
Abstract
During B and T lymphocyte maturation, V(D)J recombination is initiated by creation of DNA double-strand breaks. Artemis is an exonuclease essential for their subsequent repair by nonhomologous end-joining. Mutations in DCLRE1C, the gene encoding Artemis, cause T−B−NK+ severe combined immunodeficiency (ART-SCID) and also confer heightened sensitivity to ionizing radiation and alkylating chemotherapy. Although allogeneic hematopoietic cell transplantation can treat ART-SCID, conditioning regimens are poorly tolerated, leading to early mortality and/or late complications, including short stature, endocrinopathies, and dental aplasia. However, without alkylating chemotherapy as preconditioning, patients usually have graft rejection or limited T cell and no B cell recovery. Thus, addition of normal DCLRE1C cDNA to autologous hematopoietic stem cells is an attractive strategy to treat ART-SCID. We designed a self-inactivating lentivirus vector containing human Artemis cDNA under transcriptional regulation of the human endogenous Artemis promoter (AProArt). Fibroblasts from ART-SCID patients transduced with AProArt lentivirus showed correction of radiosensitivity. Mobilized peripheral blood CD34+ cells from an ART-SCID patient as well as hematopoietic stem cells from Artemis-deficient mice demonstrated restored T and B cell development following AProArt transduction. Murine hematopoietic cells transduced with AProArt exhibited no increase in replating potential in an in vitro immortalization assay, and analysis of AProArt lentivirus insertions showed no predilection for sites that could activate oncogenes. These efficacy and safety findings support institution of a clinical trial of gene addition therapy for ART-SCID.
Introduction
S
Early diagnosis of SCID by newborn screening using the T cell receptor excision circle assay, 2 –4 followed by definitive treatment by allogeneic HCT, leads to excellent rates of survival, exceeding 90%. 5,6 HCT from human leukocyte antigen (HLA)-identical siblings or unrelated donors results in the best outcomes, with 97% and 92% survival respectively. 7 However, T-depleted, haploidentical HCT may require preparative chemotherapy to avoid rejection and achieve B cell reconstitution. 6,8 Moreover, for recipients of HCT from donors other than HLA matched siblings, graft versus host disease (GVHD) can be a major complication with significant morbidity and mortality. 6
The DCLRE1C gene encoding Artemis was first identified in patients with T−B−NK+ SCID (ART-SCID) who had increased cellular radiosensitivity. 9,10 Artemis is an endonuclease essential for nonhomologous end-joining of DNA double-strand breaks that arise following exposure to external agents or during V(D)J recombination of T and B cell receptor genes. 9 Navajo and Apache Native Americans from the southwestern United States have a frequent founder nonsense mutation in exon 8 of DCLRE1C, Y192X. 11,12 Homozygosity for this mutation causes T−B−NK+ SCID in 1/2000 births, or 25 times the incidence of SCID in the general population. 12 –14
While HCT can cure ART-SCID, the heightened sensitivities to irradiation and alkylator chemotherapy conferred by DCLRE1C mutations are associated with increased early toxicity as well as late effects in survivors, including short stature, absent or malformed permanent dentition, and endocrinopathies. 15,16 Thus, using gene addition to autologous hematopoietic stem cells (HSC) constitutes an alternative strategy for the treatment of ART-SCID to avoid the observed risks of partially matched or unrelated HSC allotransplant. 17 –19
We previously described a mouse model of ART-SCID (Art−/−) that accurately represents both the T and B cell lymphopenia and the resistance to treatment by allogeneic mismatched HCT found in ART-SCID patients. 20,21 Using this model, Multhap et al. demonstrated restoration of functional T and B lymphocytes by ex vivo complementation of the Artemis deficiency in murine stem cells using a vector carrying murine Artemis cDNA driven by the human Artemis promoter. 22 Building upon these studies, we have now developed a novel lentiviral vector with the human Artemis DCLRE1C cDNA under transcriptional regulation of its own human Artemis promoter, designated AProArt. We used AProArt to transduce human ART-SCID fibroblasts and CD34+ HSC as well as HSC from Art−/− mice, achieving correction of radiation sensitivity in transduced human ART-SCID fibroblasts and successful in vivo and in vitro differentiation of transduced Artemis-deficient HSC into T and B cells.
Methods
Animals
Mice were housed in sterile isolator cages and fed autoclaved chow, undergoing procedures according to approved protocols at the University of California, San Francisco. Art−/− mice have been described previously, 20 and NSG mice were from the Jackson Laboratory (Bar Harbor, ME).
Lentivirus assembly, production and determination of titer
A 3750 bp DNA fragment was synthesized to include the DCLRE1C promoter (APro) from −1000 to the translational start site (NG_007276.1, NM_1033855.2), 23 the 1024 bp human DCLRE1C coding sequence and 3′ untranslated region, and a 590 bp woodchuck post-transcriptional regulatory element (WPRE). The putative protein-encoding sequence within the WPRE was modified to replace its translational start site (GCTGAcgtcctttccAtg) with a stop codon (ATCATcgtcctttccTtg) (modified bases in uppercase letters). The synthetic MfeI–KpnI fragment was cloned into lentiviral vector pCSI-CDF-CG-PRE 24 between EcoRI and KpnI to generate AProArt. A cytomegalovirus–green fluorescent protein (CMV-GFP) control vector with the same modified WPRE was generated by replacing the 5′ end of pCS-AProArt through the DCLRE1C promotor and coding sequences with a PvuI – XhoI fragment isolated from pCSI-CDF-CG-PRE, generating pCS-CMV-GFP. The AProArt lentiviral vector was produced under good manufacturing practice conditions (Indiana University Vector Production Facility, Indianapolis, IN). Briefly, 293T cells were transfected with plasmids encoding AProArt provirus and vesicular stomatitis virus envelope G protein; lentiviral supernatant was harvested 48-72 hours after transfection and concentrated by ultracentrifugation. The control GFP lentivirus was produced in-house using the same protocol. Lentiviral titering in 293T cells was performed with serially diluted lentivirus; after 3 days the 293T cell DNA was isolated for vector-specific quantitative PCR (primer sequences available on request).
In vitro assays with human fibroblasts
Immunoprecipitation and Western blotting
Skin fibroblasts from Artemis-deficient (ART-SCID) and Ligase-4 deficient patients and healthy controls were cultured in Dulbecco's modified Eagle medium with 10% fetal calf serum and antibiotics. Fibroblasts were incubated with GFP or AProArt lentivirus at multiplicity of infection 100 for 24 h. Protein was isolated 3 or 8 days post-transduction and precipitated using a rabbit anti-Artemis antibody (kindly provided by Steve Yannone, Laurence Berkeley Laboratory, Berkeley, CA). For Western blot, a chicken polyclonal anti-Artemis antibody (Abcam) was used followed by a peroxidase-conjugated donkey anti-chicken second antibody (Gallus Immunotech) to detect precipitated protein.
Proliferation
Fibroblasts were exposed to 1, 3, or 5 Gy γ-radiation 3 days after lentivirus transduction. After 24 h, the cells were labeled with BrdU for 24 h, harvested, fixed, and stained with anti-BrdU antibody (BD Biosciences) following the manufacturer's instructions. Cell cycle distribution and BrdU incorporation were determined by flow cytometry using BD FACSVerse and analyzed using FlowJo software.
γH2AX foci
Fibroblasts were seeded on chamber slides (Nunc Lab-Tek) and exposed to 4 Gy γ-radiation 3 days after transduction. After 6, 24, 48, and 168 h (7 days) the cells were fixed, permeabilized, blocked with 5% goat serum, and incubated with mouse anti-γH2AX (Ser139) (Upstate Antibodies) and rabbit anti-53BP1 (Bethyl Laboratories) overnight at 4°C. The cells were washed, incubated with fluorochrome-conjugated secondary antibodies (Jackson ImmunoResearch) for 45 min at room temperature, washed thrice, stained with DAPI, mounted in Vectashield (Vector Laboratories) and analyzed using a fluorescence microscope (Keyence).
Isolation, transduction, and transplantation of murine bone marrow hematopoietic stem cells
Femurs, tibias, and fibulas from donor wild-type (WT) or Art−/− CD45.1 mice were flushed and crushed to isolate bone marrow cells. After depleting lineage committed cells (mouse depletion kit, Miltenyi), cells were stained with c-kit and Sca-1 (clones 2B8 and D7, respectively, eBioscience) and lineage negative, Sca1 positive, c-kit positive (LSK) HSC were sorted (FACSJazz, BD Biosciences). LSK cells were plated in Rectronectin-coated 96-well plates in X-Vivo-15 (Lonza) with
Functional assays with murine lymphocytes
T cell proliferation
Splenocytes from transplant recipients were isolated and resuspended in RPMI 1640 with 10% fetal bovine serum and antibiotics. One hundred thousand splenocytes per well in a 96-well plate were stimulated with 4 or 10 μg/mL of Concanavalin-A (ConA, Sigma-Aldrich), 5 μg/mL plate-bound anti-mouse CD3ɛ (Tonbo), or medium only at 37°C for 4 days. Proliferation was measured by EdU incorporation (Click-iT EdU imaging kit, Life Technologies), following the manufacturer's instructions using a FACSVerse flow cytometer (BD Biosciences). The assays were carried out in triplicate.
Enzyme-linked immunosorbent assay for serum immunoglobulin M and immunoglobulin G antibody
ELISA using serum from transplanted mice injected intraperitoneally with 100 μg of either NP41-Ficoll or NP24-keyhole limpet hemocyanin (KLH) (Biosearch Technologies, Novato, CA) was carried out as described. 20
Transduction and differentiation of human CD34+ HSC
With informed consent cytokine-mobilized peripheral blood stem cells (PBSC) were collected from an ART-SCID patient, under an internal review board–approved protocol (Laboratory of Host Defenses, National Institute of Allergy and Infectious Diseases, National Institutes of Health), and CD34+ HSC were isolated (CliniMACS, Miltenyi Biotechnology). De-identified CD34+ cells from PBSC of healthy donors were used as control HSC.
Human HSC were prestimulated in rectronectin-coated six-well plates (1–2 × 106 cells/mL), in X-Vivo-15 (Lonza) with human SCF, Flt3L, thrombopoietin (Peprotech, all 100 ng/mL), IL-3 (Peprotech, 20 ng/mL), 1% human albumin, and gentamicin (50 μg/mL) for 24 h. AProArt or GFP lentivirus was added at a final concentration of 1 × 108 IU/mL for 8 h at 37°C. The cells were then washed and cultured overnight in fresh medium, followed by a second transduction with fresh lentivirus for 8 h. Aliquots were used for progenitor colony formation assays in semisolid medium with cytokines (Methocult, Stem Cell Technologies), 25 and 106 transduced cells were transplanted by intrahepatic injection into 150 cGy irradiated newborn to 3 day old NSG mice. The mice were bled every 4 weeks and sacrificed at 3 months of age for analysis of peripheral blood, bone marrow, and spleen for human cell chimerism by flow cytometry.
Determination of vector copy number and lentivirus integration site analysis
Lentiviral vector copy number for transduced murine LSK cells and human CD34+ cells was determined as described. 25,26 The virus integration site assay was carried out as described. 27 Briefly, genomic DNA from lentivirus-transduced cells was digested and ligated with adaptors, and two rounds of nested PCR were performed. PCR products were cloned, analyzed by Sanger or next generation sequencing (HiSeq2500, Illumina), mapped to the human genome, and examined for oncogenic potential by Gene Ontology software.
In vitro immortalization assay
The in vitro immortalization (IVIM) assay was carried out as described. 25,28 Briefly, prestimulated, lineage negative murine bone marrow cells were transduced with AProArt lentivirus; a long terminal repeats (LTR)-regulated γ-retroviral positive control (RSF91); or an intermediate positive control, a self-inactivating-lentivirus with an internal strong spleen focus-forming virus element. These gene-modified cells were expanded alongside a nontransduced negative control. After 14 days of expansion, the cultures were harvested and replated in 96-well plates with 100 cells/well. After 2 weeks, the wells were scored for immortalized clones. Differences in the incidence (Fisher's exact) and replating frequency (Mann-Whitney) were considered significant at p < 0.05.
Results
Lentiviral vector design and production
Previous studies demonstrated that high-level Artemis protein expression was associated with DNA damage, increased apoptosis, and cell cycle arrest at G1. 29 The endogenous human DCLRE1C minimal 5′ promoter (APro) was hypothesized to produce more moderate and physiologic expression of Artemis; reduced toxicity had been demonstrated in murine Artemis expression comparing APro versus the strong human elongation factor 1 alpha (EF1α) promoter. 30 Building on this experience, human Artemis cDNA driven by its own endogenous promoter was cloned into self-inactivating lentiviral vector CSII with promoter and enhancer sequences deleted from the U3 region of the 3′ LTR (Fig. 1). 24 The central polypurine tract was included to increase the virus titer and the frequency of integration into the host cell genome. This AProArt vector included a modified WPRE element to increase transgene expression in which the putative X protein start codon was mutated to a stop to reduce the risk of insertional mutagenesis. A control CMV-GFP vector was constructed with the same backbone (Fig. 1). The titer of each lentivirus was about 2 × 109 IU/mL.
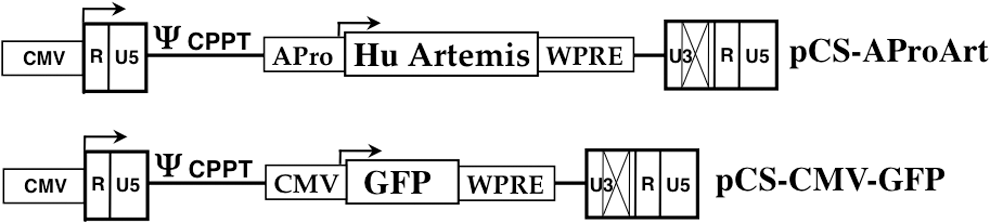
Schematic representations of AProArt and control cytomegalovirus–green fluorescent protein (CMV-GFP) lentivirus vectors. The human Artemis (DLCRE1C) cDNA (Hu Artemis), with its endogenous promoter (APro) was introduced into self-inactivating lentiviral vector pCS with promoter and enhancer sequences deleted (X) from the U3 region of the 3′ LTR to generate pCS-AProArt. A central polypurine tract (CPPT) was included in the vector backbone, preceded by the packaging site (ψ). The vector includes a modified woodchuck post-transcriptional regulatory element (WPRE) and a 5′ CMV promoter to generate full-length vector message for packaging. The control lentiviral vector with the same backbone, pCS-CMV-GFP, was constructed to express GFP under transcriptional regulation of the CMV promoter. LTR, long terminal repeats.
Human Artemis expression and rescue from radiation toxicity in ART-deficient fibroblasts
To demonstrate Artemis protein expression and function following transduction with AProArt lentivirus, we transduced control (WT), Artemis-deficient (ART-SCID) and ligase-4 deficient (LIG4) human fibroblast cell lines with AProArt or control GFP lentiviruses. Like Artemis deficiency, ligase-4 deficiency results in radiation-sensitive SCID; 31,32 therefore, LIG4 fibroblasts provided a control for specificity of the effects of AproArt transduction. At 3 days post transduction, Artemis protein was identified by Western blot in WT and LIG4 fibroblasts and in AProArt-transduced ART-SCID fibroblasts, but not in untreated or GFP-transduced ART-SCID fibroblasts (Fig. 2a). Similar expression was seen at 8 days (data not shown).
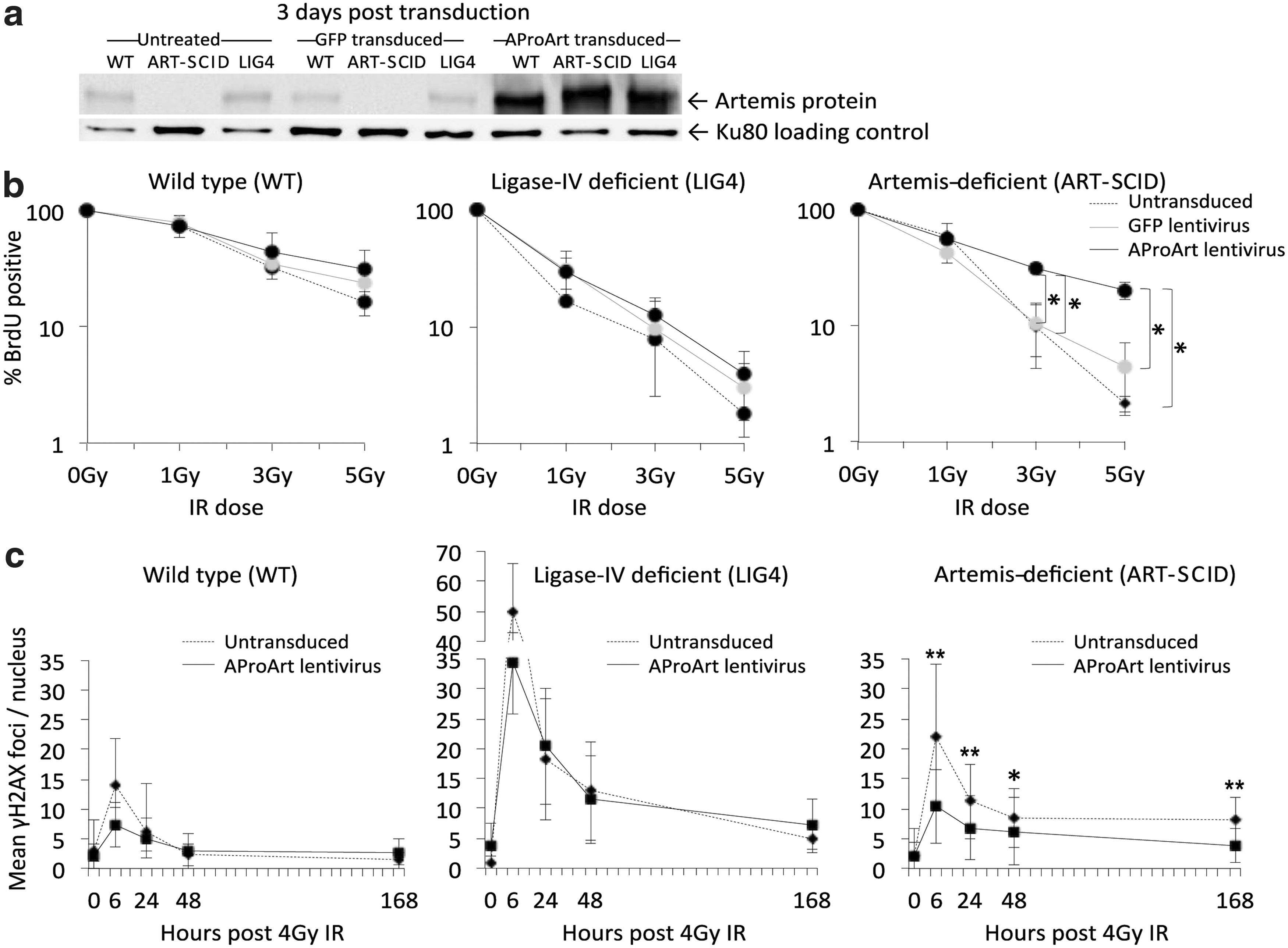
In vitro assays with human skin fibroblasts.
We also evaluated proliferation of the fibroblasts following exposure to 0–5 Gy of γ-irradiation (IR) (Fig. 2b). Without transduction, both the LIG4 deficient (middle panel) and untransduced ART-SCID (Fig. 2b, right panel) fibroblasts were radiation sensitive, exhibiting up to 10-fold reduced proliferation compared to WT (Fig. 2b, left panel) following exposure to graded IR doses. The WT and LIG4 cells showed no change in proliferation when transduced with either control vector or AProArt; however, ART-SCID cells following AProArt transduction proliferated significantly more than untransduced or GFP-transduced ART-SCID cells (p < 0.05), achieving a level comparable to that of untransduced WT fibroblasts.
Finally, we evaluated γH2AX foci as an indicator of ongoing repair of DNA damage; elevated numbers of foci persisting over time indicate incomplete DNA repair (Fig. 2c). AProArt transduction did not change the number of γH2AX foci either at peak production (6 hours) or after 1 to 7 days in either the WT or the radiation sensitive LIG4 fibroblasts (Fig. 2c, left and middle panels); in contrast, DNA repair was normalized in AProArt-transduced ART-SCID fibroblasts (Fig. 2c, right panel). The numbers of γH2AX foci in transduced ART-SCID cells were comparable to those of WT cells (Fig. 2c, left panel), at all time points post IR. Thus AProArt lentivirus specifically reversed the DNA repair defect of Artemis-deficient human fibroblasts.
Determination of optimal AProArt lentivirus concentration for transduction of human CD34+ HSC
Previous studies suggested that overexpression of DCLRE1C could cause cellular cytotoxicity. 23 Moreover, Art−/− murine HSC transduced with murine Artemis cDNA under transcriptional regulation of a strong EF1α promoter failed to engraft in Art−/− recipients. 22 To determine the optimal concentration of AProArt lentivirus for transduction of human CD34+ HSC, we examined the relationship between lentivirus concentration, transduction rates and resulting vector copy number (VCN) per cell. Human mobilized PBSC from normal, Artemis-replete donors were transduced with increasing concentrations (1.25 × 107 to 2 × 108 IU/mL) of AProArt lentivirus and cultured for 14 days in either semi-solid medium for colony forming assays or in liquid bulk cultures. With increasing AProArt lentivirus concentration, the proportion of transduced colonies increased, reaching approximately 50% at both 1 × 108 IU/mL and 4 × 108 IU/mL (Fig. 3a). With 1 × 108 IU/mL of AProArt lentivirus, an average VCN of 3 was achieved (Fig. 3b). Therefore, this concentration of lentivirus was selected for subsequent experiments.
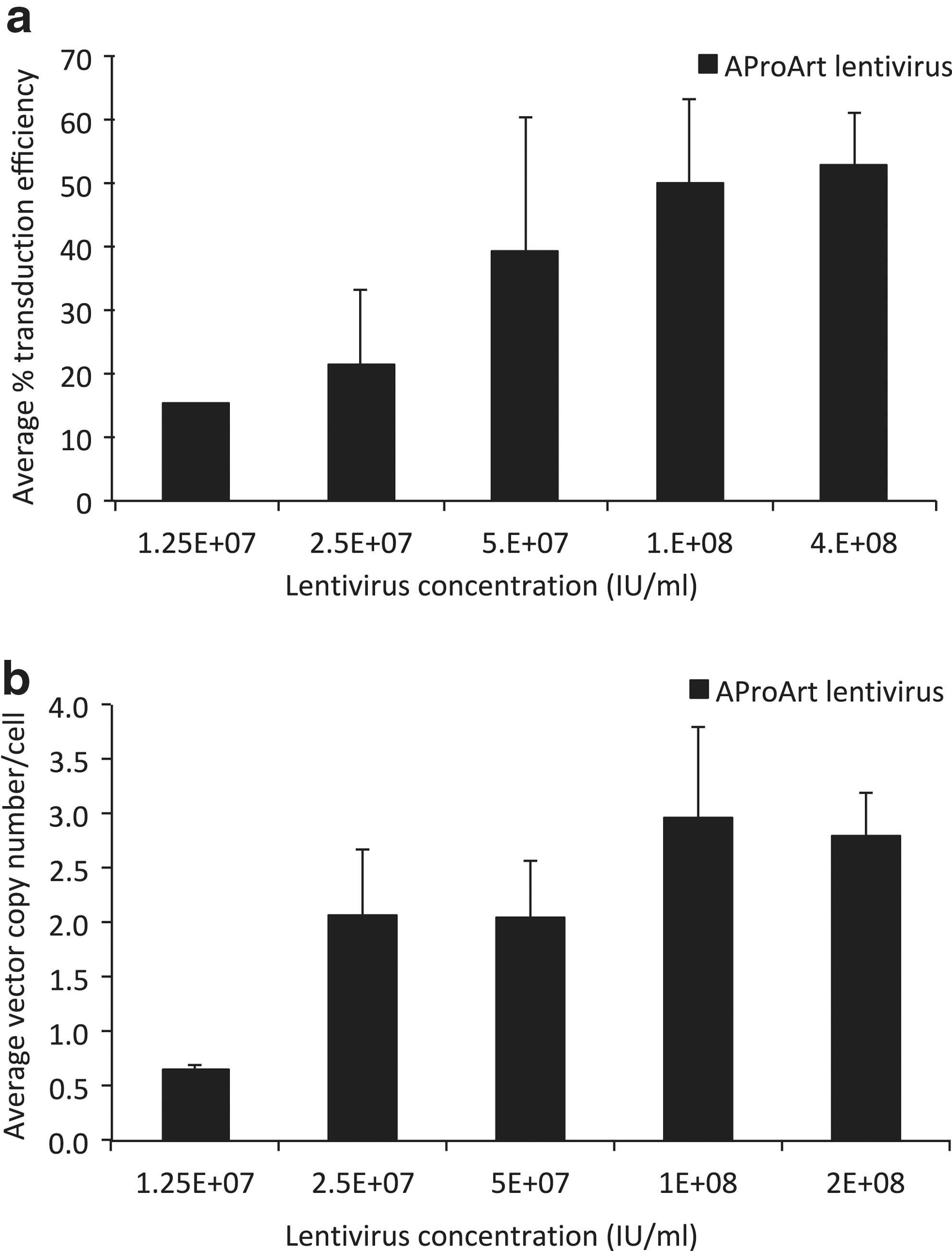
Relationship between vector copy number or transduction efficiency, and lentivirus concentration. Healthy control human peripheral blood mobilized CD34+ cells transduced with increasing concentrations of AProArt lentivirus (black bars) were cultured for 14 days.
Correction of immunodeficiency in Artemis-deficient mice
To determine whether human AProArt transduction could rescue the ability of Art−/− LSK HSC to differentiate into T and B lymphocytes, CD45.2 Art−/− mice pretreated with 500 μg of anti-ckit antibody 9 days previously, to open marrow niches, 33 were injected with either 105 CD45.1 Art−/− LSK cells transduced with AProArt, or 104 or 105 untransduced CD45.1 WT LSK cells (Fig. 4a). As evidenced by the presence of donor-derived CD45.1 lymphocytes in their peripheral blood, five of six transplanted Art−/− mice engrafted with 105 AProArt-transduced Art−/− LSK HSC and all transplanted Art−/− mice engrafted with WT donor cells (Fig. 4a, black squares). Chimerism for T, B, and myeloid lineages in Table 1 indicated multilineage engraftment.
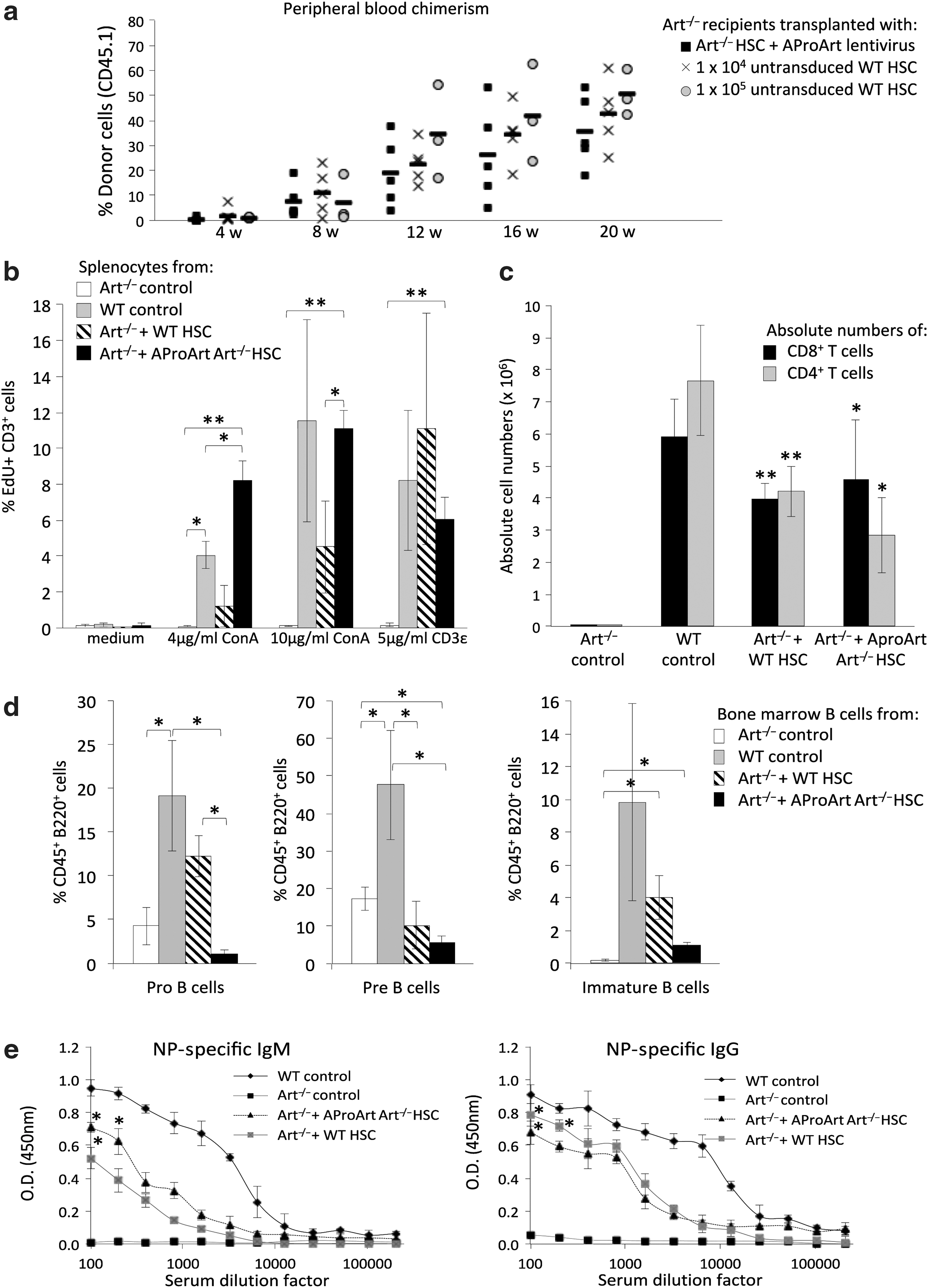
Analysis of Art−/− mice, transplanted with CD45.1 donor cells.
T, B, and myeloid chimerism (mean ± SD) in Art−/ − CD45.2 recipients of CD45.1, AProArt transduced Art−/−, or control untransduced lineage negative, Sca1 positive, c-kit positive hematopoietic stem cells
Mean ± SD of at least five mice per group.
HCT, hematopoietic stem cell transplantation; WT, wild type (control).
The post-transplant splenocytes were cultured in medium alone or with ConA (4 or 10 μg/mL) or anti-mouse CD3ɛ (5 μg/mL), with proliferation of gated populations measured by flow cytometry 4 days later (Fig. 4b). Donor-derived CD45.1 T cells in Art−/− recipients of either WT (hatched bars) or AProArt-transduced Art−/− HSC (Fig. 4a, black bars) proliferated in response to stimuli. Analysis of absolute numbers of CD4+ and CD8+ T cells in the spleens showed that Art−/− mice transplanted with WT or AProArt-transduced Art−/− HSC had comparable numbers of CD4 helper and CD8 cytotoxic T cells, significantly more than those seen in Art−/− controls (Fig. 4c).
Art−/− mice transplanted with AProArt-transduced Art−/− HSC showed B cell development in the bone marrow, as did those receiving WT HSC, while untransplanted Art−/− mice had undetectable immature B cells (Fig. 4d). To assess specific antibody production in vivo, mice were immunized 16–24 weeks following transplant with 100 μg of either NP41-Ficoll or NP24-KLH. Serum ELISA assays 42 days after immunization revealed that Art−/− mice transplanted with AProArt-transduced Art−/− HSC produced antigen-specific IgM and IgG in amounts comparable to those produced by Art−/− mice transplanted with WT HSC (Fig. 4e). Untransplanted Art−/− mice produced no immunoglobulin M or immunoglobulin G.
Colony forming assays with the AProArt-transduced Art−/− LSK HSC used for the above experiments showed a transduction efficiency of 87.5% and a mean vector copy number of 3 ± 2 per cell in positive colonies. The average VCN in the different lineages of the Art−/− mice transplanted with gene corrected cells was 3 ± 2 in T cells, 3 ± 1 in B cells, and 2 ± 2 in granulocytes.
Transduction and in vivo differentiation of human HSC
Cytokine-mobilized PBSC were obtained from an ART-SCID patient, homozygous for the Navajo founder mutation, who was 20 years post HCT and had partial T cell and no B cell reconstitution. 34 Isolated CD34+ HSC had no detectable cells of donor origin (data not shown). These ART-SCID HSC were transduced with 1 × 108 IU/mL AProArt or GFP lentivirus and injected into irradiated, newborn to 3 day old NSG mice to assess whether transduction with AProArt would correct the DCLRE1C defect (Fig. 5). CD34+ HSC from healthy donors were similarly transduced with the control GFP virus and injected into irradiated recipients. At 4, 6, 8, and 10 weeks, peripheral blood was analyzed by flow cytometry for the appearance of human lymphocytes bearing the human pan-leukocyte marker CD45, and at 12 weeks bone marrow and spleens were harvested. NSG recipients of GFP-transduced control HSC, as well as those transplanted with AProArt-transduced ART-SCID HSC, exhibited sustained human lymphocyte chimerism (Fig. 5a). The average percent of human CD45 cells detected in the periphery of mice that received gene corrected cells from 8 weeks onwards, represented 27% of the cells detected in NSG mice transplanted with WT HSC, corresponding to the 25% transduction efficiency of the ART-SCID cells. GFP lentivirus-transduced ART-SCID HSC did not differentiate into mature lymphocytes, however, reflecting their uncorrected DCLRE1C defect. AProArt transduced ART-SCID CD34+ cells gave rise to differentiated human CD3+ T and CD19+ B cells, while GFP-transduced ART-SCID CD34+ cells failed to do so (Fig. 5b). Control CD34+ cells, whether transduced with AProArt or with GFP, differentiated into T and B cells in peripheral blood, bone marrow, and spleen of NSG mice.
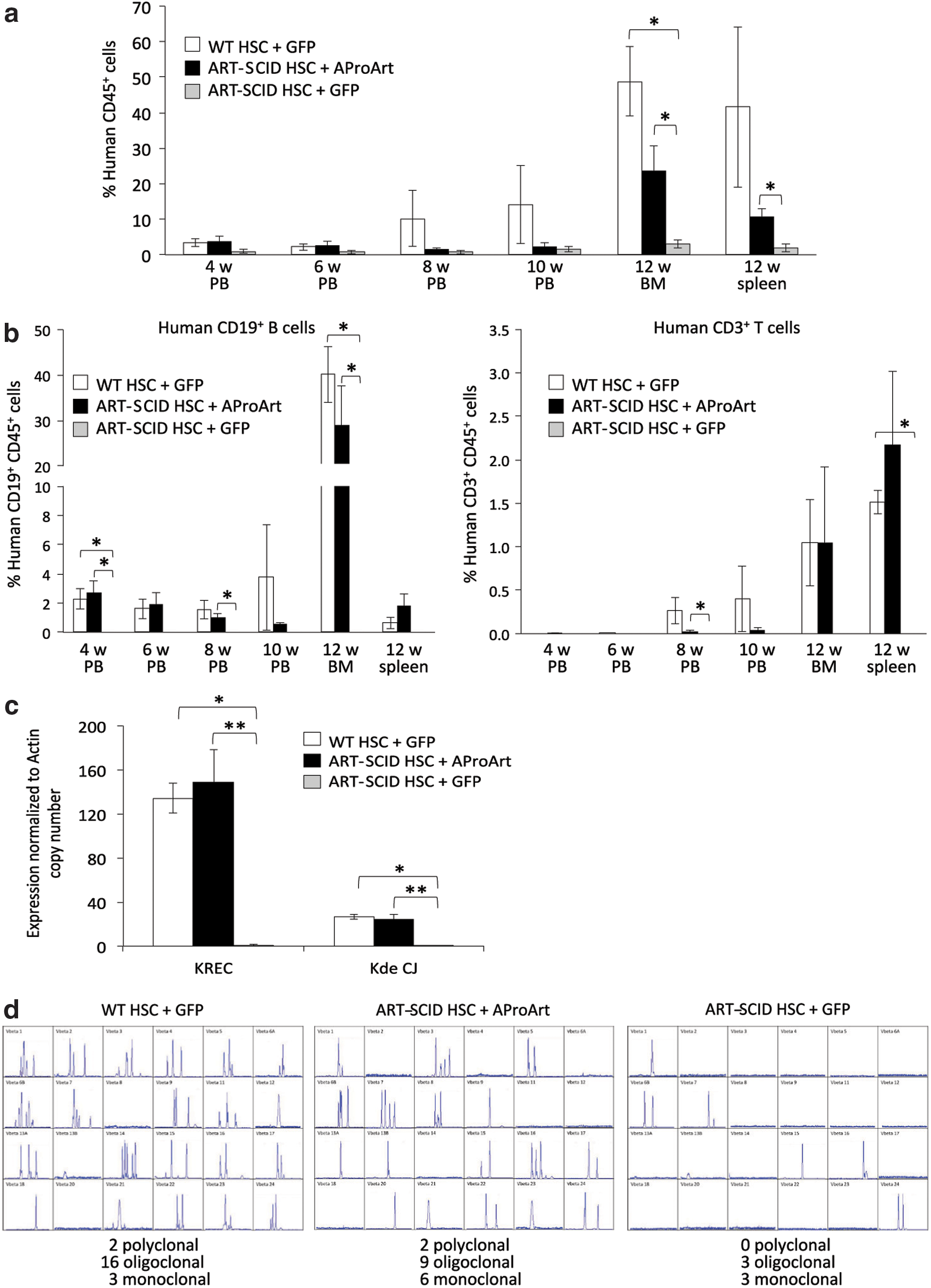
Analysis of NSG mice transplanted with human CD34+ HSC.
DNA from the bone marrow of NSG recipient mice was used to confirm de novo polyclonal B cell development by assaying for Ig κ-receptor excision circles and rearranged linear mature gene coding joints (Kde CJ), by quantitative PCR (Fig. 5c). 35 Mice receiving GFP-transduced normal HSC or AProArt-transduced ART-SCID HSC had equivalent κ-receptor excision circles and coding joints, indicating that the human CD34+ cells had differentiated into mature Ig κ expressing B cells. In contrast, mice receiving GFP-transduced ART-SCID CD34+ cells had no evidence of Ig κ rearrangement.
RNA from chimeric NSG mouse spleens was subjected to T cell receptor spectratyping to measure the diversity of human T cell receptor rearrangement (representative examples in Fig. 5d). 36 Again, mice with GFP-transduced normal or AProArt-transduced ART-SCID HSC showed several human recombinants yielding oligoclonal and polyclonal tracings within the 24 T cell receptor, V beta (TCR Vβ) families tested, while mice transplanted with GFP-transduced ART-SCID CD34+ cells had only minimal TCR Vβ rearrangement.
AProArt lentiviral vector copy number and integration site analysis
Colony forming assays with the AProArt-transduced ART-SCID HSC used for the above experiments showed a transduction efficiency of 25.4% and a mean vector copy number of 2 ± 1 per cell in positive colonies, confirming that human transduced HSC were within an acceptable integrant range for clinical application.
Lentivirus integration site analysis in bulk cultured AProArt or GFP transduced CD34+ cells from healthy donors showed a variety of insertion sites. 27 A total of 1,468 unique integration sites were recovered in AproArt-transduced samples, of which 924 (63%) were located within the promoter or genomic span of human reference gene sequences. 37 In GFP-transduced samples, a total of 1,529 unique integration sites were found, with 976 (64%) mapping to reference genes. To determine whether there were any preferred virus integration sites, the gene lists were examined using the DAVID Bioinformatics Resources 6.8, 38,39 for gene ontology analysis. Following functional annotation (Biological Process Database, ≥3-fold enrichment, Benjamini-Hochberg corrected p value <0.01), the gene list from the AProArt-transduced samples showed slight enrichment in “chromatin modification” category (GO:0016568) (fold enrichment 3; corrected p value 1.5 × 10−4), while the genes from GFP-transduced samples were overrepresented in “microtubule cytoskeleton organization” (GO:0000226) (fold enrichment 3; corrected p value 0.002) and “organelle localization” (GO:0051640) (fold enrichment 3.5; corrected p value 5.3 × 10−3). No insertion sites in the T cell oncogene LMO2 and no significant enrichment for genes with oncogenic potential were found among either AProArt or GFP insertion sites (data not shown, details available from the authors upon request).
Mutagenic potential of AProArt lentiviral vector
To assess the risk of insertional mutagenesis of the AProArt lentivirus, an IVIM assay was performed (Fig. 6). In this assay, nontransduced cells lack the ability to grow when very low seeding densities are used, while insertional mutants show a replating phenotype. The potential to induce immortalization is directly linked to the design of the integrating vectors. 40 RSF91, a gamma retroviral vector with intact LTRs, was used as a positive control to transduce murine lineage negative cells. From previous assays, it had been shown that at VCN >3 and transduction efficiency >80% (Fig. 6, level Q1), RSF91 showed a replating phenotype in 90% of the cases. Furthermore, 75% of these immortalized clones had a replating frequency >3.17 × 10−4. In the current assays, RSF91 scored positive seven of eight times, with a mean replating frequency of 1.61 × 10−3. A second positive control lentiviral vector, with a spleen focus-forming virus promoter/enhancer element (Iv-SF), was able to immortalize cells in two out of three assays. In contrast, while cell growth was detected following transduction with the AProArt lentiviral vector, it was not distinguishable from background proliferation (mean replating frequency 4.67 × 10−7). Therefore, the AProArt vector showed a significantly improved safety profile in the IVIM assay, with respect to both the incidence and replating frequency of the insertional mutants (p < 0.01).
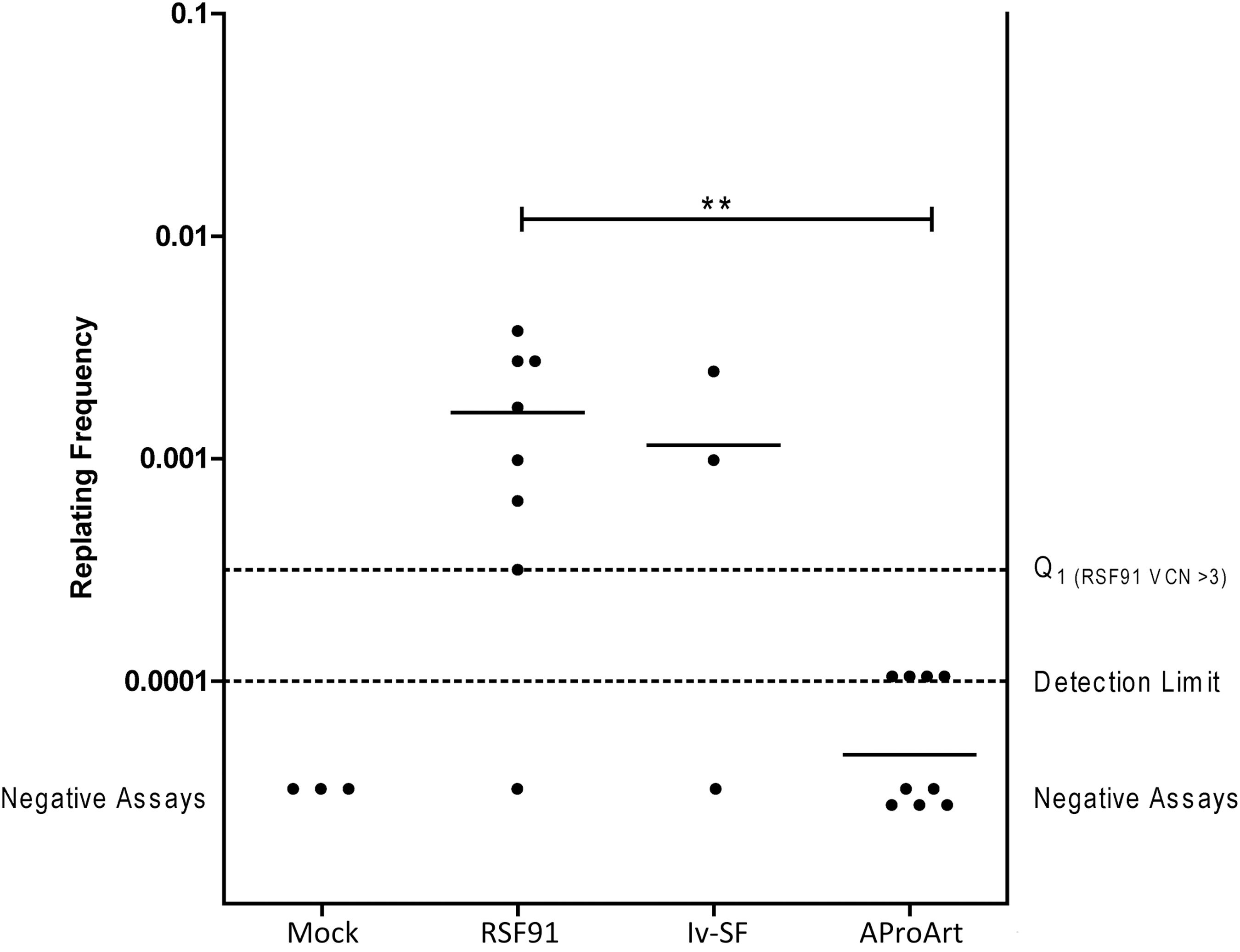
In vitro replating frequency to assess immortalization potential. Replating frequency of nontransduced mock cells (n = 3), positive control samples, RSF91 (n = 8) and lv-SF (n = 3), and the test vector AProArt (n = 9). Bars, mean repopulating frequency; Q1, lower detection range between background proliferation and the first quartile of the RSF91 clonal expectation value at viral copy number 3. Negative assay points manually inserted into the graph. *p < 0.01.
Discussion
A novel lentivirus, AProArt, expressing human DCLRE1C cDNA encoding Artemis under transcriptional control of the endogenous human DCLRE1C promoter, was constructed and evaluated for ex vivo correction of ART-SCID. Expression of AProArt complemented in a specific manner the radiation sensitivity of Artemis-deficient human skin fibroblasts. This vector, achieving a mean of one to three copies per cell in transduced HSC, also restored T and B cell immunity in a murine Art−/− model and enabled human ART-SCID CD34+ HSC to differentiate into mature T and B cells in NSG mice.
Although further study is required, AProArt integration site analysis to date revealed no predisposition for vector insertion into oncogene loci. Furthermore, no integration-mediated toxicity or transformation was observed in animals or in an in vitro insertional mutagenesis assay.
Children with SCID of all genotypes are susceptible to severe infections and typically do not survive beyond the first year of life without immune restoring treatment that has most often been allogeneic HCT, although enzyme replacement has been possible for adenosine deaminase deficiency (ADA) deficiency and gene addition therapy has been successful for ADA and X-linked common gamma chain deficiency. 41 –43 For most patients with SCID, the only available treatment has been allogeneic HCT. The best outcomes occur with a matched sibling donor, because with such donors no conditioning with alkylating chemotherapy is necessary and the majority of patients reconstitute both T and B cell immunity. 6 However, when a matched sibling is unavailable, as is the case for >80% of patients, 15 and an alternative donor is used (i.e., a haplocompatible parent or a matched unrelated volunteer) without conditioning, rejection of the graft can occur, there is a significant risk of graft versus host disease (GVHD), only T cell immunity is typically restored and most patients require life-long gamma globulin therapy to replace B cell function. If chemotherapy with high dose alkylating agent is given as conditioning prior to HCT, T and B cell immunity are usually restored and gamma globulin therapy discontinued; however, this conditioning confers higher peritransplant risks in patients with infections, and the potential late effects of high dose alkylating agents in infants and young children with SCID have not been well characterized. 6,43
ART-SCID is the most difficult form of SCID to cure with HCT. 32,34 In contrast to patients with other SCID genotypes, ART-SCID patients receiving a matched sibling HCT rarely achieve B cell reconstitution, and T cell immunity is often not fully reconstituted. 15,36,43 When haplocompatible related donors are used without conditioning in ART-SCID, normal T cell immunity is even more difficult to achieve, no B cell reconstitution is seen, and there is an increased incidence of graft rejection that can be overcome only with high doses of immunosuppressing alkylating agents. 15,32,34,36,43 Graft resistance may in part be due to NK cell–mediated graft rejection, since NK cells are not deficient in ART-SCID. 16 Use of unrelated allogeneic donors without conditioning in ART-SCID is associated with very poor B cell reconstitution and an increased risk of GVHD. 7 Finally, because Artemis is essential for nonhomologous DNA double-strand break repair, patients with ART-SCID are highly sensitive to the high doses of alkylating agents required to overcome graft rejection and secure B cell reconstitution. They have poorer survival, increased infections, abnormal dental development, short stature, and increased endocrinopathies in comparison to children exposed to no or limited alkylators or children with SCID genotypes not associated with a global DNA repair defect, such as RAG-deficient SCID. 15,34,43
With the increasing adoption of newborn screening for SCID, affected children are being diagnosed in the neonatal period and treated before the onset of life-threatening infections with much improved outcomes compared to those identified and treated later with active infections. 2,6 However, for young babies with ART-SCID lacking an HLA-matched sibling, the risks of toxicity from high dose alkylator preparative regimens to achieve B cell reconstitution and prevent rejection are even more profound. Thus, the use of autologous HSC corrected with gene therapy would be of great benefit, as it would eliminate the risk of GVHD and the need for high dose alkylators to overcome rejection.
Methods to open marrow niches without either alkylating agents or ionizing radiation are currently not available for clinical use. Elimination of HSC from marrow niches by anti-ckit antibody has shown promising results in the RAG−/− mouse model 33 as well as our Art−/− mouse model. The anti-human ckit antibody is undergoing evaluation in a clinical trial that has just been initiated and depending on results won't be available for gene therapy trials for several years. Currently, the only clinically available agent for opening marrow niches is busulfan. Both published 6 and unpublished data from our own center suggest that very low exposures to busulfan (∼25% of the marrow ablative exposure), can achieve T and B cell engraftment and reconstitution in patients with T−B−NK+ SCID. This is further supported by the experience with gene therapy for ADA SCID 41 and X-linked SCID, 42 in which very low doses of busulfan can achieve T and B cell immune reconstitution. Moreover, we have preliminary data (unpublished) to indicate that Artemis-deficient murine HSC are more susceptible to busulfan than wild type HSC, which would suggest that even low dose exposure busulfan could be sufficient to be effective in a gene therapy clinical trial for ART-SCID. In the future, studies comparing low dose busulfan to anti-ckit antibody may determine the optimal treatment for ART-SCID and other patients with SCID receiving hematopoietic cell therapy.
Adverse events from lentiviral gene transfer for ADA deficient and X-linked SCID have been mostly minimal to date, 44 in contrast to the insertional mutagenesis and leukemia that complicated the original X-linked SCID gene therapy mediated by gamma retrovirus vectors with intact LTR promotors. 45 Our use of a self-inactivating lentiviral vector with the autologous Artemis promotor should assure a low risk of insertional mutagenesis. Thus, risks of gene transfer for Artemis deficiency compare favorably with the known adverse consequences arising from the natural history of the disorder and from its standard treatment. The successful application of gene-corrected autologous HSC for children with ART-SCID may have a major positive impact on their survival and quality of life compared to what is currently achieved with allogeneic HCT.
Our results demonstrate the ability of the lentivirus vector AProArt to provide sufficient Artemis protein expression to correct radiation sensitivity and allow successful differentiation of hematopoietic stem cells to reconstitute T and B lymphoid lineages. Therefore we believe that gene therapy using this lentiviral vector can be clinically feasible as a treatment for human ART-SCID.
Footnotes
Acknowledgments
These studies were supported by a research grant from the California Institute of Regenerative Medicine TR3-05535 and the National Institutes of Health National Institute of Allergy and Infectious Diseases, Division of Intramural Research and Warren Magnusen Clinical Center. We thank Steve Yannone, Laurence Berkeley Laboratory, for anti-Artemis antibody and Yanning Wang for technical expertise. We also thank the ART-SCID patients who volunteered to donate cells for these studies.
Author Disclosure
No competing financial interests exist.