
Abstract
In order to support the clinical application of hematopoietic stem cell (HSC) gene therapy for mucopolysaccharidosis I (MPS I), biosafety studies were conducted to assess the toxicity and tumorigenic potential, as well as the biodistribution of HSCs and progenitor cells (HSPCs) transduced with lentiviral vectors (LV) encoding the cDNA of the alpha-iduronidase (IDUA) gene, which is mutated in MPS I patients. To this goal, toxicology and biodistribution studies were conducted, employing Good Laboratory Practice principles. Vector integration site (IS) studies were applied in order to predict adverse consequences of vector gene transfer and to obtain HSC-related information. Overall, the results obtained in these studies provided robust evidence to support the safety and tolerability of high-efficiency LV-mediated gene transfer and above-normal IDUA enzyme expression in both murine and human HSPCs and their in vivo progeny. Taken together, these investigations provide essential safety data to support clinical testing of HSC gene therapy in MPS I patients. These studies also underline criticisms associated with the use of currently available models, and highlight the value of surrogate markers of tumorigenicity that may be further explored in the future. Notably, biological evidence supporting the efficacy of gene therapy on MPS I disease and its feasibility on patients' HSCs were also generated, employing clinical-grade LVs. Finally, the clonal contribution of LV-transduced HSPCs to hematopoiesis along serial transplantation was quantified in a minimum of 200–300 clones, with the different level of repopulating cells in primary recipients being reflected in the secondary.
Introduction
T
Hematopoietic cell transplantation (HCT) from healthy compatible donors can generate a stable source of the functional IDUA enzyme through the tissue-engrafting donor-derived cell progeny. 3 Although HCT has been shown to increase life expectancy and improve some clinical disease manifestations in MPS I patients, residual disease burden is typically observed in the majority of the transplanted children at long-term follow-up, with high variability between patients. 4 Preservation of cognitive function at the time of HCT, a younger age at transplantation, and a normal leukocyte IDUA enzyme level obtained post-HCT are major predictors for superior cognitive development and better long-term outcomes in most organ systems, including the skeleton. The long-term prognosis for patients with MPS I receiving HCT could be further improved by reducing the age at HCT through earlier diagnosis, preferring non-carrier donors, and achieving complete donor chimerism. 5 Moreover, preclinical work in animal models of MPS I, as in the case of other LSDs, 6 –9 unequivocally demonstrated the superior benefit associated to the transplantation of lentiviral vector (LV)-transduced hematopoietic stem cells (HSCs) expressing supra-normal IDUA enzyme levels instead of wild-type (WT) HSCs in correcting disease manifestations. The superior outcome of HSC gene therapy is due to greater enzyme delivery to the affected tissues, including the brain. 6,7 In line with these promising preclinical data is the emerging clinical experience of HSC gene therapy in LSDs. 10,11
In order to develop a successful clinical translation of HSC gene therapy in MPS I patients, biosafety studies are necessary. The document “Guideline on the non-clinical studies required before first clinical use of gene therapy medicinal products” (EMEA/CHMP/GTWP/125459/2006) defines scientific principles and provides guidance to applicants developing gene-therapy medicinal products in order to facilitate a harmonized approach in the EU. Based on these guidelines, the toxic and tumorigenic potential of IDUA gene transfer by LVs into murine hematopoietic stem and progenitor cells (HSPCs) was assessed in vivo in a murine HSPCs into a murine recipient transplantation setting (mouse/mouse). Importantly, the study design was adapted to advanced and internationally recognized safety requirements and to the intrinsic limitations in survival of the disease mouse model by including the use of secondary transplantation and high-throughput vector IS analysis. In vivo biodistribution and differentiation of IDUA overexpressing human HSPCs transduced with clinical-grade LVs and the potential inadvertent transmission of LV sequences to murine cells were also studied in immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (human/mouse). To meet regulatory requirements, these studies were conducted in a Good Laboratory Practice (GLP) setting, taking advantage of the GLP-accredited SR-TIGET test facility.
These assessments demonstrated that high-efficiency LV-mediated gene transfer and above-normal IDUA enzyme expression are well tolerated in murine and human HSPCs and their progeny in vivo. The reported results also contribute to the ongoing discussion on the best modalities to be employed for monitoring LV integration side effects. Finally, the data support the efficacy of gene therapy on MPS I disease and characterize the clonal contribution to hematopoiesis of LV-transduced HSPCs along serial transplantation.
Materials and Methods
Large-scale LV production
Large-scale VSV-pseudotyped IDUA LV concentrated stocks were produced by MolMed S.p.a. (Milan, Italy) by transient quadritransfection of 293 T-cells followed by purification through endonuclease treatment, anion exchange chromatography, gel filtration, and finally sterilizing and aseptic filling. 10 The vectors used for these studies were produced following full good manufacturing practice guidelines (Lot # LV13008 for the biodistribution study and Lot # DSP-10 for the toxicology study). The lot was characterized for potency, purity, and safety aspects. The physical titer of HIV-1 gag p24 capsid protein was measured by enzyme-linked immunosorbent assay (ELISA; Perkin-Elmer, Applied Biosystems, Foster City, CA). The infectious titer was determined on a human T-cell line transduced with serial dilutions of vector, calculating the integrated vector by qPCR. For the transduction of murine cells, LV was concentrated by TIGET personnel through centrifugation in Amicon Ultra-4 and Ultra-15 columns (Millipore, Darmstadt, Germany). After concentration, the titer was assessed again as described above.
Mouse studies
Idua–/– mice on a C57BL/6 background 12 were imported into the animal facility as a kind gift from Prof. J.M. Heard, and were maintained in germ-free conditions. Heterozygous offspring was screened and intercrossed to obtain an inbred strain. Identification of IDUA–/– mice was performed as described. 12 All procedures were performed according to protocols approved by the Animal Care and Use Committee of the Fondazione San Raffaele del Monte Tabor (IACUC 573) and communicated to the Ministry of Health and local authorities according to Italian law.
Ophthalmoscopic examination
An ophthalmoscopic examination was performed using an indirect ophthalmoscope and a slit lamp when necessary. Preliminary observation was performed at the moment of the midriatic instillation. Mydriasis was induced by instilling one drop of 0.5% tropicamide (Visumidriatic) solution into each eye. Animals were manually restrained during the examination. The following were examined: conjunctiva, cornea, sclera, anterior chamber, iris, lens, vitreous body, and fundus.
Transduction and transplantation of murine hematopoietic progenitors and total bone marrow and engraftment evaluation
Six-week-old WT or MPS I mice were sacrificed with CO2, and the bone marrow (BM) was harvested by flushing the femurs and the tibiae. HSPCs were purified for lineage selection as described. 6 Transduced or untransduced cells (5–7 × 105 cells/mouse) were injected via the tail vein into 7- to 9-week-old MPS I mice 4 h after lethal irradiation (a total of 750 cGy divided into two administrations). Transduced cells were also used for clonogenic assays, performed by plating 1.5 × 104 hematopoietic progenitors in a methylcellulose-based medium (MethoCult M3434; Stemcell Technologies, Inc., Milan, Italy). Fourteen days later, colonies were scored, collected as a pool and as a single colony, and lysed for qPCR analysis for the detection of LV sequences. The remaining cells were cultured for 12–14 days as described 6 for the evaluation of IDUA activity. For secondary transplantation, BM was retrieved from primary recipients, red blood cells were lysed, and cells (5–7 × 106 cells/mouse) were injected via the tail vein into 8-week-old MPS I mice 4 h after lethal irradiation (a total of 750 cGy divided into two administrations; Phase II; group A went into D, B into E, and C into F).
Five weeks after transplantation, the transduced cell engraftment was determined by IDUA activity assay on peripheral blood mononuclear cells (PBMCs) and vector copy number (VCN) quantification on colonies obtained by colony forming unit (CFU) assay from PBMCs (same procedure applied on lineage-negative cells, plating 100–200 μL of peripheral blood [PB] after red blood-cell lysis).
Hematologic and cytofluorimetric analysis
Hemocytometric analysis was performed with a Vet abc hemocytometer (scil, Gurnee, IL), collecting PB from the tail vein with anticoagulant (EDTA). The measured parameters were: hemoglobin, hematocrit, total red blood-cell count, mean cell volume, mean cell hemoglobin, mean cell hemoglobin concentration, platelet count, and total leukocyte count.
Cytofluorimetric analysis of PB was performed with a LSR II cytofluorimeter at termination. Approximately 50 μL of PB was collected from the tail vein in EDTA-coated vials. Blood was blocked with Fc Block 1:100 for 10 min at room temperature, and then it was stained with antibodies against the surface markers CD3, B220, and CD11b. Red blood cells were lysed, and the percentage of CD3, B220, and CD11b positive cells was analyzed by flow cytometry. Flow cytometry analysis was also performed on splenocytes, thymocytes, BM cells, and leukocytes from the liver in mice where abnormalities were observed. Staining was performed as follows: approximately 250,000 cells were washed and resuspended in blocking buffer (phosphate-buffered saline [PBS] with 2% fetal bovine serum [FBS], Fc Block 1:100), and then a tissue-specific antibody mix was added (thymus: anti-CD4, CD3, and CD8; spleen, liver, and BM: same antibodies used for PB). After 20 min at 4°C, cells were washed, resuspended in PBS with 2% FBS, 1% paraformaldehyde for cytometry analysis. In all, 10,000 events were acquired with FACS LSR II (BD Biosciences, Franklin Lakes, NJ) using DIVA software and then analyzed by FCS Express.
Transduction of human HSPCs
Human HSCPs (UCB CD34+) were purchased from Lonza (Verviers, Belgium; product code 2C-101). BM samples from MPS I patients were collected under ethical approval REC 08H1010/63 in the United Kingdom; CD34+ cells were freshly isolated and frozen for subsequent use. Soon after thawing, cells were incubated in retronectin-coated (T100A; Takara, Otsu, Japan) non-tissue culture-treated wells (Falcon; BD Labware, Franklin Lakes, NJ) at a concentration of 1 × 106 cells/mL in CellGro® SCGM medium (CellGenix, Freiburg, Germany) with cytokines (IL-3, 60 ng/μL; TPO, 100 ng/μL; SCF, 300 ng/μL; Flt3-L, 300 ng/μL; PeproTech, Rocky Hill, NJ) for 24 h of prestimulation. Cells were than mock-transduced or transduced with IDUA-LV at a multiplicity of infection (MOI) of 100 for 14 h. Cells were washed for 10 h in CellGro medium with the addition of cytokines, and underwent a second round of transduction under the same conditions as the first round. Next, the transduced cells were collected for proper characterization (viability and purity), and were transplanted in NSG mice as described below. The transduced cells were also plated for clonogenic assay (800 cells/mL in methylcellulose medium; human MethoCult; Stemcell Technologies). After 14 days, colonies were counted and collected for molecular analysis. The remaining cells were plated in Iscove's modified Dulbecco's medium, 10% FBS with cytokines (IL-3, 60 ng/μL; IL-6, 60 ng/μL; SCF, 300 ng/μL), and cultured for a total of 14 days for the evaluation of IDUA activity and VCN. 13
NSG mice transplantation and engraftment evaluation
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice 14,15 were obtained from Charles River Laboratories International (Wilmington, MA) and maintained in the animal facility according to protocols approved by the Animal Care and Use Committee of the Fondazione San Raffaele del Monte Tabor (Milan, Italy; IACUC 573) and communicated to the Ministry of Health and local authorities according to Italian law. Seven- to nine-week-old female mice were sub-lethally irradiated (210 cGy) 6 h before the intravenous injection of 3 × 105 untransduced or transduced CD34+ cells. Twelve weeks after the transplantation, the mice were killed, and hematopoietic (BM, spleen, and thymus) and non-hematopoietic organs (liver, brain, and gonads) were collected after intracardiac saline perfusion. According to material availability, priority was given to flow cytometry analysis, VCN determination, and human-to-mouse genome ratio. Flow cytometry analysis was performed as follows: 250,000 cells were washed and resuspended in blocking buffer (Miltenyi Buffer, Fc Block 1:100), and then a human-specific antibody mix was added. 13 After 20 min at 4°C, cells were washed, resuspended in PBS with 2% FBS, 1% PFA for cytometry analysis. In total, 10,000 events were acquired with FACS LSR II (BD Biosciences) using DIVA software and then analyzed by FCS Express. Only animals with >1% CD45+ human cells were considered engrafted.
Quantitative PCR
Genomic DNA was extracted from human CD34+ liquid culture samples and from murine HSPC liquid culture and hematopoietic colony pool samples with a QIAamp DNA Blood Mini Kit (Qiagen, Venlo, Netherlands), and from murine tissues with the same kit, with a Blood Micro Kit, or with a DNeasy Blood & Tissues Kit (Qiagen) after o/n digestion with proteinase K (Roche, Bern, Switzerland).
In the toxicology study, vector copies per genome were quantified by TaqMan analysis as described. 16 In the biodistribution study, LV sequences were detected by qPCR on total genomic DNA and normalized on the human TERT gene. Absolute quantifications were plotted on standard curves prepared with serial dilutions of genomic DNA from the CEMA301 T-cell clone containing one copy of HIV per cell. VCN was then calculated by the following equation: (ng LV ng–1 endogenous DNA) × (n of LV integrations in the standard curve). 16 Engraftment of human cells was determined as ratio between the number of human and murine cells present in the tissues. 13
IDUA activity assay
Enzymatic activity was evaluated using a fluorimetric method as described. 6
Clinical chemistry on serum samples
Samples were collected at necropsy into tubes for serum without anticoagulant. For each serum sample, the following clinical chemistry parameters were evaluated using the Advia 1650 instrument: alanine aminotrasferase, aspartate aminotrasferase, alkaline phosphatase, glucose, albumin, total protein, blood urea nitrogen, creatinine, and triglycerides.
Histopathology
The tissues listed below were fixed using standard fixation procedures in 10% neutral-buffered formalin. They were then paraffin embedded and sectioned at 4 μm, and stained with hematoxylin and eosin. Phase I tissues: aorta (thoracic), brain, cecum, colon, duodenum, epididymides, eyes, gall bladder, heart, ileum, jejunum, kidneys, liver, lungs, mandibular lymph nodes, mesenteric lymph nodes, ovaries, spinal cord, spleen, sternum with BM, stomach, testes, thymus, and lesions. Phase II tissues: liver, mandibular and mesenteric lymph nodes, spleen, sternum with BM, thymus, and lesions. An arbitrary score (from 0 to 5) was given by the investigators on the basis of the percentage of vacuoles observed as a surrogate marker of efficacy. Lesions observed were graded using the industry standard semi-quantitative approach, with grades from 0 (no lesion) to 5 (maximum severity/distribution of the lesion). The study was ethically reviewed and carried out in accordance with Animals (Scientific Procedures) ct 1986 and the GSK Policy on the Care, Welfare, and Treatment of Animals.
Linear amplification mediated–PCR and sequencing
Linear amplification mediated (LAM)-PCR was performed on ∼300 ng of DNA extracted from cultured cells or murine tissues. Briefly, 100 cycles of linear PCR pre-amplification of vector-genome junctions were made, followed by magnetic capture of the biotinylated target DNA by streptavidin-coupled magnetic beads, hexanucleotide priming, restriction digestion using MluCI, HpyCHIV4 and AciI enzymes, and ligation to a restriction site–complementary linker cassette. The ligation product was then amplified by two nested PCR with primers specific for the vector long terminal repeat (LTR) and the Linker cassette sequences. LAM-PCR amplicons were separated on Shimadzu MultiNA Microchip Electrophoresis System to evaluate PCR efficiency and the bands pattern for each sample. Primers and PCR thermal protocols used have been described previously. 17 LAM-PCR products were then purified by AmpureXP beads and quantified with a Qubit™ Fluorometer (Thermo Fisher Scientific, Pittsburgh, PA). Forty ng of PCR product was reamplified with Fusion-LTR (AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNNNNNNNNNNXXXXXXXXACCCTTTTAGTCAGTGTGGA) and Fusion-LC (CAAGCAGAAGACGGCATACGAGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNNNNXXXXXXXXgatctgaattcagtggcacag) primers, containing eight nucleotides (X) tags allowing the samples to be barcoded on both the LTR and the Linker cassette side of the amplicons, specific sequences that allow paired end sequencing with the Illumina MiSeq System, and a random 12 nucleotides (N) sequence to increase cluster separation. PCR was performed using Qiagen TAQ DNA Polymerase under the following conditions: 95°C for 2 min, and 95°C for 45 s, 58°C for 45 s, and 72°C for 1 min, for 12 cycles, followed by a further 5 min incubation at 72°C. Barcoded LAM-PCR products were quantified by fluorometric quantification and assembled into two libraries in equimolar ratios, and avoiding repetition of identical pairs. The two libraries contained LAM-PCRs generated from in vitro and primary mice, and secondary mice, respectively.
Common insertion site analysis
Significant common insertion sites (CIS) were identified by using a sliding-window approach 18 and the Grubbs test for outliers for IS analysis. 19 For the sliding-window method, the R package (latest update of August 2012) was exploited using the function “Cluster” that returned the original input list of ISs with additional annotation fields such as “CIS max order” that represents the maximum number of integrations contained in each CIS and “Cluster ID” that represents a genomic window in which one or more CIS intervals are clustered. For the gene-centric method, the annotation of RefSeq genes was used (General Feature Format file downloaded from UCSC Genome Browser, release MM9, NCBI Build 37), and the average transcript length for each gene symbol was computed. Then, the Grubbs test was performed for outliers to compare the integration frequency for each targeted gene with respect to the average frequency for all the targeted genes of the data set.
Statistics
The assumptions of equality of variance for each group and normally distributed data were tested using Bartlett's test and the Shapiro–Wilk test before performing analysis of variance (ANOVA or Kruskall–Wallis test). Survival was assigned a log or rank transformation. If a statistically significant difference (or trend) was identified, the mean was reported with an asterisk against the relevant group(s), indicating the level of significance (e.g., *p ≤ 0.05 or **p ≤ 0.01).
Results
Assessment of the toxic and tumorigenic potential of IDUA LV-transduced murine HSPCs: study design
The study was designed to assess the toxic and tumorigenic potential of murine homozygous defective (Idua–/–) HSPCs, isolated as lineage negative (Lin–) BM cells, transduced with a LV encoding the human IDUA enzyme, active in murine cells, 20 under the control of the human phosphoglycerokinase (PGK) promoter (defined as the test item [TI]), in the MPS I mouse model 21 (defined as the test system [TS]). To represent a future clinical product better, LV produced in large-scale clinical grade conditions and a clinically relevant minimum target vector integration dose were prospectively employed. Both the tested cell dose (5–7 × 105 cells/mouse, equivalent to 25–35 × 106 cells/kg) and the target vector integration load (2–3 × 108 integrations/kg) exceeded the dose of human CD34+ cells and LV integrations that are generally administered in humans. 11 The control item (CI) for the study was untransduced and manipulated equivalent cells. To favor the engraftment of the transplanted cells, myeloablative pretransplant conditioning based on total body irradiation was administered to young adult recipients (both sexes). Previous experiments excluded the use of busulfan as a conditioning agent in this mouse model because of the death of recipients due to a severe short-term hemodynamic imbalance after transplant. TI and CI were administered by intravenous injection, which is the same as in the clinical setting, according to the study design presented in Fig. 1A. The control system (CS) was represented by untreated MPS I mice (group C). All mice were monitored for survival on a daily basis. Clinical signs, food consumption, and body weight were measured weekly up the end of the study; additional pharmacodynamics endpoints to support hematopoietic reconstitution were measured at 5 weeks post-transplant. Due to the limited survival of MPS I mice caused by disease burden (by 10 months, roughly 50% of the animals usually die), the maximum feasible duration of the post-transplant follow-up was 6 months. At the end of this observation time, transplanted animals were killed, and dedicated evaluations were performed, including clinical chemistry, hematology, flow cytometry, and macroscopic and microscopic examination of organs to assess toxicity or tumorigenicity associated with the use of the TI.
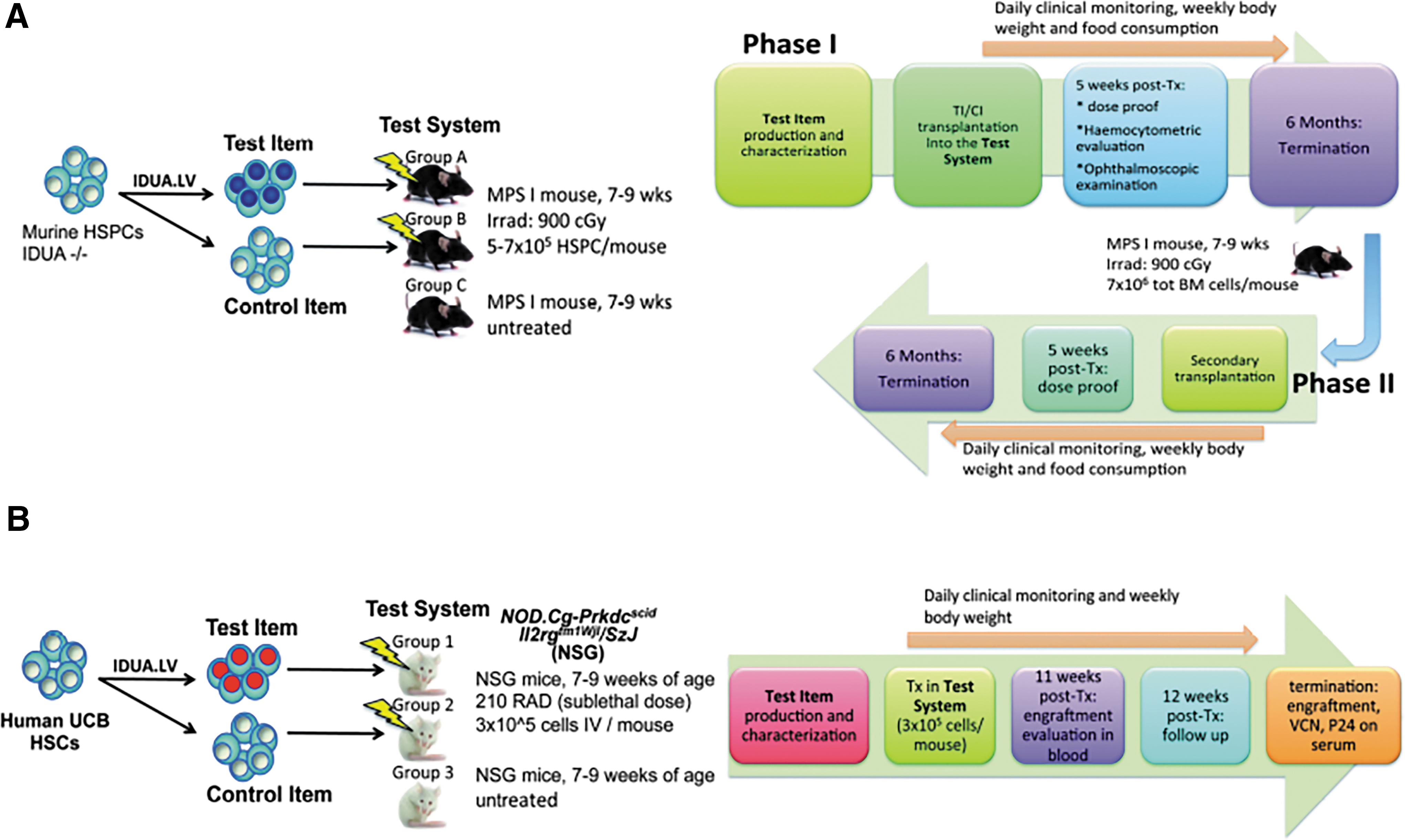
Study design.
In order to study the effect of LV transduction on hematopoietic cells further, BM was transplanted primarily from treated mice into secondary recipients. Secondary-transplanted mice were evaluated at 5 weeks and 6 months after transplant for proof of engraftment and for the occurrence of malignancies affecting liver and lymphohematopoietic tissues (Fig. 1A). Six months after this secondary transplantation, Phase II animals were killed and underwent a full macroscopic evaluation and collection of tissues as in Phase I. In order to obtain additional information on the genotoxic potential intrinsic to the TI, if any, 22 –26 and to study the clonal composition of the engrafted cells and the size of the cell population that participated in the hematopoietic reconstitution of the transplanted mice, IS analysis was also performed on the BM and spleen of the TI group in primary recipients and on the BM of secondary recipients.
Lack of evidence of toxic and tumorigenic potential of IDUA LV-transduced murine HSPCs in primary and secondary transplant recipients
HSPCs were isolated from murine BM by Lin– selection and, for TI only, transduced with IDUA-encoding LVs as described. 16 Multiple batches were produced. The homogeneity and consistency of the TI and CI batches were shown by (i) a similar purity of Lin– and (ii) the comparable clonogenic potential measured in all batches of TI and CI, with low variability (Fig. 2A). All TI batches were efficiently transduced, with a VCN ranging between 5.4 and 11 copies/genome. Enzymatic activity was very high (M = 3026.5 ± 675.2 nmol/mg/h, approximately 35-fold the WT level) in the in vitro progeny of the transduced cell (Fig. 2B).
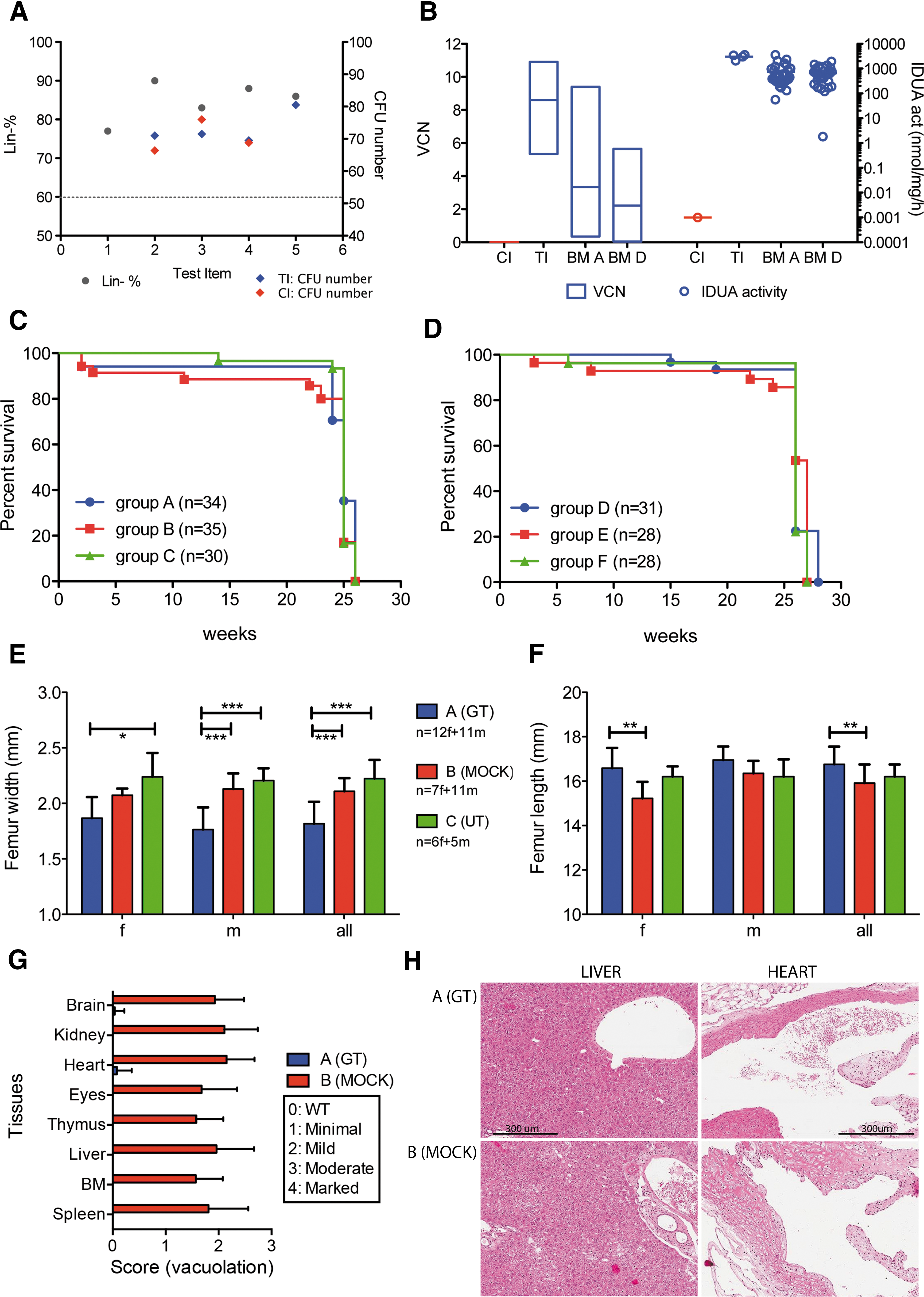
TI characterization, survival, and phenotype rescue of transplanted mice.
Both the primary and secondary transplantations were well tolerated by recipient mice, with only 4–9% inter-current deaths (ICDs) recorded in the first month after the procedure. This mortality rate was similar among the different experimental groups receiving conditioning and transplant (Fig. 2C and D), and considered as a consequence of irradiation conditioning and engraftment failure. The majority of mice survived until the end of the study, with 3–11% ICDs between week 11 and week 24. The mortality rate was similar between treatment groups according to a log-rank (Mantel–Cox) test (Fig. 2C and D). Body weight and food consumption were recorded weekly in all the mice. As expected, due to the use of conditioning, a body weight loss (−4% to −5% relative to baseline) was observed in irradiated groups in the first week after treatment, followed by a weight gain between weeks 2 and 4 (+3% to +8%). Body weight then stabilized for the remainder of the observation period. There was no effect of treatment on food consumption.
High VCN values were measured on bulk colonies obtained from the PB of mice from groups A and D both at week 5 (average VCN ± SD in A: 3.8 ± 1.8; D: 3.8 ± 2.7) and at termination (average VCN ± SD in A: 3.4 ± 1.9; D: 2.2 ± 1.4), demonstrating robust engraftment of gene-modified cells following serial transplant (Fig. 2B). VCN on single CFUs was evaluated only on a few randomly selected mice with an average VCN comparable to the VCN of the pool of CFUs. The sustained engraftment of gene-corrected cells resulted in reconstitution of the defective IDUA activity at and above WT levels in most of group A and group D mice, both in PBMCs (up to 50-fold above corresponding WT levels) and MNCs (up to 15-fold above corresponding WT levels). Hemocytometric analysis on PB showed that control (group C) animals and mice receiving their BM (group F) had higher counts for some parameters compared with groups A and B and their BM recipients (groups D and E), respectively (p < 0.001 for female and p < 0.01 for male with one-way ANOVA, Dunnett or Kruskall–Wallis, Dunn test; Supplementary Table S1; Supplementary Data are available online at
Negative p24 ELISA testing on sera from Phase I mice (below the analytical sensitivity of the ELISA test, <3.125 pg/mL) excluded the presence of circulating aberrantly generated recombinant replication-competent LV. Clinical chemistry showed minimal treatment-related changes in group A (aspartate aminotransferase, urea nitrogen, alanine aminotransferase, and glucose in respect to groups B and C). There were no significant differences between males and females.
Ophthalmoscopic examination, performed on Phase I mice as a standard toxicology observation, showed no treatment-related findings at examinations performed at 5 weeks and at the end of the study. Lens opacity was observed in some animals in groups A and B at the end of the observation period, possibly related to irradiation.
No statistically significant differences in absolute or relative to body weight weights were observed between groups (one-way ANOVA test; Supplementary Table S2). In some early decedents of groups A, B, D, and E, an enlarged thymus, and sometimes also spleen, were observed macroscopically. These enlargements correlated with the lymphomas observed microscopically, also present in additional tissues (aorta, heart, kidneys, liver, lungs, lymph nodes, ovaries, sternum BM, and thymus). In Phase I of the study, seven tumors were observed only in irradiated mice (groups A and B); four of these lesions were lymphohematopoietic in nature, of which one vector-negative lymphoma (VCN = 0) was observed in group A and three lymphomas were observed in group B. Cytofluorimetric analysis of the blood, thymus, spleen, and liver confirmed an expansion of CD3+ cells in the BM and PB of these animals bearing lymphomatous lesions. In addition, one adenoma was observed in the lung of a mock-treated animal, and one hemangioma was observed in the ovary of one gene-therapy animal. In Phase II of this study, seven tumors were found, distributed in all three groups, all receiving irradiation conditioning. Six tumors, found in groups D and E, were lymphohematopoietic (Supplementary Table S2). Pathological findings in other tissues included bilateral lens degeneration in the eyes and an increased incidence and severity of testicular and ovarian atrophy. Overall, these changes were considered to be related to irradiation conditioning and genetic background.
Unmanipulated group C MPS I animals presented multifocal accumulation of vacuolated macrophages in all examined tissues (the aorta, heart, brain, spinal cord, cecum, colon, duodenum, ileum, jejunum, stomach, gall bladder, liver, lung, lymph node, spleen, sternum BM, thymus, kidney, testes, epididymides, ovaries, and eyes). Notably, the width and length of the femur demonstrated normalization of skeletal defects in group A mice compared with mock-transplanted and untreated Phase I animals (Fig. 2E–F). Moreover, the transplantation of gene-corrected HSPCs/BM resulted in treatment-related effects in both primary and secondary recipients, with full recovery or reduction of histologic changes that characterize the disease in this animal model (Fig. 2G and H for primary recipients; data not shown for secondary).
Evaluation of the safety and efficacy of the treatment by IS analysis
The LV genomic integration profile was analyzed in the in vitro progeny of the TI (four independent transductions), on the spleen and BM MNCs harvested from 32 primary mice, and on the BM MNCs retrieved from 28 secondary recipient mice. To amplify the vector/genome DNA junctions from genomic DNA of transduced cells, LAM-PCR was used. PCR efficiency was evaluated by migrating the second exponential PCR products on Shimadzu MultiNA or Perkin-Elmer LabChip systems: samples showed on average an oligoclonal pattern. To identify the vector IS, the PCR products were sequenced on the MiSeq Illumina platform and mapped on the mouse genome (version mm9, July 2007) by a dedicated bioinformatics pipeline. 27 After filtering for quality and collisions, 28 from the different samples, >45 × 106 sequencing reads containing a valid LTR sequence were retrieved that mapped in 6,941 non-redundant genomic positions (Supplementary Table S3). On average, the BMs and spleens of primary transplanted mice yielded 163 ± 100 non-redundant IS (ranging from 24 to 475 IS). Spleen samples produced more IS (140 ± 101 IS, range 19–464 IS) than BM samples (52 ± 22 IS, range 12–105 IS). The BMs from secondary transplanted mice on average yield 28 ± 11 IS (range 6–42 IS; Supplementary Table S4). Overall, the distribution of IS in in vitro cultured cells (n = 1,328), BM and spleen of primary (n = 5,170), and BM of secondary (n = 779) transplanted mice was very similar and reflected the classic LV integration profile, with a marked preference to integrate within gene bodies and no bias for regulatory regions (Fig. 3A). As a safety readout, CIS were identified 22,25,26,28 –32 by the sliding-window approach 18 and the Grubbs test for outliers for IS analysis. From the pool of the TIs and all primary and secondary recipients, 135 CIS (and the nearest genes) were found that were targeted at a frequency significantly higher than expected. After multiple testing correction of p-values, only two CIS were identified and were targeted by 9–16 IS (Supplementary Table S5). None of the CIS identified targeted genes known to be involved in insertional mutagenesis or cancer genes, suggesting a “neutral” bias intrinsic to the vector in mouse cells. GREAT analysis in the three main gene ontologies (GO)—biological process (BP), molecular function (MF), and cellular component (CC)—showed similar biases for gene classes involved in chromatin remodeling, RNA, processing, and genes with hematopoietic functions, and no enrichment for gene classes involved in oncogenesis in in vitro or in vivo data sets. The data set of genes targeted in the BM of secondary mice did not provide any significant overrepresentation of gene classes (Supplementary Fig. S1).
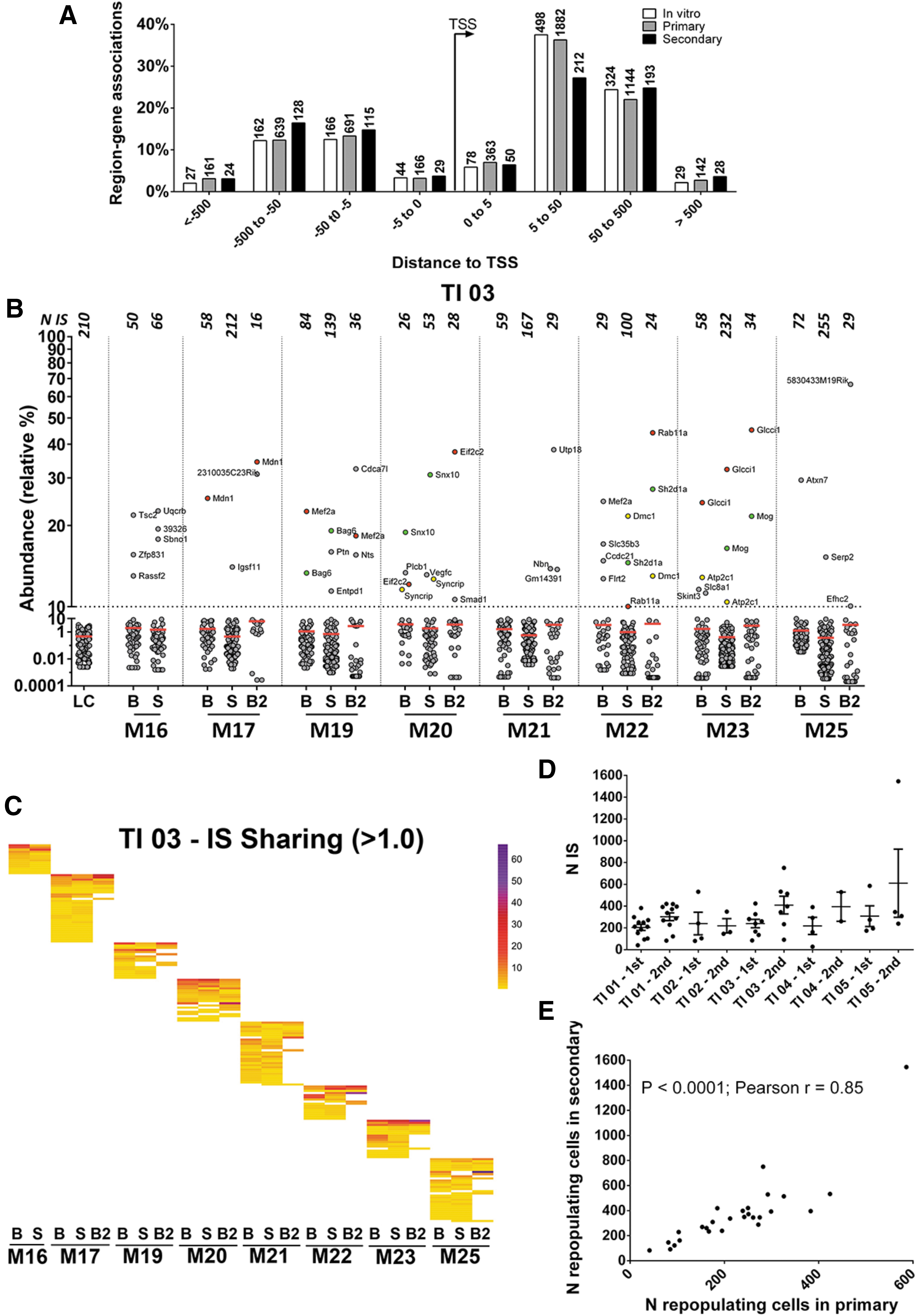
LV IS distribution around gene transcription start sites (TSS).
Given the semi-random nature of LV integration, independently transduced cells will harbor IS in different positions of their genome, a stable genetic mark that allows independent cell clones and their progeny to be distinguished. High-throughput IS analysis allowed the identification of thousands of vector-marked cell clones and estimated their relative abundance in tissues by measuring the number sequence reads obtained from the next-generation sequencing runs with respect to the total number of sequences (Fig. 3B and Supplementary Fig. S2). Among the 6,915 IS analyzed, only 149 (1.8%) showed a relative percentage >10%, distributed in all mice, 97 of which were present in more than one tissue. The IS with an abundance >10% did not show a preferential targeting for specific gene classes or CIS. On the other hand, samples with low numbers of integrations showed the highest number of dominant integrations and the highest abundance (Supplementary Fig. S3), suggesting that in conditions of oligoclonal reconstitution, marked fluctuations in abundance are more frequent. The IS retrieved in the different tissues of primary and secondary recipient mice were also used to analyze the clonal composition and HSPC marking levels. The BM and spleen from primary mice shared on average 20% of the total IS of the mouse. Moreover, 27/28 secondary transplanted mice shared on average 7.5% of the IS with the primary mice (Fig. 3C and Supplementary Fig. S4). In this experimental setting, the simultaneous detection of identical IS in the spleen and BM of mice at 6 months after transplant implies that hematopoietic cells must have divided and contributed to the repopulation of the BM and spleen tissues. Similarly, the sharing of identical IS between primary and secondary mice imply that vector-marked HSPCs repopulated the secondary mice. To estimate the number of repopulating cell clones in primary and secondary mice, the Lincoln–Petersen estimator was used. This method assumes that the number of IS shared between different samples should be proportional to the number of IS in the whole vector-marked cell population. 33 The results of this analysis showed that mice transplanted by each TI were repopulated by 200–300 cells, while secondary recipients were repopulated by 200–600 cells (p = 0.0004 by two-tailed paired t-test; Fig. 3D and Supplementary Table S6). The relative number of repopulating cells in primary and secondary recipients (n = 27 mouse pairs) correlated significantly (p < 0.0001; Pearson r = 0.85; R 2 = 0.73), indicating that the different levels of repopulating cells in primary recipients were reflected in the secondary recipients (Fig. 3E).
Assessment of the biodistribution of LV-transduced, IDUA overexpressing human HSPCs: study design
The objectives of the study were to determine how, after transduction with IDUA-encoding LV, human CD34+ cell (TI) engrafts differentiate into mature progeny and distribute to lymphohematopoietic murine organs, and to compare this to similar control untransduced, equally manipulated CD34+ cells (CI). It was expected that circulating differentiated cells, such as monocytes, might reach non-hematopoietic tissues and became a resident population. Thus, as a secondary objective, distribution of human cells in non-hematopoietic tissues, including the gonads, was assessed. To represent any potential future clinical product better, the LV was produced on a large scale using clinical-grade conditions, and a clinically relevant minimum target vector integration dose was prospectively employed. To achieve the latter goal, and to increase the sensitivity of the system by achieving greater engraftment probability, umbilical cord blood (UCB) was chosen as the preferred HSPC source. Both the tested cell dose (equivalent to approximately 15 × 106 cells/kg) and the target vector integration load (0.5 × 108 integrations/kg) were representative of the dose of human CD34+ cells and LV integrations that are generally administered in humans in clinical trials, and are similar to those likely to be used in any potential future clinical testing. Two rounds of LV transduction at a MOI of 100 with 60 h of in vitro culture were used to ensure high levels of CD34+ cell transduction. 10,34 The CI for the study was represented by untransduced and equally manipulated cells. The TS was represented by NSG murine mice (7–9 weeks old), which represent one of the most valid models for establishing human-mouse chimera by transplantation of human HSPCs. 14,35 The CS was represented by untreated NSG mice Before transplantation, mice were pre-treated with a sub-lethal dose of irradiation (210 cGy) to provide depletion of endogenous BM cells allowing the engraftment of human cells (Fig. 1B). TI and CI were administrated by intravenous injection, which is the most efficient route for systemic distribution of hematopoietic cells in the NSG mouse model and the same route that is employed in the clinical setting.
The treated mice were monitored daily for survival and clinical signs, while body weight was monitored once per week up to 12 weeks after transplantation. Eleven weeks after transplantation, PB samples from transplanted mice were drawn to determine human cell engraftment, and the percentage of cells in different lineages was measured by flow cytometry. At the end of the study, the engraftment, the differentiation to specific lineages, and the persistence of transduced human cells in lymphohematopoietic organs (BM, spleen, and thymus) were assessed. The gonads, liver, and brain were also analyzed, after extensive perfusion to eliminate contaminating blood, in order to evaluate the distribution of human cells in non-lymphohematopoietic organs. Animals were also tested for the presence of HIV p24 in mouse serum in order to test for the presence of RCL.
LV-mediated IDUA above normal expression does not affect human HSPC biodistribution and differentiation in vivo
One batch of TI and one of CI were prepared 35 and coded with a unique identification number. TI and CI properties are shown in Supplementary Table S7. After transduction (or mock-transduction), cells were tested for viability and immunophenotype (CD34 expression) by flow cytometry. Both batches passed the acceptance criteria (viability ≥60%, CD34+ ≥50%) and were released for infusion. IDUA LV transduced cells did not differ significantly from mock-treated control cells in clonogenic potential, as shown by the CFU assay. The VCN measured on the 14 day liquid culture progeny of the TI was 3.54, with a 79% transduction efficiency evaluated by scoring individual colonies from CFU assay by qPCR. This level of transduction supported sustained IDUA expression and activity, up to >70 fold above the mock-transduced (normal cord blood cell) levels. Animals were randomly allocated to treatment group 1 (mice treated with the TI) and to control groups 2 (mice treated with the CI), and 3 (mice left untreated; Fig. 1B). Only one ICD was recorded in the first month after treatment in group 1 recipients. This mortality was considered a consequence of irradiation conditioning. Differences between the survival curves of each group were not statistically significant (log-rank Mantel–Cox test). The body weight recorded at day 1 and weekly until the end of the study (12 weeks) showed no difference among groups using a two-way ANOVA. PB analysis performed 1 week before termination (week 11) showed human CD45+ cell engraftment at similar levels in all mice in groups 1 and 2 in PB (49% on average in both groups 1 and 2; Supplementary Table S8). Statistical analysis (Student's t-test) showed that the myeloid (CD13+) and lymphoid (CD19+ and CD3+) progeny of these cells was present in the PB at similar levels between group 1 and group 2 mice. At termination, 12 weeks after transplantation, human CD45+ cell engraftment was confirmed on hematopoietic tissues (BM, spleen, thymus) and the liver of all mice in groups 1 and 2, with no significant difference between mice in either group (Student's t-test). The majority of human cells detected in the PB, BM, spleen, and liver were CD19+ B cells, while in thymus, the majority of human cells were CD3+ T cells, as expected from the NSG host. Statistical analysis (Student's t-test) showed no significant difference in human cell differentiation in tissues between group 1 and group 2 mice. The small differences between groups observed in CD19+, CD13+, and CD3+ cells can be considered within normal variation (Supplementary Table S9).
Engraftment and biodistribution of human cells were also measured by qPCR (Supplementary Table S10). The high sensitivity of the qPCR method was proven by the estimation of human cell engraftment levels in the BM, with values closely matching the flow cytometry data (Fig. 4A and C). Importantly, in support of the intended mechanism of action and target disease, human cells were detected in the brain of TI-treated animals; TI cells were also detected in the gonads, consistent with the presence of a resident population, progeny of the transplanted cells, or residual blood contamination (Fig. 4B). The biodistribution of transduced and mock-transduced human cells at non-hematopoietic sites was comparable, demonstrating that cells are not influenced by transduction with IDUA LV. The average VCN measured in all examined hematopoietic and non-hematopoietic organs (normalized to human genomic DNA) was comparable with the VCN measured on the TI prior to administration (Fig. 4C). The variability in the VCN measured likely reflected the polyclonal nature of the TI engrafting in vivo and differences in the composition of the engrafted/resident cells in the different organs. Importantly, these findings indicate that the LV distributes together with the human cells in all the tested organs, and that the detectable vector genomes remain associated with the human genomes in stable proportions.
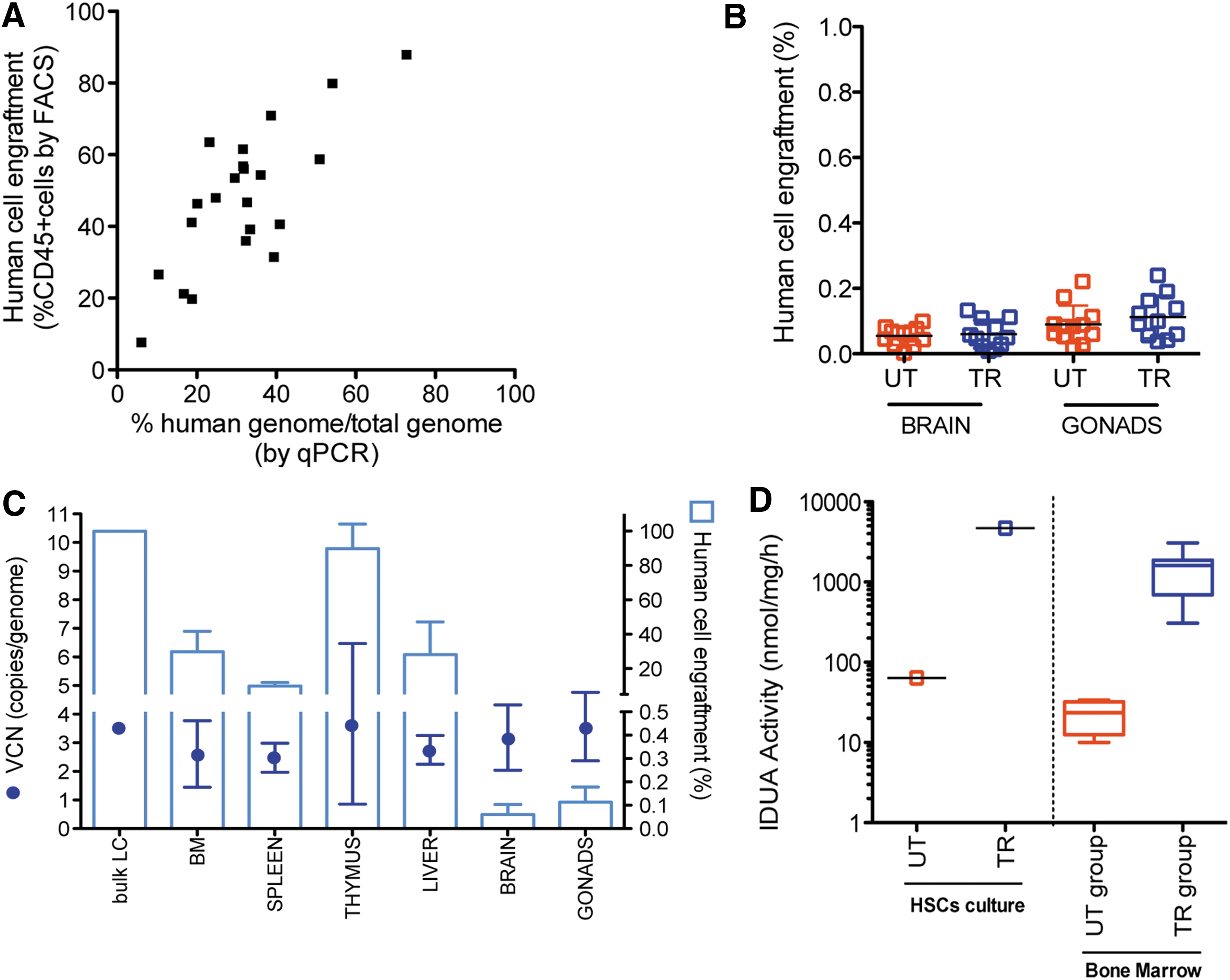
In vivo engraftment and VCN of transplanted mice. Molecular analysis of qPCR was performed on the organs (BM, spleen, thymus, liver, brain, and gonads) of NSG mice transplanted and sacrificed 12 weeks later.
Notably, persistent high expression and activity of IDUA, in a range similar to that observed in the liquid culture progeny of the TI, was proven at termination on the BM of group 1 mice. The average of IDUA activity in the BM of group 1 mice was 1,558.82 ± 891.39 nmol/mg/h (n = 11), 70 times greater than the activity measured in the BM of group 2 mice (22.70 ± 10.18 nmol/mg/h; Fig. 4D).
Transduction of HSPCs from MPS IH patients is feasible
Thanks to the active collaboration with patient referral centers, there was an opportunity to receive BM cells from five MPS IH patients undergoing sedation and BM aspiration before donor HCT, upon collection of informed consent. MPS IH CD34+ HSPCs were transduced with large-scale IDUA LV and a clinically validated protocol at high efficiency, comparable to what was observed on health donors' (HD) CD34+ cells (Fig. 5A). Consistently, sustained IDUA expression and activity above untransduced HD levels were measured in the transduced MPS IH HSPC in vitro progeny. Importantly, no IDUA activity was detectable prior to transduction in MPS IH patient cells (Fig. 5B). Moreover, when transplanted in NSG mice with the settings previously described, transduced patient cells showed long-term repopulation capacity in the BM, spleen, and thymus comparable to those of equally transduced HD HSPCs (Fig. 5C).
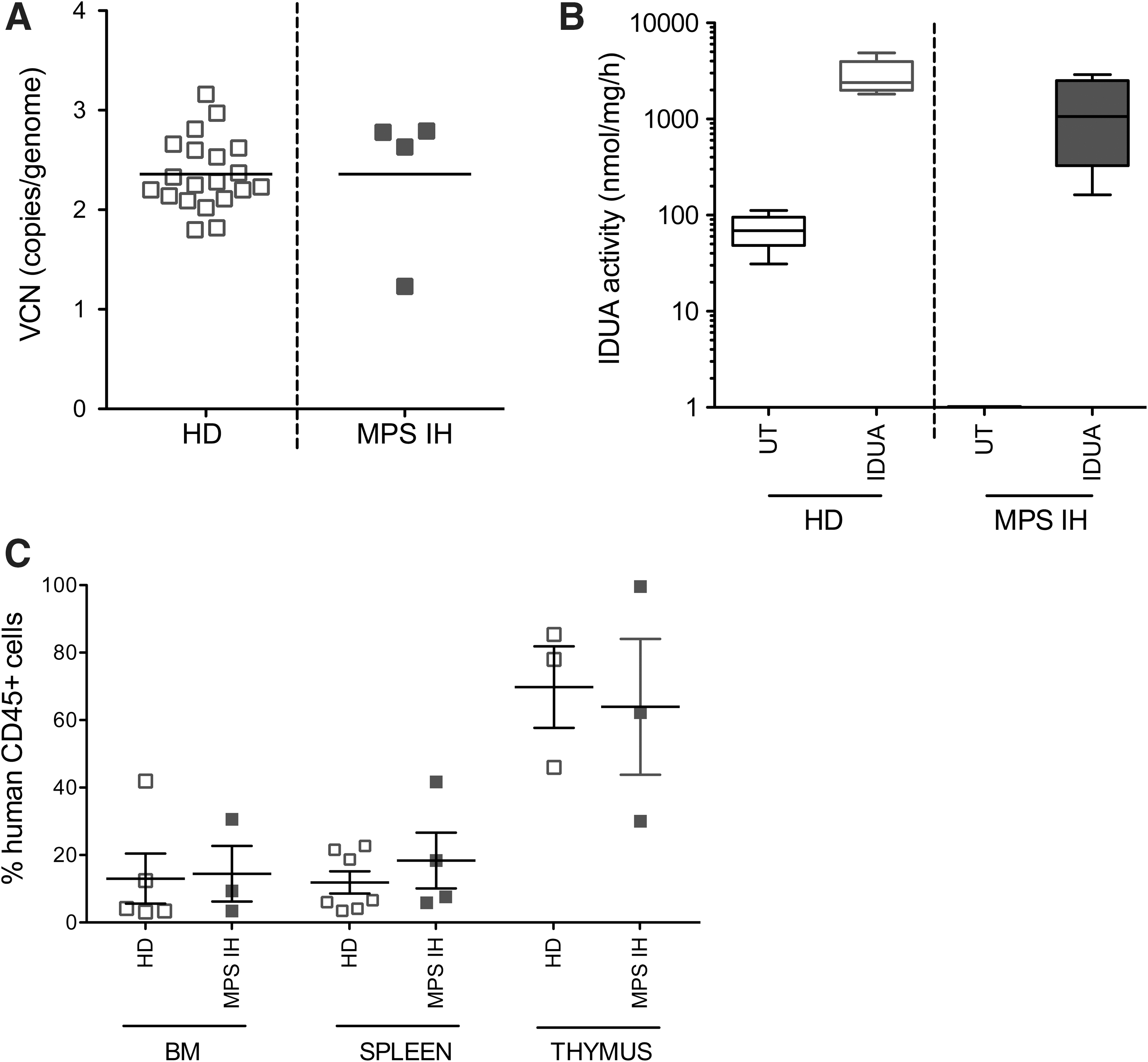
Transduction of MPS IH patient HSPCs. VCN
Discussion
The design of preclinical studies for testing the safety, tumorigenicity, and biodistribution of HSC-based gene-therapy medicinal products is under continual review, and has become increasingly relevant in light of the benefit exerted by this approach in humans in early-phase clinical trials. In order to protect the safety of patients, it is crucial not only to identify the most informative studies and modalities for adverse-effect detection in preclinical models, but also to adapt standard drug protocols to the challenging setting of gene-therapy drug products. Here, the final results are described of a rigorous process of study planning and execution that allowed the assessment of the safety, tumorigenic potential, and biodistribution of IDUA LV-transduced HSPCs, which could support the clinical development of HSC gene therapy of MPS I. In particular, appropriate TIs and TSs were selected for toxicology and biodistribution studies, and standardized methodologies and settings were developed for biosafety GLP studies so that they were appropriate for gene-therapy mechanisms and endpoints. The results obtained in this setting demonstrate the safety of IDUA LV transduction of HSPCs, but also highlight the main limitations of the currently employed testing modalities for LV-integration side-effect monitoring. Biological evidence supporting the potential efficacy of human gene therapy for MPS I disease and semi-quantification of the clonal contribution of LV-transduced HSPCs to hematopoiesis after serial transplantation were also obtained.
Murine HSPCs transduced with IDUA LV were chosen as TI and MPS I mice as TS for a toxicology and tumorigenicity study, based on feasibility and comparability criteria. Indeed, toxicology studies aim to apply the most appropriate and sensitive models to evaluate any possible risk of tumorigenicity in a preclinical context, as demanded by international guidelines. Humanized chimeric mouse models, due to their intrinsic limitations (i.e., insufficient survival of the human graft in the mouse for the evaluation of tumorigenicity) do not support such a long-term evaluation, which is instead feasible in the mouse/mouse transplant setting. Importantly, MPS I mice recapitulate most of the features associated with IDUA deficiency in humans, thus also rendering them a valuable mimic of the clinical situation. Of note, Lin– cells can be considered equivalent to the human CD34+ progenitor cells used in the clinical setting. CD34+ cells derived from UCB transduced with IDUA LV were chosen as the TI in the biodistribution study. UCB-derived CD34+ cells represent a reliable model for assessing any detrimental effects of high-level transduction and sustained high-level IDUA expression on CD34+ cell engraftment and lineage differentiation. Indeed, engraftment levels are particularly high and homogenous in NSG mice when UCB cells are employed. Moreover, since young children (<30 months of age) represent the most likely target patient population for potential further development of this concept in an MPS I clinical trial, CD34+ cells from UCB represent the cell source close to the real target of the clinical application because commercially available BM-derived CD34+ cells are usually obtained from adult donors. Of note, when selected experiments were repeated employing MPS I patient samples, similar results were obtained. The NSG mouse model was selected as the TS for the biodistribution study, since, being immunodeficient, it allows the high-level engraftment of human cells after HSPC transplantation. 14,15
The most appropriate pretransplant regimen was also selected in order to obtain sustained engraftment of the transduced cells, long-term survival of the transplanted mice, and the most neutral setting for detection of transduced cell-related adverse effects. Pilot studies, conducted in WT, MPSI and NSG mice comparing different regimens, allowed the selection of total body irradiation at myeloablative and sub-myeloablative doses as the conditioning regimen for toxicology and biodistribution studies, respectively. Dedicated studies analyzing the actual need and the extent of immunosuppression in association to myeloablation in the human gene-therapy setting are ongoing, considering that many MPS IH children are CRIM– and receiving enzyme-replacement therapy. Irradiation at myeloablative doses led to sustained engraftment of the transduced murine HSPCs in the BM of mice treated by gene therapy in the toxicology study. However, these mice, in both Phase I and Phase II, developed tumors at a higher incidence compared with mice not receiving pretransplant conditioning. This finding, likely due to background oncogenesis caused by irradiation of the hematopoietic cells of the host surviving after the administration of the conditioning, could represent an impediment for the detection of a possible tumorigenic potential of vector integration into the transplanted HCSs.
The choice of the most informative timing for analysis of the genotoxicity and tumorigenicity of the LV-transduced HSPCs is also critical. Indeed, the onset of adverse events such as tumors associated to senescence in control and treated populations could mask the possible effects of the TI. Thus, studies covering a long period could be replaced by serial transplantation designs, which may allow the long-term fate of the genetically manipulated cells to be monitored independently from aging of the hosts. Serial transplantation also allows for a more challenging repopulation setting, possibly contributing to unravelling any abnormal behavior of the TI. In the present case, this design was also chosen in order to overcome the limited survival of the mutant strain. Even in this setting, however, the above-mentioned background oncogenesis represented the main finding as far as neoplasms and abnormal proliferations are concerned.
These limitations may indicate the need for surrogate markers of tumorigenicity (i.e., IS analysis). In the present study, IS analysis performed on in vitro–cultured cells, BM, and spleen tissues from primary mice and BM from secondary recipients allowed a comprehensive analysis of the safety of the treatment, as far as genotoxicity is concerned. Actually, the analyses did not evidence any genotoxicity in this study.
Having evaluated the neutrality of LV marking, the IS retrieved in the different tissues of the same mice and secondary animals was used to analyze their clonal composition and HSC marking levels. Remarkably, transduced HSPCs were shown to participate in the long-term hematopoietic reconstitution of primary and secondary recipients. The size of the cell population participating in the hematopoietic reconstitution of transplanted mice was estimated on average as 200–400 cells, which corresponds to 0.02–0.04% of the transplanted Lin– cells (5–7 × 105).
Of note, the mouse study also allowed confirmation of the efficacy of gene therapy on disease-related manifestations, previously shown in proof-of-concept studies, in a stringent and rigorous setting and employing clinical-grade LV.
In the biodistribution study, sustained engraftment of transduced human HSPCs was observed. The biodistribution of human cells in target and non-target organs was comparable between the mice receiving transduced IDUA, overexpressing HSPCs, and control cells. The range of VCN measured in the ex vivo samples from animals that received the TI was comparable to the pretransplant sample and similar between analyzed tissues. This indicates that the long-term repopulation and multilineage differentiation potential of human HSPCs was not affected by gene transfer or by a sustained level of IDUA enzyme overexpression. In this setting, it was also shown that the LV remained strictly associated to the human cells in stable proportions, thus confirming the absence of significant vector shedding and/or generation of replication competent recombinants. 13 Moreover, supporting efficacy data on the maintenance of sustained levels of enzyme overexpression upon repopulation of the host and in vivo differentiation of the transduced human HSPCs were recorded. Overall, the study design for biodistribution turned out to be feasible and informative. However, the inherent heterochimeric nature of the model makes it less informative regarding LV integration side-effect monitoring, which was thus not applied in this setting. Indeed, it could have been significantly affected by the artificial expansion and differentiation of the human graft into a different species, possibly providing different and non-physiological stimuli, and by its limited survival in time.
Lastly, IDUA LV transduction of MPS I patient cells with a clinically validated protocol was shown to be feasible in laboratory scale and, importantly, as efficacious as in healthy donors in restoring IDUA activity and raising it above HD levels. Importantly, transduced IDUA overexpressing patient cells could engraft in NSG mice similarly to healthy donors cells, confirming their preserved functional features in vivo.
Overall, these studies, designed and optimized for addressing the specific requirements of HSC gene therapy for a lysosomal enzyme deficiency in a standardized GLP setting, provide strong and robust evidence for the lack of any significant toxic or tumorigenic effect of LV-mediated IDUA transduced cells. Furthermore, the above-normal IDUA enzyme expression level that results from LV transduction does not significantly affect normal-cell longevity, development, or function. These studies also underline the limitations and criticisms associated with the use of currently available methodologies, that is, for recipient mice conditioning, and highlight the value of surrogate markers of tumorigenicity that may be further explored in the future. Of note, this experimental setting also provided relevant biological information, including tracking of HSPCs repopulating clones in vivo along serial transplantation.
Footnotes
Acknowledgments
We would like to thank Paola Albertini, Mark Goodwin, and Edoardo Stradella for the quality-assurance work; Di Gallo Guido for the ophthalmoscopic examination; Laura Lorioli for critical discussion during manuscript revision; and the GSK R&D Rare Disease Unit for the review of this manuscript. This study was funded by Telethon Foundation.
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.