently with 1,000 LP/cell lentiCRISPRv2:EGFRt vector control, PD-L1gRNA-EGFRt, or IL-10gRNA-EGFRt virus. Flow cytometry for epitope tags was used to determine the percent of cells double positive, and assays performed for each factor as above.
Detection of LPS/IFN-γ-induced factors
To determine the longitudinal expression of interleukins, cytokines, chemokines, and growth factors elicited by LPS/IFN-γ stimulation, GM-CSF-differentiated macrophages were seeded on day 6 at 200,000 cells/well in a 24-well plate. On day 7, macrophages were stimulated with 100 ng/mL of LPS, 20 ng/mL of IFN-γ or LPS + IFN-γ for 18 h in 500 μL of media. Conditioned media was collected at 18 h (day 1) and replaced with fresh media without cytokines. Media was harvested every 24 h for a total of 10 days. Cytokine release was detected using the Luminex Human 30-plex cytokine kit (Life Technologies).
Animal studies
All mouse studies were conducted with the oversight of the Seattle Children's IACUC (Protocol #15181) and efforts were made to minimize use in accordance with institute policies. Animals were euthanized following the appearance of symptoms secondary to tumor engraftment, including cachexia, lethargy, hind-limb paralysis, or when they reached 80% of their original body weight. Eight-week-old male NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJ (NSG) mice were purchased from the Jackson Laboratory. Ketamine/xylazine-anesthetized animals were immobilized in stereotactic apparatus (Stoelting), a 0.5 cm incision made on the skin covering the skull, and a burr hole drilled 2 mm lateral and 0.5 mm anterior to the bregma. A total of 200,000 wild-type or GFP-ffluc-expressing U87 cells were injected in a 2 μL volume at a rate of 1 μL/min at 2.5 and 2.25 mm beneath the dura, 1 μL at each location. After wound closure, the mice received lactated Ringer's solution for fluid recovery and buprenorphine as an analgesic. Surgery for injection of GEMs was similar to that for U87s, except that 150,000 GEMs were injected in a 3 μL total volume at three steps of 2.5, 2.35, and 2.25 mm below the dura. Bioluminescent imaging of GEMs or U87s expressing ffluc was conducted three times weekly. Mice were anesthetized with isoflurane, injected with 150 μL of a 28.57 mg/mL solution of D-luciferin (Perkin Elmer) intraperitoneally or subcutaneously in the scruff. Bioluminescent images were collected with a Xenogen IVIS Spectrum Imaging System (Perkin Elmer), and Living Image Software (Perkin Elmer) was used to analyze the data.
Immunohistochemistry of human CD45+ cells in brain tumor xenografts
After reaching the defined experimental endpoint (above), animals were deeply anesthetized with 4% isoflurane, the chest cavity opened, and 15 mL of PBS perfused through the heart and vasculature followed by 15 mL of 10% neutral buffered formalin. Mouse brains were harvested, and were then formalin-fixed and paraffin-embedded using standard protocols. To identify injected GEMs within the tumor xenograft, brain sections were immunostained with anti-human CD45 (clone HI30; BioLegend) at a 1:100 dilution and detected with the iVIEW DAB Detection Kit on the Ventana Ultra automated platform. Images were acquired on a Nikon Eclipse Ci with a 20 × PlanApo objective (0.75NA) with a DS-Ri1 color camera. Tile scanned images were stitched together using Nikon Elements software.
Isolation and flow cytometry of human cells from brain xenografts
Following euthanasia and perfusion as described above, the brain was dissected and tumor with 1 mm of the surrounding normal tissue isolated. Dissociation of the brain tumor was performed with the human Tumor Dissociation Kit (Miltenyi), followed by removal of mouse cells with the Mouse Cell Depletion Kit (Miltenyi), according to the manufacturer's protocols. Single-cell suspensions of human cells were stained for flow cytometry as described above.
Statistics and reproducibility
Unless otherwise stated, all experiments were performed a minimum of three times using macrophages isolated from different donors. Results were analyzed with Prism software (GraphPad), using unpaired t-tests or one-way analysis of variance followed by Dunnett's multiple comparisons test as appropriate. Statistical significance (p < 0.05) is denoted with an asterisk.
Results
Monocyte-derived macrophages can stably express lentivirally encoded genes
The use of lentivirus to stably transduce T cells for cancer immunotherapy has become routine in recent years. In contrast, no standard system for infection of myeloid cells exists. Recent reports have demonstrated that DCs derived from monocytes using GM-CSF and IL-4, or pro-inflammatory macrophages differentiated using GM-CSF alone, can be transduced at high efficiency when LP are packaged with Vpx.
14,26
Prior studies reported that DCs can be successfully transduced without Vpx if they are exposed to high MOI earlier in the differentiation process.
25,27
To determine if Vpx is necessary for successful transduction of monocyte derived macrophages, this study packaged epHIV7:GFP-ffluc, a lentiviral backbone currently in use in clinical trials (NCT01683279, NCT02311621), with and without Vpx using a standard lentivirus production protocol. CD14+ cells isolated from healthy donor PBMCs were differentiated to DCs or macrophages using GM-CSF and IL-4, or GM-CSF only, respectively. Three days after isolation and initiation of differentiation, cells were counted and re-plated in fresh media containing cytokines and GFP-ffluc-encoding lentivirus packaged with or without Vpx. Cells infected with virus that did not contain Vpx also received 100 μg/mL of protamine sulfate to aid viral entry. Following 6 days of in vitro differentiation, cells were analyzed by flow cytometry. Using an MOI of 15 as demonstrated in DCs
25
and 100 μg/mL of protamine sulfate, it was observed that 20% of DCs and 60% of macrophages expressed lentivirally encoded GFP (Supplementary Fig. S1A and B; Supplementary Data are available online at
Because all subsequent experiments evaluated GEM expression of varied viral payloads that cannot be normalized using traditional flow cytometry–based titering, viral dose was standardized based on the amount of virus-associated p24, where 1 ng of p24 is equivalent to 1.25 × 107 LP. To determine the minimal dose of Vpx-containing virus necessary to achieve high transgene expression consistently, both pro-inflammatory macrophages differentiated in GM-CSF (Fig. 1A, representative of three independent experiments) and anti-inflammatory macrophages differentiated in M-CSF (Supplementary Fig. S2A) were transduced on day 7 of differentiation with a range of LP/cell. GFP expression was evaluated after an additional 7 days in culture (Fig. 1A and Supplementary Fig. S2A). It was found that the two phenotypes have similar transduction efficiencies following infection with Vpx-containing virions at all doses tested, nearing 100% at concentrations as low as 250 LP/cell. Importantly, infection with 250 LP/cell following differentiation with GM-CSF did not significantly impact viability (Fig. 1C). Virions packaged without Vpx had very poor transduction efficiency in both GM-CSF (5.09%) and M-CSF (14.6%) differentiated macrophages at the highest concentration tested (1,000 LP/cell; Fig. 1A and Supplementary Fig. S2A).
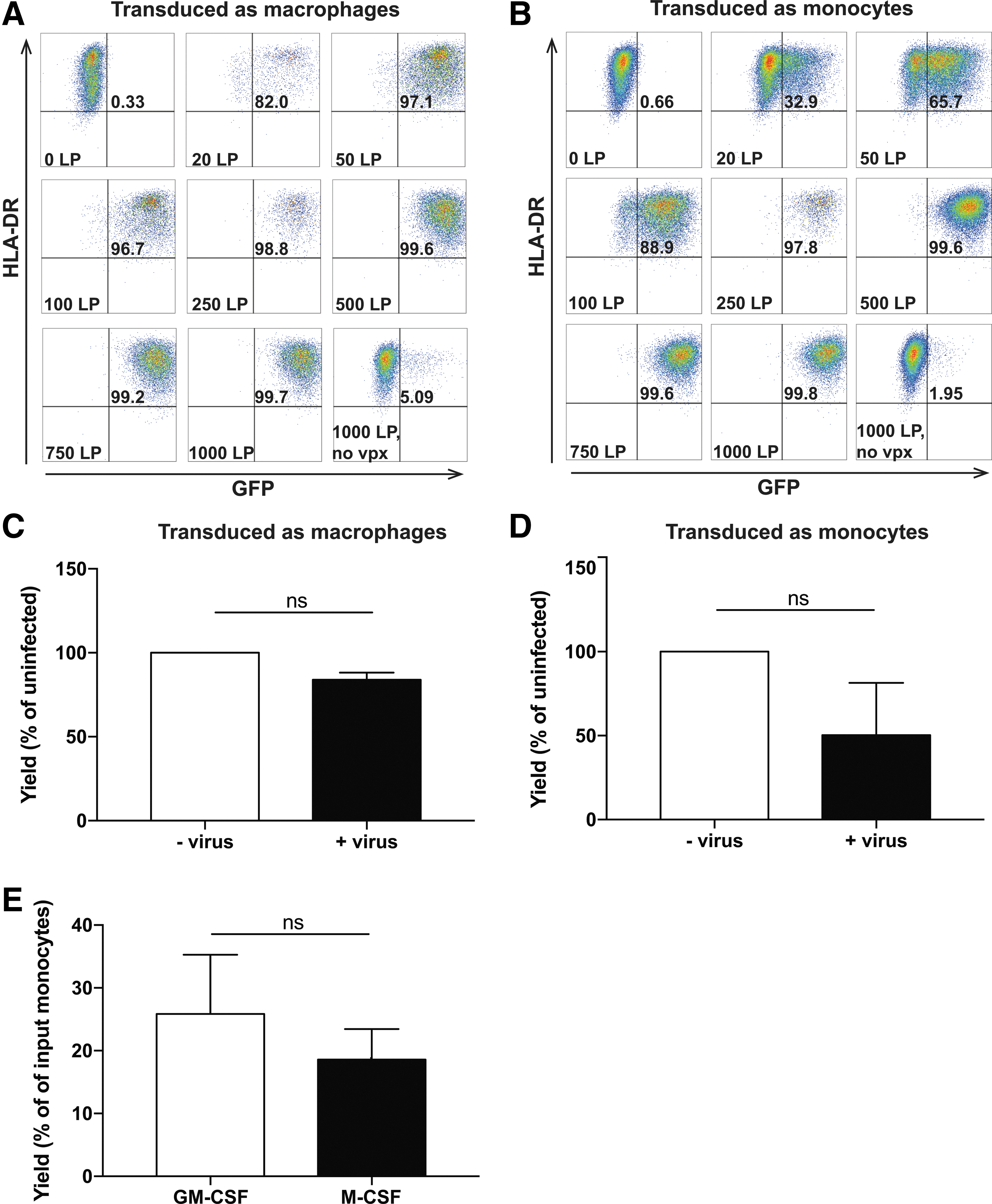
Granulocyte macrophage colony-stimulating factor (GM-CSF)-differentiated macrophages and monocytes can be infected with viral protein X (Vpx)+ lentivirus in a dose-dependent fashion. (
One of the challenges of treating patients with relapsed disease is the time between apheresis to delivery of an autologous cellular therapy, which can be several days to weeks for CAR T cell products. 28 To determine if the time and cost of developing a clinical monocyte-derived cell product for patients could be reduced, the feasibility of concurrent transduction and differentiation of freshly isolated CD14+ monocytes was tested. Transduction of monocytes at the time of selection and induction in GM-CSF or M-CSF (day 0) resulted in similar dose-dependent proportions of GFP+ macrophages as consecutive differentiation and transduction (Fig. 1B and Supplementary Fig. S2B). However, this approach had a significant impact on macrophage viability. Although differentiation in GM-CSF or M-CSF without virus resulted in approximately 20–30% yields relative to the original number of CD14+ cells (Fig. 1E), differentiation in combination with infection with 250 LP/cell further reduced yields by 49.7% in GM-CSF, and 67.4% in M-CSF differentiated macrophages (Fig. 1D and Supplementary Fig. S2C, respectively).
Successful clinical development of a transduced adoptive cellular therapy must balance efficacy and safety. High transgene expression and transduction efficiency is often associated with a high rate of lentiviral integration into the host cell genome. It is important to control the integration rate because increased vector copy numbers increase the likelihood of insertional mutagenesis or toxic levels of transgene synthesis. To determine the number of lentiviral copies per cell, genomic DNA was isolated from macrophages that had been transduced on day 0 of differentiation, or after 7 days of differentiation with Vpx-containing lentivirus encoding GFP-ffluc. In a representative experiment, the number of lentiviral copies per cell was found to be 34 and 22 when cells were transduced with 250 or 100 LP/cell, respectively, as macrophages (Supplementary Fig. S3A), and 18 and 10 when the cells were transduced as monocytes (Supplementary Fig. S3B).
GEMs express myeloid cell surface markers and respond to LPS/IFN-γ stimulation
To determine if viral infection would impact differentiation of monocytes to pro- or anti-inflammatory macrophages, the phenotype of both GM-CSF and M-CSF GEMs was analyzed. Monocytes were transduced with epHIV7.2 lentivirus encoding the red fluorescent protein mCherry at a viral concentration of 250 LP/cell, and concurrently differentiated the cells in GM-CSF or M-CSF. GEMs analyzed for mCherry expression using flow cytometry 7 days later were 100% positive, regardless of differentiation conditions (Fig. 2A). GEMs were further analyzed by flow cytometry for surface expression of the common myeloid markers CD163, a scavenger receptor; HLA-DR (major histocompatibility class-II), which presents antigens to CD4 T cells; CD80, a co-stimulatory signal for T cells; PD-L1, a T cell inhibitory protein; CD11b, an integrin responsible for cell adhesion; and CD16, an Fc receptor (Fig. 2B–G). As expected, a higher fraction of M-CSF macrophages expressed the canonical anti-inflammatory macrophage marker CD163 than GM-CSF macrophages did 29 (90% vs. 5%; Fig. 2B). Importantly, GEMs are responsive to stimulation with the potent Toll-like receptor 4 (TLR4) agonist, LPS, and the pro-inflammatory cytokine, IFN-γ, as evidenced by their upregulation of proteins associated with improved antigen presentation, HLA-DR and CD80, as well as PD-L1, which is consistent with previous findings 30 –32 (Fig. 2C–E). Transduction with Vpx-containing virus did not have a significant impact on the expression of many of these markers, and CD16 was unaffected by any treatment (Fig. 2G). Interestingly, however, it was found that viral transduction resulted in a 60% decrease in CD11b expression in M-CSF differentiated GEMs (Fig. 2F), and a threefold increase in CD80 in unstimulated, GM-CSF differentiated GEMs (Fig. 2D). The impact of increased CD80 expression on antigen presentation will be the subject of future investigations.

Genetically engineered macrophages (GEMs) express standard myeloid cell surface markers and respond to lipopolysaccharide (LPS)/interferon gamma (IFN-γ) stimulation. GM-CSF or M-CSF monocyte-derived macrophages were infected on day 0 of differentiation with 250 LP/cell of lentivirus encoding mCherry. On day 6, cells were treated with fresh media or LPS/IFN-γ for 18 h then analyzed by flow cytometry. Macrophages were found to be 100% positive for mCherry expression (
Because pattern recognition receptors such as TLR3 and TLR7 recognize viral RNA, and respond by upregulating cytokines and chemokines to mount an immune response, 33 it was important to determine if viral infection impacted GEM secretion of canonical pro-inflammatory cytokines. To evaluate this, monocytes were infected on day 0 with 0 or 250 LP/cell mCherry Vpx+ lentivirus and differentiated in GM-CSF. Media was collected at 24 h and 7 days post transduction for Luminex analysis of TNF IL-12, and type I interferons (Supplementary Fig. S4). All three analytes fell below the limit of detection for the assay, suggesting that lentiviral transduction does not induce pro-inflammatory cytokine secretion.
Genetically engineered macrophages persist in mouse glioblastoma model
Evidence supporting the antitumor effect of pro-inflammatory macrophages comes from several mouse studies where locally or systemically administered TLR agonists overturn the tumor supportive program of TAMs and restore immune surveillance. 10,34 –36 An ideal GEM would be engineered to release factors sustainably that are elicited by classical LPS/IFN-γ activation and stimulate both the innate and adaptive immune systems. 37 Having demonstrated that GM-CSF-differentiated GEMs are responsive to LPS/IFN-γ, the feasibility of employing activated macrophages as a cellular therapeutic that is injected directly in to the brain was tested. Specifically, this study determined if stimulated and unstimulated GEMs would survive in the TME, impact morbidity, or support tumor growth. To assess the behavior of GEMs directly, without the confounding effects of other immune cells, GEMs were evaluated in the T and B lymphocyte-deficient NOD-SCID gamma (NSG) mouse using an intracranial glioblastoma xenograft model that has frequently been used for preclinical evaluations of cellular immunotherapies. 4,38 –42 In these experiments, 200,000 U87 cells were injected intracranially and allowed to establish tumor for 6 or 7 days (Figs. 3A and 4A). For the initial experiment, freshly isolated monocytes were transduced with 250 LP/cell epHIV7:GFP-ffluc and differentiated toward a pro-inflammatory phenotype with GM-CSF for 6 days (Fig. 3A). On day 5 after differentiation and transduction, half of these GEMs were stimulated with LPS/IFN-γ for 18 h. A total of 150,000 stimulated or unstimulated GEMs were injected into the established tumor, and bioluminescence was monitored three times per week.
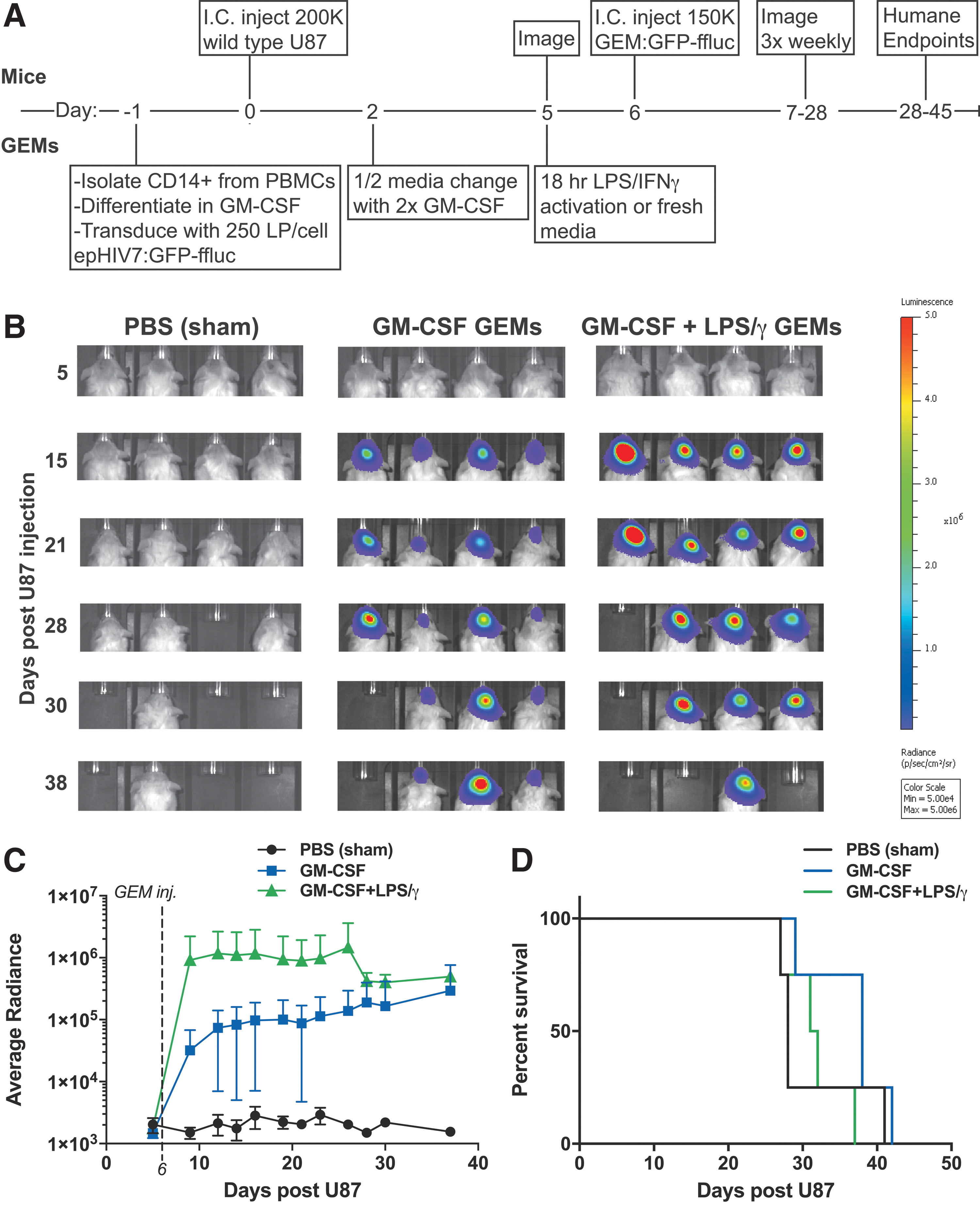
GEMs persist within a glioma xenograft model. (

GEMs do not affect tumor growth in a glioma xenograft model. (
The imaging revealed that once implanted, both GM-CSF-differentiated, unstimulated and GM-CSF-differentiated, LPS/IFNγ-stimulated GEMs have a stable luciferin signal throughout the life of the animal (Fig. 3B and C). Importantly, GEMs do not have a detrimental effect on animal survival, with no significant differences between groups (Figs. 3D and 4D), nor did the animals display outward signs of distress, suggesting that even LPS/IFN-γ-stimulated GEMs are well tolerated. Furthermore, GEMs do not enhance the growth of ffluc-expressing U87 tumors (Fig. 4B and C). Despite being a rare cell population, GEMs can be detected within the tumor mass by immunostaining for human CD45 (Fig. 4E–G) and can be recovered following enzymatic dissociation of tumor tissue (Supplementary Fig. S5). Collectively, the data suggest that GEMs are capable of the long-term expression of transgenes in vivo, and neither support tumor growth nor reduce the survival of tumor-bearing animals. Future experiments will evaluate GEMs that constitutively express activation signals to determine whether they are capable of supporting an endogenous antitumor response or whether safety concerns arise when they interact with other immune cells.
CRISPR targeting of TAM immunosuppressive genes in GEMs
Given that our intracranial glioblastoma model lacks a complete immune system and that GEMs were not armed with functional payloads and do not express detectable levels of inflammatory cytokines (Supplementary Fig. S4), an impact on tumor growth or overall survival in xenograft-bearing animals was not expected. However, human clinical trials from the 1980s and 1990s evaluating IFN-γ and/or LPS ex vivo–stimulated macrophages for the treatment of patients with solid tumors also showed no survival benefit, despite tumor-directed antibody-dependent cell-mediated cytotoxicity (ADCC), and increased TNF secretion in vitro following an 18 h IFN-γ stimulation. 43 It was hypothesized that the failure of macrophages to provide a survival benefit in the setting of a complete immune system is due to: (1) diversion of macrophages by tumor cells to a pro-tumor or anti-inflammatory phenotype, (2) absence or ineffectiveness of other cytotoxic immune cells, and (3) lack of persistent inflammatory LPS/IFN-γ-induced cytokine release, as seen in vitro following 18 h stimulation (Supplementary Fig. S6). In fact, the use of a strong TLR4 agonist such as LPS might have unintended anti-inflammatory consequences such as sustained IL-10 secretion (Supplementary Fig. S6, page 1) or increased PD-L1 surface expression (Fig. 2E), two factors also known to be expressed by TAMs due to tumor-secreted signals. 44 Therefore, using CRISPR/Cas9-mediated gene editing, 21,22 the aim was to disrupt GEM expression of these genes that contribute to immune evasion. Using a lentiviral vector encoding both Cas9 and guide RNA, several guide RNAs 21 were screened using the Surveyor assay to find a sequence that interrupts either the PD-L1 or IL-10 genomic loci. Successful interruption is indicated by the generation of a cleavage product following incubation of the amplified and reannealed genomic regions with the surveyor endonuclease, which cleaves at mismatched base pairs 45 (Supplementary Fig. S7A and B, arrows). When introduced to GM-CSF-differentiated macrophages, disruption of the IL-10 locus resulted in a 60% reduction of LPS/IFN-γ-induced IL-10 secretion (Fig. 5A). Following infection of GM-CSF-differentiated macrophages with a virus encoding Cas9 and a PD-L1 guide RNA, surface PD-L1 expression as a result of LPS/IFN-γ stimulation was reduced by 40% (Fig. 5B and C). This is consistent with the findings of other investigators who report that the CRISPR system often modifies only one allele. 46,47
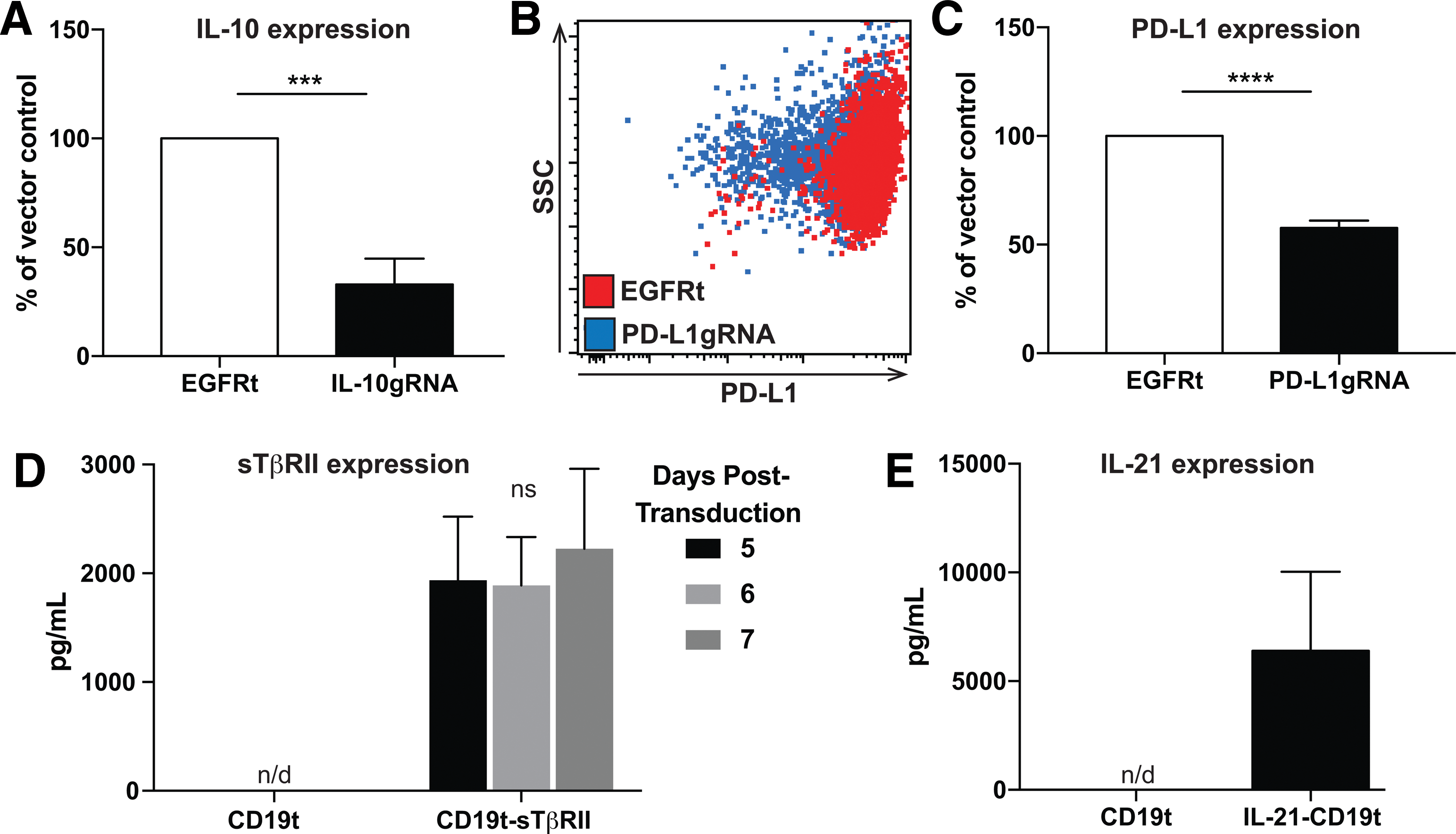
GEMs can be engineered to resist the immunosuppressive tumor microenvironment and support an antitumor response. (
GEM secretion of anti-inflammatory blockades or activating cytokines
This study also evaluated whether GEMs could be programmed to resist the suppressive milieu in the TME and support the activation, proliferation, and survival of cytotoxic antitumor immune cells. For example, TGF-β is a well-described tumor-derived factor that prevents a cytotoxic immune response in a variety of ways. 48 –52 It was demonstrated that GEMs can be engineered to secrete nanogram quantities of the sTβRII (Fig. 5D) to function as a decoy receptor for TGF-β and reduce TGF-β signaling, as previously described. 19 GEMs were also engineered to secrete IL-21 (Fig. 5E), a cytokine normally expressed by CD4+ T cells that activates NK and T cells, supports ADCC, and shifts the polarization of TAMs toward an M1 phenotype. 53 –55 Recombinant IL-21 is currently in clinical trials either as a cancer monotherapy, or in combination with tyrosine kinase inhibitors or therapeutic antibodies. 53,56 The observation that macrophages can be engineered to secrete as much as 6 ng/mL of IL-21 suggests that impaired cytotoxic immune cell functions may be restored in the presence of IL-21-expressing GEMs. Together, these data demonstrate that GEMs can be engineered to express reduced levels of anti-inflammatory proteins, or produce soluble factors that either interfere with immunosuppressive signaling or support antitumor immune responses.
GEMs can express genes from multiple viruses
Due to the multiple challenges that arise as a result of the immunosuppressive milieu of the TME, this study sought to engineer macrophages that both resist the tumor microenvironment, through CRISPR-mediated gene disruption, as well as secrete sTβRII and IL-21. To this end, a truncated CD19 sequence was inserted into the epHIV7.2 vector (Supplementary Fig. S7C), and the epitope tag EGFRt was added to replace the puromycin sequence of the lentiCRISPRv2 vector (Supplementary Fig. S7D). It was found that both epitope tags can be detected on macrophages infected with lentivirus encoding the respective sequences (Supplementary Fig. S7E and F). When GEMs are infected with both viruses, 30–45% cells were double positive by flow cytometry (Fig. 6A and E). Furthermore, evaluation of PD-L1 expression on cells expressing both CD19t and EGFRt shows a 70% reduction in PD-L1 (Fig. 6C); these GEMs are also uncompromised in their expression of sTβRII (Fig. 6D). Interestingly, IL-21 seems to prevent LPS-induced IL-10 expression. GEMs encoding IL-21 alone reduce IL-10 expression by 76.4%, and when IL-21 is expressed in combination with IL-10gRNA, expression drops by 87.3%. These are significantly lower levels of expression than seen in the IL-10gRNA + CD19t vector control (38.5%; Fig. 6F). IL-21 expression was unchanged when IL-10 expression was targeted (Fig. 6G).
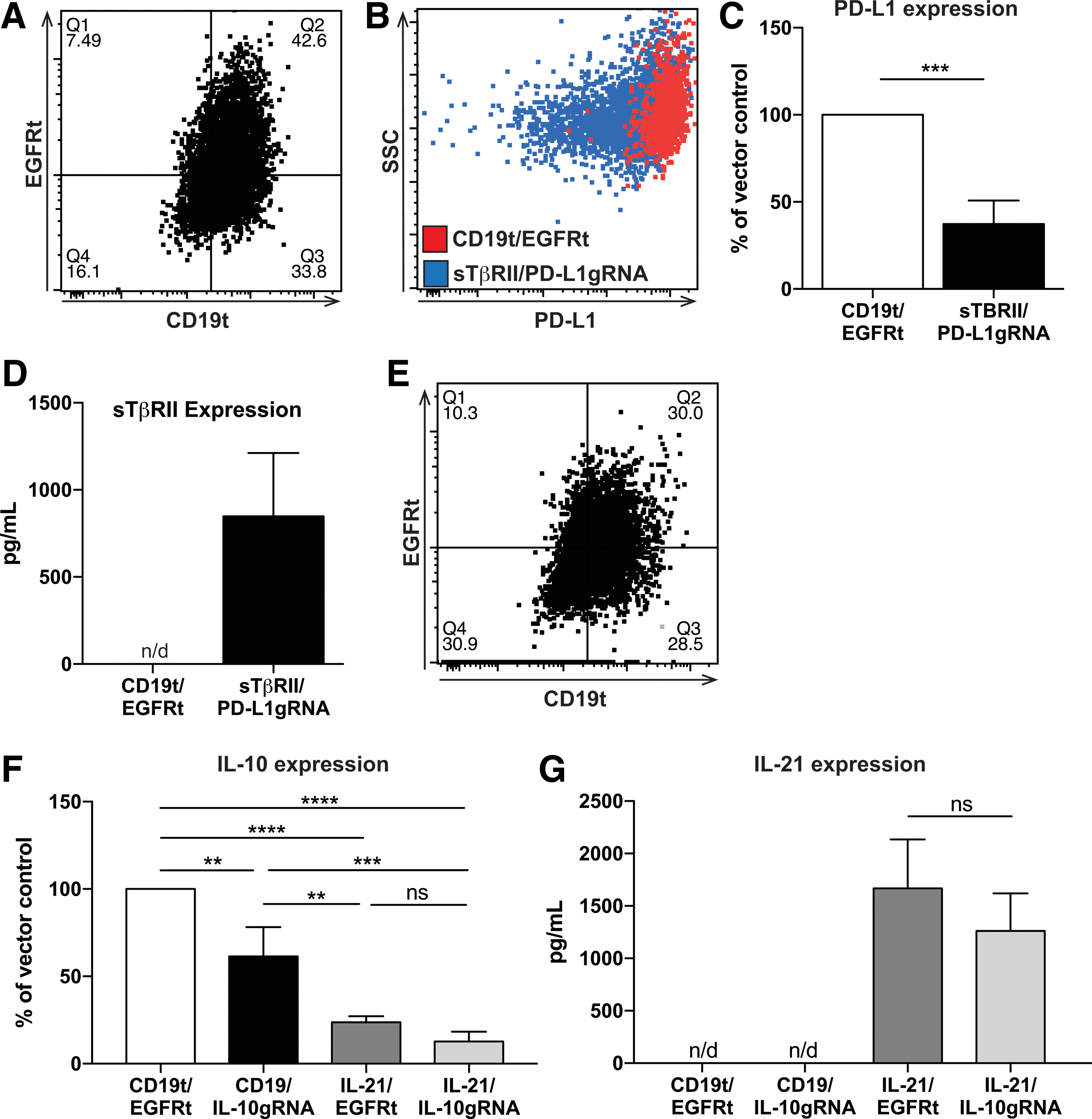
GEMs can be engineered to have multiple functions. (
Discussion
This study reports the validation of a lentivirus-based method to engineer primary human monocytes and macrophages with the goal of generating a novel cellular immunotherapy for solid tumors. Previous studies report 80% infectivity rate when transducing DCs with 600 ng of p24 per 500,000 cells. 57 As demonstrated above, virus packaged with Vpx achieves nearly 100% efficiency at 250 LP/cell, equating to 10 ng of p24, per 500,000 cells, 60 times less virus. This approach causes a higher number of genomic integration events than are observed in some engineered T cells used clinically (Dr. Michael Jensen, pers. commun.). However, because the GEMs do not divide, as observed in the in vivo studies demonstrating a lack of GEM expansion, it is not anticipated that insertional mutagenesis will have an impact on the safety of a clinical product. Furthermore, if functions are found to be affected or that the insertion rate exceeds Food and Drug Administration (FDA) regulations, vector copy number will be reduced by sorting for median gene expressers.
The flexibility of this approach has allowed CRISPR-mediated gene interruption for the important immunosuppressive factors IL-10 and PD-L1, and overexpression of sTβRII and IL-21, secreted proteins that will prevent intrinsic inhibitory signals and support the antitumor functions of NK and cytotoxic T cells. Interestingly, when co-expressing using two viral vectors, it was found that a significant percentage of GEMs express the epitope tags for both viruses, and that CRISPR-mediated reduction of IL-10 was significantly augmented when GEMs also express IL-21.
This method of lentiviral modification of primary human macrophages may serve as a novel type of cellular immunotherapy, which can be generated in as little as 7 days from a patient's blood, using a clinically approved lentiviral backbone. GEMs may be generated from a patient's monocyte population that is currently discarded during the preparation of therapeutic TCR or CAR T cells, reducing the time and cost associated with developing new infrastructure for a clinical product. It is anticipated that in the clinic, GEMs will be directly implanted into the tumor site immediately following surgical tumor resection. This approach has advantages over intravenous infusion, and non-specific systemic approaches that have been previously used in clinical trials, maximizing the likelihood that GEMs will interact with tumor cells and alleviating concerns that they may become trapped in narrow capillary beds during peripheral circulation.
The tumor microenvironment prevents immune responses through multiple redundant mechanisms, 6,8 and it is hypothesized that reversing immune suppression may be more effective at inducing antitumor activity than a small molecule, antibody, or cellular therapy designed against a single target. The intracranial xenograft model will allow the adjuvant properties of GEMs to be evaluated when used with other cellular therapies such as CAR T cells, and will be subsequently evaluated in an immune-competent model to determine the ability GEMs to function in a complex TME.
The concept of employing macrophages for antitumor therapy originated in 1974 when Fidler et al demonstrated that ex vivo stimulated macrophages could suppress pulmonary metastases in a B16 melanoma model. 58 Subsequent studies confirmed these findings and laid the groundwork for several clinical trials that tested the safety and efficacy of adoptively transferred macrophages for the treatment of patients with various cancers, including ovarian, colorectal, and renal carcinomas. 43 Dose escalation studies of autologous macrophages activated with IFN-γ or LPS/IFN-γ prior to infusion showed that the therapy was safe, inducing only flu-like symptoms, but had no clinical benefit. 43 The failure to improve patient outcomes could be due to temporally limited expression of LPS/IFN-γ-induced factors, such as IL-12, TNF, or IL-6, which support the cytotoxic function of effector cells. 37 Additionally, tumor-derived factors such as TGF-β, M-CSF, IL-4, and IL-10 may alter the polarization of the adoptively transferred macrophages, modifying their production of cytokines to promote the function of Tregs while inhibiting tumor-specific T cell respsonses. 59
Evidence supporting the efficacy of persistently polarized macrophages comes from recent animal studies in which adoptively transferred or tumor infiltrating macrophages, engineered to inhibit NFκB-mediated alternative activation or to overexpress IFN-α, overturn an immunosuppressive TME and inhibit tumor growth. 60,61 Additionally, reversing the anti-inflammatory phenotype of endogenous macrophages with an Ang2/VEGF bi-specific antibody, 62 antagonists of M-CSF, 12,63 or agonists of CD4011 or TLR pathways 10,34,36,64 reduces tumor progression by inhibiting tumor-supportive behavior and restoring antitumor immune responses. Notably, these antitumor mechanisms may be either direct, such as the induction of tumoricidal activity by macrophages themselves, 10,11,60 or indirect, through support of NK 36,60 and T cell-mediated cytotoxicity 36,64 or via activation of DCs. 65 Furthermore, studies demonstrating TLR3-mediated repolarization of tumor myeloid cells from GBM patients suggest that their inherent plasticity supports their potential to initiate local immune activation. 66 These findings suggest that macrophages can induce multifaceted changes to the way the immune system responds to tumor, and underscore the power of harnessing this cell population to restructure the TME.
In support of this hypothesis, several studies that evaluate the impact of innate immune cell activation in the TME indicate that this is an important component to clinical efficacy for patients with solid tumors. For example, DC-based cellular therapies are the first to receive approval by the FDA based on their ability to induce antitumor immune responses in patients with castration-resistant prostate cancer. 67 Other experimental studies seek to boost immunity using adjuvants, 68 live oncolytic viruses, 69 or viruses engineered specifically to infect tumor-resident antigen-presenting cells. 70,71 These approaches likely result in significant innate immune cell activation, including their production of pro-inflammatory mediators, and support of cytotoxic immune cell functions in the tumor. Although these approaches demonstrate the impact of enhancing innate and adaptive immune cell crosstalk on clinical efficacy, the mechanisms underlying pleiotropic effects on the TME and antitumor immunity are not well defined. In contrast to treatments that broadly activate innate immune cells, GEMs deliver specific immune-supportive factors, an important consideration for locations such as the brain, where extensive inflammation may be detrimental. Furthermore, the precision of GEMs will allow elucidation of the contributions of individual factors to overcoming immunosuppression in the TME, informing the development of the next generation of immunotherapies.
Footnotes
Acknowledgments
The authors would like to thank Kristen Haberthur, Adam Johnson, Stephanie Mgebroff, Cindy Chang, Joshua Lieberman, and Michael Baldwin for their technical expertise and helpful feedback. This work was supported by the Pediatric Bain Tumor Research Fund, the Pediatric Cancer Research Foundation, and the Steven Higgins Brain Tumor Research Fund.
Author Disclosure
C.A.C., K.W.M., and N.A.P.L. are inventors on “Genetic Engineering of Macrophages for Immunotherapy.” Patent Application Number: PCT/US2016/050552. The remaining authors have no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.