
Abstract
Acute myeloid leukemia (AML) still represents an unmet clinical need for adult and pediatric high-risk patients, thus demanding advanced and personalized therapies. In this regard, different targeted immunotherapeutic approaches are available, ranging from naked monoclonal antibodies (mAb) to conjugated and multifunctional mAbs (i.e., BiTEs and DARTs). Recently, researchers have focused their attention on novel techniques of genetic manipulation specifically to redirect cytotoxic T cells endowed with chimeric antigen receptors (CARs) toward selected tumor associated antigens. So far, CAR T cells targeting the CD19 antigen expressed by B-cell origin hematological cancers have gained impressive clinical results, leading to the possibility of translating the CAR platform to treat other hematological malignancies such as AML. However, one of the main concerns in the field of AML CAR immunotherapy is the identification of an ideal target cell surface antigen, being highly expressed on tumor cells but minimally present on healthy tissues, together with the design of an anti-AML CAR appropriately balancing efficacy and safety profiles. The current review focuses mainly on AML target antigens and the related immunotherapeutic approaches developed so far, deeply dissecting methods of CAR T cell safety improvements, when designing novel CARs approaching human studies.
Introduction
A
Nowadays, allogeneic HSCT remains the only curable option for adult and pediatric intermediate- and high-risk AML, as the immune-based principle of graft versus leukemia (GvL) effect has the potential to eradicate LSCs. The estimated 3-year survival rate after human leukocyte antigen (HLA)-matched sibling allogeneic HSCT in first remission varies from >70% in children and adolescents to almost 64% and 53% in adults aged 21–40 and 41–60 years, respectively, dropping to 45% in patients older than 60 years of age. Improvements made in HLA typing and matching have led to a higher number of unrelated HSCT in recent years, with a 3-year estimated survival rate of around 50% in childhood and young adults to <40% in the elderly. 7 However, the risk of transplant-related mortality (TRM) remains high because of the possibly severe side effects of HSCT such graft-versus-host disease (GvHD) and opportunistic infections. As the TRM in adolescents and adults receiving unrelated donor transplantation is still near 30%, elderly or unfit patients are often not considered for HSCT. The challenge of novel therapies for AML is therefore to match efficacy and safety, targeting LSCs to prevent clonal evolution or survival of leukemic cells while reducing treatment-related toxicity.
Targeted Therapy in AML: An Overview
The rationale behind conventional chemotherapies for AML employed in the last four decades has been conceived to be highly cytotoxic against AML blasts. However, current novel insights into pathophysiology and genetic and molecular characterization better defined the biological underpinnings of this disease, suggesting that we are possibly dealing with multiple distinct entities. 4,8 The uncovered AML heterogeneity relies on the existence of different deregulated pathways, and in the last decade, several novel targeted drugs have been designed, moving AML treatment toward a more individualized and tailored approach. 9
The power and potentiality of a targeted individualized therapy can be seen for example with acute promyelocytic leukemia (APL). By targeting the promyelocytic leukemia/retinoic acid receptor-alpha (PML-RARα) protein, these agents have improved the cure rate from 30% to 90%. 10
With the advances obtained in next-generation whole-genome sequencing, several recurrent somatic mutations have been identified. In a recent study among 200 patients with de novo AML, an average of 13 genetic mutations were found, including mutations in signaling pathway genes, DNA-methylation-related genes, chromatin-modifying genes, transcription factor genes, tumor suppressor genes, and cohesin-complex genes. 11 Some of these molecular altered pathways, such as NPM-1, FLT3, and CEBPA mutations, have already been included in the AML classification systems, implementing the prognostic risk categories traditionally defined only based on cytogenetic abnormalities. The identification of altered genes, and thus the resultant heterogeneity of the leukemic cells of an individual patient, has not only improved stratification and treatment decisions, but has led to the development of non-genotoxic, “more targeted” drugs with synthetic inhibitors. 12
Beside these new drugs, which act at a molecular level, immunotherapeutic approaches aimed at augmenting the immune-system functions to enhance tumor-specific cytotoxicity while avoiding systemic toxicities associated with chemotherapy have gained impressive results in the last decades. Indeed, the knowledge of both the innate and adaptive (humoral and cellular) primary components of the immune system has offered fertile ground to develop more advanced strategies. Concerning active immunotherapy (i.e., treatments with AML vaccines and cytokines), which is aimed at boosting the patient's immune system to react against leukemia cells, consistent results have not been yet achieved, and the real benefit of these therapies is still to be determined. 13 On the other hand, immunotherapy with antibodies or other immune-system components were conceived to provide passive immunity against AML. In particular, antibody-based therapeutics represent a continuous growing field of research, starting from unconjugated monoclonal antibodies (mAbs), selected on the basis of altered expression of a lineage-specific human differentiation antigen on the surface of leukemic cells, and moving toward more effective systems, such as mAbs conjugated with cytotoxic payloads (drugs, toxins, or radioisotopes) and bi- or tri-specific mAbs (Fig. 1). Moreover, evidence of a T-cell mediated curative effect known as GvL in the context of HSCT has prompted the use of immune effector cells for the treatment of hematological malignancies. One of the major challenges in the last decade is developing adoptive cellular therapies inducing GvL in the absence of GvHD, an allogeneic reaction of donor T cells against normal host tissues. Thus, from the classical use of donor lymphocyte infusions or infusion of ex vivo–expanded tumor-infiltrating lymphocytes, research has focused on innovative techniques of genetic engineering to equip cytotoxic T cells with chimeric antigen receptors (CARs) or artificial T-cell receptors (aTCRs) in order to redirect them specifically toward selected tumor-associated antigens (TAA). 14,15 Employing CARs, target recognition is dependent on a single-chain variable region domain of a monoclonal antibody (scFv), while aTCRs are conceived to increase the affinity of the aTCR beyond that expressed by naturally occurring T cells in response to an identical epitope. As an alternative to T cells, other cellular sources for the immunotherapy of AML are represented by natural killer (NK) cells and dendritic cells (DC), and their clinical application has been deeply reviewed elsewhere. 16,17 In the era of advanced medicine, adoptive cell transfer of ex vivo expanded and manipulated effector T cells has already reached the clinic, gaining successful results for the targeting of both solid and hematological malignancies. 18,19 The identification of an ideal target cell surface antigen, which is highly expressed on the resistant tumor cells but minimally expressed on healthy tissues, represents one of the main requirements when designing an immunotherapeutic strategy. The main targeted AML antigens, also conceived to recognize AML-LSCs, are represented by CD33, CD123, CD44v6, Lewis Y (LeY), CD47, CD96, TIM-3, MUC-1, CD32A, and CD25 (Table 1). 20,21 Most of them are currently the subject of CAR-mediated targeting, and they will be described more in detail below. The current knowledge on the complexity of AML and the earliest clinical evidence from studies exploiting single agents clearly push the scientific community to move toward combination strategies involving specific targeted agents at different target levels with standard chemotherapeutic drugs known to have activity in AML. In this sense, numerous combination regimens are under investigation at either the preclinical or clinical level, involving both molecular pathways inhibitors and immunotherapeutic strategies. 22

Acute myeloid leukemia (AML) antigen-specific immunotherapeutics in clinic. Different targeted immune-based strategies are illustrated. ADC, antibody-drug conjugate; RAIT, radioimmunotherapy; BiTE, bi-specific T-cell engagers; DART, dual-affinity re-targeting; aTCR, artificial TCR; CAR, chimeric antigen receptor; CD33, cluster of differentiation 33 (Siglec-3); HuM195 or Lintuzumab, humanized monoclonal antibody 195; 225-Ac, actinium-225; 90Y, yttrium-90; Bi213, bismuth-213; SGN-CD33A, anti-CD33 monoclonal antibody-drug conjugate; rGel, recombinant gelonin; CD123, cluster of differentiation 123 (IL-3 receptor α); CSL362, KHK2823, anti-CD123 monoclonal antibodies; SL-401, recombinant IL3 fused to diphtheria toxin; CD47, cluster of differentiation 47 (integrin associated protein Ig superfamily); Hu5F9-G4, CC-90002, anti-CD47 monoclonal antibodies; CD117, cluster of differentiation 117 (c-Kit or tyrosine-protein kinase kit); LOP628, LMB-2, anti-CD117 monoclonal antibody-drug conjugate; CD25, cluster of differentiation 25 (IL-2 receptor α); ADCT-301, anti-CD25 monoclonal antibody-drug conjugate; FLT-3, fms-like tyrosine kinase 3; IMC-EB10, FLYSYN, anti-FLT3 monoclonal antibodies; CXCR4, C-X-C chemokine receptor type 4; BMS-936564, anti-CXCR4 monoclonal antibody; WT-1, Wilms tumor 1. Color images available online at
AML associated antigens with detailed expression on AML, leukemic stem cells, hematopoietic stem/progenitor cells and other normal tissues
AML, acute myeloid leukemia; LSC, leukemic stem cell; HSC, hematopoietic stem cell; HSPC, hematopoietic stem/progenitor cells; NA, not assessed; ND, not detected.
Anti-Myeloid Cars: Implementation Toward the Clinic
So far, CAR T cells redirected toward the CD19 antigen have been applied for the treatment of acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (B-CLL), and B-cell non-Hodgkin lymphoma (NHL), with impressive and dramatic clinical results, 23 paving the way for the application of the CAR technology also to treat solid tumors and other hematological malignancies such as AML.
However, difficulties in translating this approach to other tumors are mainly related to concerns regarding target antigen choice. Since no widely expressed tumor-specific AML antigens have been described so far, the emphasis has been on the identification of AML TAAs, being highly overexpressed on the surface of myeloid malignant cells, with no or little expression on normal cells. Therefore, given the autoreactivity risk against normal and vital tissues expressing the target antigen, predictive safety studies concerning each target antigen are mandatory.
For some of these TAAs, the feasibility of T-cell redirection with CARs has already been proved in a few clinical trials, such as in the case of LeY and CD33 targets. However, efficacy and safety issues emerged from these first attempts, highlighting the need for further evaluation for a proper clinical application of this strategy. 24,25
LeY antigen (CD174) is a difucosylated type 2 blood group–related carbohydrate antigen with unknown function, whose expression has been documented in a wide range of malignancies, including AML, 26 and it has also been associated with a poor prognosis in some cancer types. However, expression of LeY on the normal cell compartment has been reported on mature neutrophils as well as on plasma cells and native CD34+ hematopoietic precursor cells, 27 while being absent on normal circulating T lymphocytes and malignant B and T cells. A Phase I study employing a second-generation LeY-CAR for patients with relapsed/refractory LeY-positive AML was performed in four patients after a fludarabine-conditioning regimen. Although two of four patients experienced a reduction in their residual disease, all patients eventually relapsed, and a slight CAR T-cell expansion and persistence was observed, 24 together with limited CAR expression levels ranging from 14% to 38%. Modest adverse events occurred, reported as a mild neutropenia, spontaneously resolved in only one patient. No downregulation of LeY antigen has been detected following CAR T-cell infusion, indicating that this antigen could also be suitable for long-term control of the disease. Moreover, CAR T-cell trafficking has been detected within both bone marrow and other disease sites, highlighting the possibility of eradicating central and peripheral disease reservoirs equally. However, the clinically observed tolerance toward LeY CAR T cells may be attributable to the modest CAR T-cell expansion and persistence, suggesting a more careful evaluation of the potential side effects arising from LeY targeting healthy cells. This issue is of particular importance, since serious adverse events (SAE) have been reported in the past by using the CAR T-cell approach. An extremely detrimental SAE is represented by CAR T lymphocyte–mediated recognition of low antigen positive cells through the known “on target–off tumor” effect. Its severity depends on whether the targeting of the chosen antigen could be well tolerated by the organism, with completely different outcomes, as in the case of B-cell aplasia after anti-CD19 CAR T-cell infusion 28 or the death of treated patients when dealing with both anti-Erbb2 and -CAIX CARs. 29 Actually, the potency of CAR-redirected T cells has been documented to be superior compared with the mAbs from which the same CARs have been derived, 30 indicating that comparison between mAbs already used in the clinic and CARs is not exhaustive of the potential damaging effects following CAR T-cell infusion in the patients.
This concept is of particular importance when dealing with AML TAA whose expression is shared by normal myeloid cells, as in the case of the myeloid-specific antigen CD33 (SIGLEC-3), 31 a transmembrane receptor belonging to the SIGLEC family, expressed on early hematopoietic progenitors, myelomonocytic precursors, and blood monocytes, but also on some lymphoid cells and resident tissue macrophages such as Kupffer cells. 32 However, CD33 expression on AML blasts and LSCs renders CD33 a good candidate to be considered for CAR T-cell-mediated targeting, as preclinically demonstrated by the authors' group and others, both in vitro and in vivo. 32,33 Moreover, a clinical trial employing anti-CD33 CAR-redirected cytokine-induced killer cells started, and a case report of a 41-year-old patient with refractory AML has also been described. 25 After a 13-day culture period, anti-CD33 autologous CAR cytokine-induced killer (CIK) cells were infused in a total of 1.12 × 109 cells without any conditioning chemotherapy regimen and with a CAR percentage of ∼40% surface expression. A potent in vivo cytotoxic activity was obtained against CD33+ blasts, since a marked reduction of the disease resulted after the infusion of the CAR+ cells in the early treatment stage. However, 9 weeks after CAR T-cell infusion, the patient experienced a conspicuous disease progression due to the gradual accumulation of CD33+ blasts. Even in the absence of an evident clinical response, no uncontrollable clinical toxicities have been encountered after CAR T-cell infusion, this being an encouraging result for the application of an anti-CD33 CAR treatment in AML. The only documented significant adverse effect has been a cytokine storm characterized by high fever due to the release of high interferon gamma, interleukin 6, and tumor necrosis factor alpha levels observed in the early stage after CAR T-cell infusion. Despite the clinical experience with anti-CD33 CAR T cells in the first treated patient not reporting the emergence of “on target–off tumor” effects, the possibility of causing a significant myelotoxicity is far from being ruled out. Therefore, among the strategies actually available in order to manage CAR T-cell-related side effects, the one adopted by Kenderian et al. 32 conceives a transient expression of anti-CD33 CAR by means of mRNA CAR electroporation in order to minimize the risk of a long-term myeloablation. The CAR employed in this study was derived from gentuzumab-ozogamicin (an anti-CD33 monoclonal antibody conjugated with calicheamicin) and engineered with a 4-1BB costimulus, resulting in high CAR expression levels after electroporation. Anti-CD33 mRNA CAR T cells demonstrated potent but transient in vitro effector functions, also showing a potent in vivo anti-leukemic effect in combination with lympho-depleting chemotherapy known to enhance mRNA-modified CAR T-cell persistence. 34 Compared with stable retrovirally transduced anti-CD33 CAR, in which a single in vivo injection of lower numbers of CAR T cells were sufficient to eradicate AML blasts in NSG mice, the mRNA CAR-based platform required multiple infusions of an higher number of CAR T cells, but equally resulted in improved leukemic control and mice survival.
Another suitable antigen for AML targeting is represented by CD123, the α subunit of the human interleukin-3 receptor (IL-3R), which binds IL-3 with low affinity, and together with the β subunit (CD131) forms the functional heterodimeric high-affinity IL-3R. A wide CD123 overexpression on AML blasts and LSCs has been documented not only in ∼75–89% of AML patients, but also in other hematologic malignancies, rendering CD123 an appealing target antigen to be considered for immunotherapeutic strategies employing redirected T cells. 35 Moreover, a significant correlation was observed between CD123 levels and the number of leukemic blasts at diagnosis being associated with a negative prognosis. 36 On the other hand, high-level expression of CD123 has been identified on plasmacytoid DC and basophils, while low-level detection was observed on monocytes, eosinophils, endothelial cells, 37 and myeloid DC. 38
The authors' group and others have shown the feasibility of redirecting T lymphocytes with second- and third-generation CARs toward CD123 AML-expressing cells, with robust effector functions both in vitro and in vivo. 38 –41 However, signs of hematopoietic damage have been reported in different settings, as in the case of myeloid hematopoietic toxicity described by Kenderian et al. in two different mouse models, 32 or the severe impairment of normal hematopoiesis in NSG mice, when CD34-selected fetal liver cells were engrafted as a source of human HSCs. 42
This preclinical evidence raises concerns for the use of anti-CD123 CAR T cells without conceiving an adequate rescue strategy. In light of these perspectives, the group of Lihua Budde (City of Hope) is recruiting patients for a Phase I clinical trial with anti-CD123 autologous CAR T cells, following lymphodepletion for patients with relapsed or refractory AML (NCT02159495). In this trial, anti-CD123 CAR T cells will be used as a salvage therapy, serving as a “bridge to transplant.” As a reactive measure to control CAR T cells over time, the anti-CD123 CAR proposed contains a truncated EGFRt, which acts as an inducible suicide gene after cetuximab administration. 43 In this way, dispensation of the proactive drug results in the termination of the therapeutic efficacy of anti-CD123-redirected T cells, with kinetics that remain to be determined in humans.
Moreover, the feasibility, safety, and efficacy of autologous T cells modified with mRNA CD123 CAR will be determined in an open-label pilot study lead by the group of Saar Gill (Abramson Cancer Center; NCT02623582). In addition to mRNA-based vectors or inducible suicide gene strategies, which both end up with the expiration of the therapeutic potential of redirected T cells, another way of ameliorating the therapeutic index of anti-CD123 CAR platforms could rather rely on CAR design strategies. In this way, variation of the CAR binding affinity with the intent of potentiating the discrimination between CD123 overexpressing AML cells and low expressing healthy cells is currently under evaluation by the authors. 44 This approach proved to be beneficial in several tumor contexts, in which empirical evaluation of the best CAR affinity variant was essential and extremely dependent on both the disease context and the choice of the CAR-Ag pairs studied. 45
CAR design strategies represent an excellent modality for decreasing or increasing the potency of the desired CAR, with a technology applied also to AML-related antigens, as in the case of folate receptor-β (FRβ).
FRβ was primarily found on myeloid-lineage hematopoietic cells, 46 with an expression resulting in approximately ∼70% of AML patients, further increasable on AML blasts following all-trans retinoic acid administration. 47 Among the normal cells sharing the expression of FRβ, there are HSCs in which FRβ is present at very low levels, but also bone marrow myeloid differentiating cells, with the highest levels reached on bone marrow and peripheral blood CD14+ monocytes. At the same time, FRβ is lacking in circulating T cells, B cells, NK cells, and granulocytes. 48 Redirection of CAR T cells against FRβ was first documented in 2015 by Lynn et al., in which limited CAR T cell functionality was shown both in vitro and in vivo, with the potential of controlling AML tumor growth only in the minimal residual disease setting. 49 Therefore, in order to improve CAR T-cell antitumor activities, an affinity maturation of the first CAR antigen binding domain was conceived, reaching the KD of 2.48 nM compared with the original 57 nM, with a tenfold affinity divergence. CAR affinity maturation allowed for efficient elimination of FRβ+ cell lines and primary AML cells both in vitro and in vivo, with an efficacy significantly higher when compared with the low CAR affinity construct. High-affinity CAR-redirected T cells were indeed able to control leukemia growth in mice with large, palpable subcutaneous AML tumors, a setting of high tumor burden, where the low affinity CAR proved to be ineffective in redirecting T-cell effector functions. As demonstrated here as well as in other contexts, CAR affinity is known to affect CAR-redirected T-cell effector functions potently, with the general consensus being that higher affinity reduces activation threshold by increasing T-cell activity at low antigen levels. Indeed, elimination of FRβ+/CD34− bone marrow–derived healthy cells were detected with the high-affinity CAR, accompanied by eradication of FRβ+/CD14+ monocytes, highlighting the need to increase the safety level of this new CAR platform. Therefore, an mRNA-CAR approach was proposed instead of lentiviral transduction, with the idea that short-term CAR expression could be of benefit for determining tumor-cell destruction, while avoiding long-term myeloid loss. Expression of high-affinity CAR was similar between letiviral and mRNA electroporated conditions, with the latter declining in approximately 8 days, together with a similar weakening of CAR T-cell antitumor activities. As previously observed with the anti-CD33 mRNA CAR, multiple doses of anti-FRβ CAR were necessary to delay tumor growth significantly in vivo compared with the stably genome integrated CAR construct.
Another viable target for CAR T-cell therapy of AML is the isoform variant 6 of the adhesion molecules CD44. Beside epithelial tumors, CD44v6 is overexpressed in a number of hematological malignancies, including multiple myeloma and AML. Importantly, it has been shown that in AML CD44v6 contributes to the leukemia-initiating phenotype in immunocompromised mice, suggesting its key role in LSC biology. 50 Accordingly, anti-CD44v6 CAR T cells mediated potent anti-leukemia effects against autologous AML in xenograft models. Concerning potential off-tumor expression, CD44v6 is only expressed on monocytes and keratinocytes. Importantly, however, while monocytes are targeted and depleted in vivo by anti-CD44v6 CAR T cells, keratinocytes are spared. Keratincoytes sparing by anti-CD44v6 CAR T cells is the subject of intense investigation. Nonetheless, the co-expression of a suicide gene might obviate any potential concern of prolonged moncoytopenia following CD44v6-directed CAR T-cell therapy of AML or of skin adverse reactions. Table 2 summarizes the current safety strategies adapted to the anti-AML CARs described above.
Current safety strategies of anti-AML CARs
CAR, chimeric antigen receptor; LeY, Lewis Y; FRβb, folate receptor-β.
Future Directions in Anti-AML Car Design Strategies
When dealing with AML-related target antigens, in order to ameliorate CAR T-cell mediated responses, several strategies can be considered, each one acting at a different level.
Even if in AML no cases of CAR design strategies have so far been considered, except those regarding CAR affinity variation, another interesting approach in the field could be represented by the creation of a dual-CAR platform in order to enhance the targeting specificity and the safety profile. 51 At the same time, in order to address better the limitations associated with the poor clinical outcome encountered so far with anti-AML CARs, several observations can be taken into consideration. Regarding efficacy concerns, the degree of in vivo CAR T-cell expansion should occur at levels sufficient to determine a sustained control of disease, together with adequate cell surface CAR T-cell expression levels. 52 In this way, several efforts can be considered to overcome these limitations, such as the use of alternative costimulatory domains.
Moreover, other possible reasons for treatment failure, despite CAR T-cell persistence and antigen stimulation, could be represented by CAR T-cell unresponsiveness due to the strong immunosuppressive environment in the bone marrow of AML patients and the possible generation of myeloid malignant cell-escape variants following antigen loss. These drawbacks are currently the subject of preclinical research, such as engineering strategies to equip CAR T cells with an intrinsic resistance to tumor-immune evasion mechanisms, 53 but also co-administration of antibodies directed toward immune inhibitory checkpoint molecules in order to boost the CAR T-cell-related responses. 54
AML in Vivo Mouse Modeling: Evolution Toward a More Reliable Preclinical Evaluation of Anti-AML Car T-Cell Efficacy and Safety
Despite the clinical successes obtained in patients affected by B-cell malignancies with CAR T-cell targeting the CD19 antigen, some CAR T-related effects (i.e., CRS and TLS) have not been accurately predicted by preclinical in vivo testing. This drawback suggests that in the era of advanced molecular medicine, which aims to offer more personalized and less toxic alternative therapeutic strategies, the implementation of preclinical in vivo efficacy and safety assessment of novel immunotherapeutic approaches is mandatory.
Concerning AML, preclinical models must be able to recapitulate the disease heterogeneity and complexity realistically. Below the evolution of xenograft mouse model systems developed in the last decades for the preclinical characterization of AML disease and their contribution to the current and future investigation of new treatment strategies is discussed (Fig. 2).
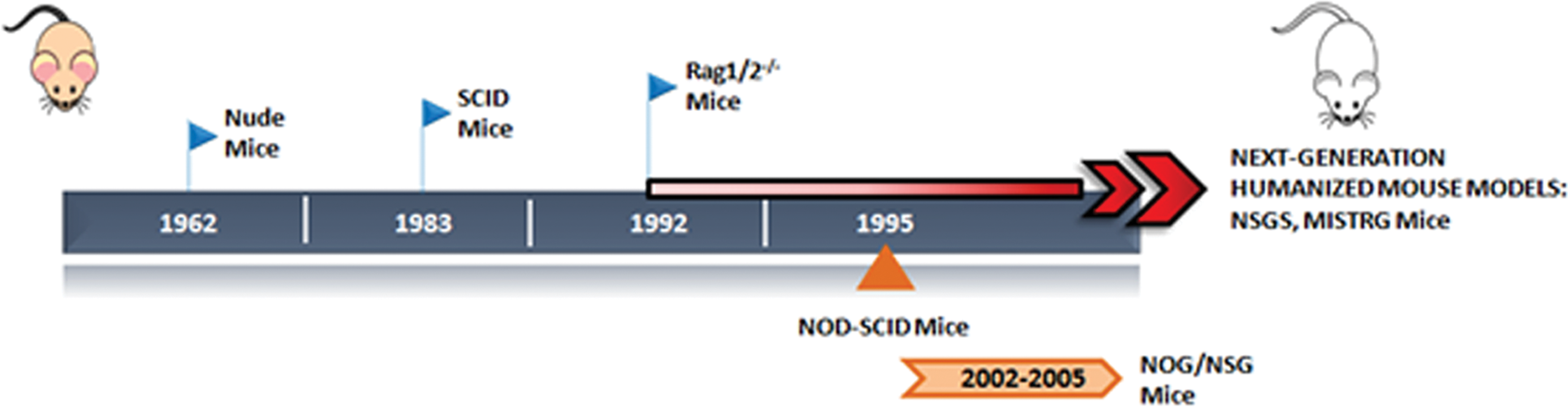
Evolution of xenograft mouse models for normal and malignant human hematopoiesis. Nude mice or severe combined immunodeficiency (SCID) mice were the first immunodeficient strains developed. RAG1/2–/– and nonobese diabetic (NOD)-SCID mice were then introduced successively, moving toward more immunocompromised mice. NOG and NSG (NOD-SCID strains lacking the IL-2Rγ) represent the more advanced strains, which totally lack the murine immune system. Next-generation strategies (NSG-S, triple transgenic expressing human IL3, GM-CSF and SCF and MISTRG, genetically humanized for M-CSF, IL-3, GM-CSF, and TPO) have been developed to humanize mice by the addition of human genes for the expression of human cytokines, which ensures a better engraftment of human myeloid cells. Color images available online at
Nude mice and subsequently severe combined immunodeficient (SCID) mice, deficient in mature B and T cells, were the first immunodeficient strains adopted in the early attempts of AML xenografting. 55 AML xenografts in SCID mice allowed for the identification and characterization of the so-called SCID leukemia-initiating cells (SL-ICs). 56 To characterize SL-ICs better and to improve AML engraftment rates, mice with a higher immunodeficiency have been generated. Nonobese diabetic mice with severe combined immunodeficiency (NOD-SCID) were established in 1995. 57 Using NOD-SCID mice, Bonnet and Dick observed that SL-ICs after transplantation were able to proliferate, differentiate, and renew themselves, reinitiating the disease in secondary transplanted mice, proper features of LSCs. 58 These observations suggested that AML is organized in a hierarchical manner, resembling the normal hematopoietic system, with LSCs at the top of the hierarchy initiating and maintaining the disease. Also NOD-SCID mice carried some limitations, such as the spontaneous development of thymic lymphoma, considerable radiosensitivity, and detectable NK cell activity, 57 thus prompting the generation of further immunocompromised strains by inactivating the IL2Rγ. These include the NOD-SCID IL-2Rγnull NSG 57 and NOG mice, 59 in which the Il2rγ is completely null or truncated in the intracellular tail, respectively, resulting in the complete abrogation of NK cell activity in the absence of both T-/B-cell leakiness and spontaneous thymic lymphoma. The enhanced engraftment granted by these new strains has allowed for the detection of smaller LSC populations in cell compartments with a more mature phenotype, 60 suggesting that leukemogenesis can occur in multipotent HSCs, as well as in more committed myeloid precursors. This gain in the identification of AML-LSCs paves the way for better characterization of the disease, together with a more accurate evaluation of novel therapies, such as anti-AML CARs, aimed at eradicating AML by specifically targeting these cells. As an example, by using this model, anti-CD33 and anti-CD123 CAR-redirected cytokine-induced killer cells were tested for their potentiality in controlling AML in secondary transplantation. 41
While the increased level of immunodeficiency allows for better engraftment rates and prevention of xenograft rejection, it is known that cytokines, chemokines, and growth factors are active players in supporting cellular development, differentiation, survival, and function. Therefore, considering the lack of cross-reactivity of mouse cytokines, next-generation humanized mouse models foresee the expression of human soluble factors. As an example, NSGS mice are NSG mice transgenic for the expression of human stem cell factor, granulocyte–macrophage colony-stimulating factor (GM-CSF), and IL-3. 61
By using this latter mouse model, the combination of the immunodeficiency condition and human cytokines support offers the main advantage of improved engraftment of primary AML cells and pre-leukemic myeloid cells by lowering the number of injected cells compared with the above-mentioned mouse models. 61 Moreover, NSGS are better recipients for those AML subsets that failed to engraft by using the most popular immunodeficient strains.
Rongvaux et al. recently generated the MISTRG mice, Rag2-IL2Rγ-deficient mice with human versions of M-CSF, IL-3/GM-CSF, SIRPA, and TPO. First conceived for studies on innate immune responses to infectious agents, MISTRG are also suitable in the AML context by promoting increased differentiation of human myeloid and monocytic cells 62 as well as by favoring the engraftment of less aggressive good risk AML. 63
By using these new models, which are more prone to the development of a competent and more active human immune system, we are now moving from the “host versus graft” issue toward the “graft versus host” one. Therefore, further optimizations are needed to induce tolerance of the human immune system for the mouse host.
Recently, another important achievement on the preclinical evaluation of AML treatments has been represented by the work of Wunderlich et al., who optimized a “5 + 3” induction chemotherapy treatment, combining Ara-C and doxorubicin in humanized mouse models. 64 This chemotherapy xenograft model paves the way for a proper examination of the potential benefit of combining standard-of-care treatment to most innovative targeted therapies (both molecular and cellular driven) on residual chemotherapy-resistant cancer cells.
Concerning the safety profile, preclinical assessment of the potential toxicities related to the treatment with anti-AML CARs is limited by the fact that the expression of the human target-antigen is restricted to tumor cells, thus not resembling the physiological expression on normal tissues. This limitation hampers the correct evaluation of potential “on target–off tumor” toxicities due to the non-cross-reactivity between human CARs and mouse antigens, prompting research to develop more reliable mouse models.
As an example, in another pathological context such as treatment of metastatic solid cancers, a CAR recognizing the VEGF-2 murine antigen has been developed, providing important considerations about treatment toxicity on murine normal tissues. This information was then employed as a background knowledge about the anti-VEGF-2 CAR T-cell safety profile for the design of the clinical trial (NCT01218867). 65
An alternative strategy to evaluate properly an “on target–off tumor” effect by CAR T cells on normal tissues expressing lower levels of TAA compared with the overexpressing tumor relies in the generation of “genomically” humanized mice. This system operates by transferring human genes into the mouse genome through targeted integration of a human sequence into the equivalent region of the mouse genome (i.e., CEA and HER-2 transgenic mouse models). 66,67
Overall, in the field of onco-immunology research, we are facing a new concept of mouse modeling, which acts by considering the global human environment surrounding the tumor rather than studying the efficacy of novel therapies solely on cancer cells that have been isolated from their pathophysiological context. These novel implementations will be fundamental for future targeted therapies' safety evaluation and will be pivotal in bridging the current translational gap to the clinic.
General Conclusions
Although major clinical results have been observed so far by CAR T-cell-mediated targeting of the CD19 antigen for the treatment of ALL, B-CLL, and NHL, much more challenging issues need to be considered for proper clinical translation of this approach for AML.
Particular attention has to be paid in the optimal balance between efficacy and safety due to the paucity of TAA discriminating between normal and leukemic tissues. In this sense, since effects on targeting myeloid normal tissues are detrimental, further implementations on safety of CAR T-cell designs still be the bottleneck for future clinical studies. In parallel, considering the immunosuppressive myeloid microenvironment, optimization of CAR T-cell potency cannot be scotomized. Only the tuning of both aspects will likely give rise to successful anti-AML strategies. To address this point, several innovative strategies have been developed (i.e., suicide genes, mRNA CAR, third-generation CARs, and a combination with immune checkpoint molecules), and they are currently the subject of first human clinical trials.
Footnotes
Acknowledgments
The authors deeply thank the parents' committees “Quelli che … con LUCA onlus,” “Comitato Maria Letizia Verga,” and “Stefano Verri” for their generous and constant support.
This work was supported by grants from AIRC Molecular Clinical Oncology 5 per mille 2010 “Innate immunity in cancer. Molecular targeting and cellular therapy,” 9962; AIRC IG Grant 2015 “Novel leukemia treatment by the use of Chimeric Antigen Receptors (CARs),” 17248; “Libera Le Ali” 2011 project, Fondazione Just Italia. S.A. and M.C.R. are fellows of the University of Milano-Bicocca, Milan, Doctoral Program in Molecular and Translational Medicine (DIMET). A particular and great gratitude goes to “Quelli che … con LUCA onlus” that generously supported S.T.'s fellowship and funded the project.
Author Disclosure
No competing financial interests exist for any of the authors.