
Abstract
Novel therapies with chimeric antigen receptor (CAR)-transduced T cells (TCs) sparked new hope for patients with relapsed or refractory CD19-positive leukemia or lymphoma even after stem cell therapies. This review focuses on CARs recognizing the B cell antigen CD19. Both retroviral and lentiviral vectors are used, encoding various anti-CD19 CAR constructs comprising costimulatory molecules such as CD28, CD137/4-1BB, and OX40 either alone (second-generation CARs) or in combination (third-generation CARs). Current, up-to-date published studies on anti-CD19 CAR therapy for acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL), and non-Hodgkin lymphoma (NHL) with observed side effects are discussed and an outlook on 58 ongoing trials is given. Clinical responses were achieved in up to 81% of ALL, 50% of CLL, and 40% of NHL patients. Factors with potential influence on the clinical outcome might be the design of the vector, the preconditioning regimen, and the number and quality of transfused CAR TCs. The applicability of clinical CAR TC therapy might include relapse after allogeneic stem cell transplantation (alloSCT), and ineligibility for or “bridging” until alloSCT. In summary, CAR therapy represents a highly promising treatment option even in heavily pretreated patients.
Introduction
T
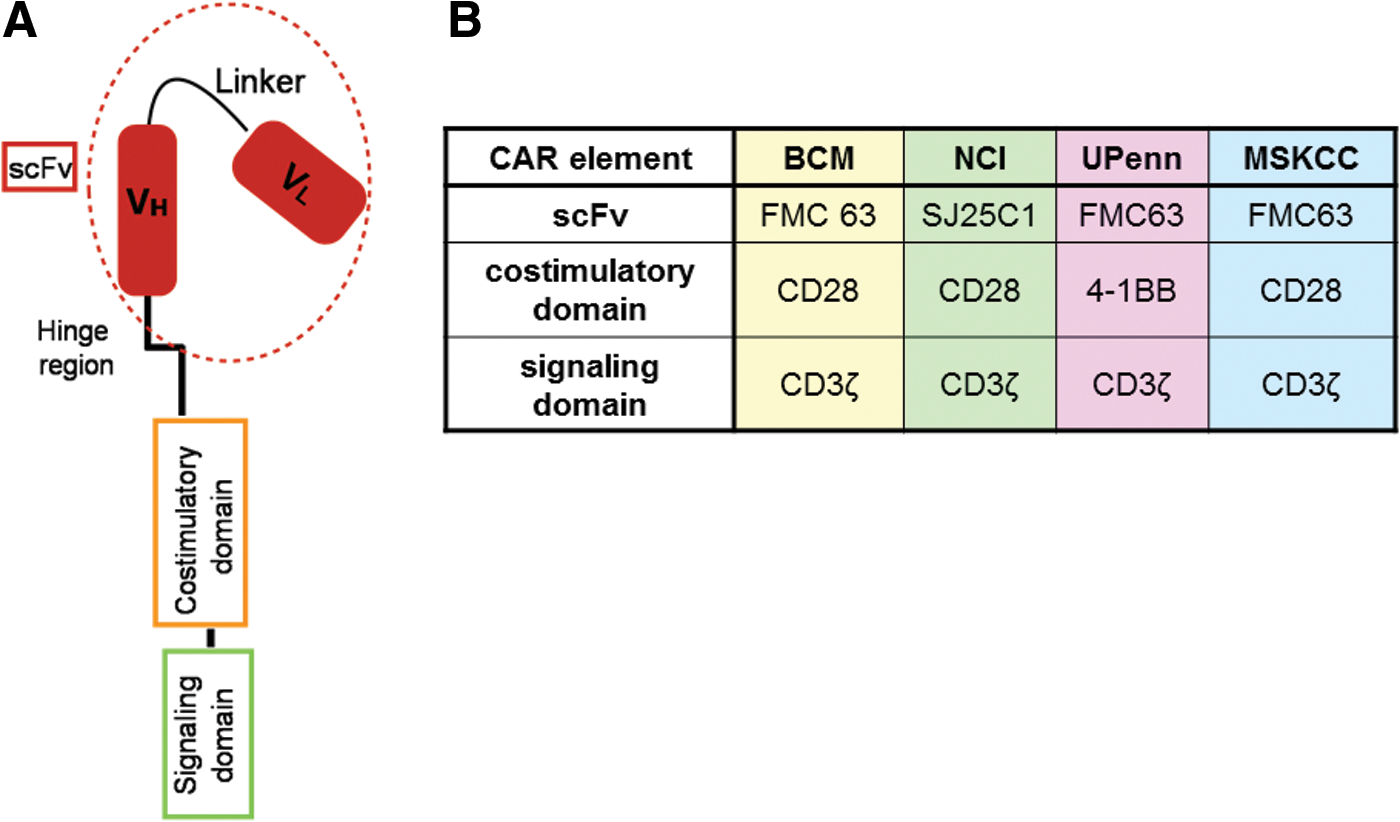
The advent of monoclonal antibodies in anticancer treatment has been a milestone on the way toward effective targeted immunotherapy. However, dependence on additional effectors for ultimate target elimination, short in vivo persistence, as well as restricted biodistribution have limited the efficiency of monoclonal antibodies in the clinical setting. On the other hand, T cells (TCs) are able to exert almost autonomously effector functions, to activate and recruit additional players of the immune system, and to traffic to distant sites within an organism. In addition, TCs mediate durable responses due to long-term persistence. Chimeric antigen receptors (CARs) are composed of an extracellular antigen-specific recognition domain that is derived from a single-chain variable fragment (scFv). CARs are linked via a hinge and transmembrane segment to an intracellular domain providing a TC activation signal (Fig. 1A). Thus, TCs engineered to express CARs combine the antigen-binding properties of a monoclonal antibody with the activating and effector function of a TC. In contrast to physiologic T cell receptors (TCRs), CARs are able to recognize unprocessed extracellular antigens and therefore can act in an HLA-independent manner. In anticancer treatment this is of importance considering that tumor escape via HLA downregulation is common. Furthermore, not only proteins but also carbohydrates, glycosylated proteins, and proteoglycans 6,7 represent suitable CAR antigens, thus expanding significantly the range of potential antigen targets.
CD19, a transmembrane glycoprotein that functions as s central response regulator of B cells, 8 represents a good target tumor antigen given that it is expressed on B cells during all stages of their differentiation with the exception of hematopoietic stem cells, is downregulated on plasma B cells, and maintained on cells that have undergone neoplastic transformation. 9,10 In fact, CD19 is expressed on more than 95% of the cells in B cell hematologic malignancies. 11 Early preclinical xenograft studies demonstrated that anti-CD19 CAR TCs exert significant in vivo tumor cytotoxicity and leukemia eradication in mice transplanted with a human CD19-positive cell line. 12,13 Subsequently, clinical trials analyzing anti-CD19 CAR TCs in patients with B cell malignancies were initiated (Table 1). The process of anti-CD19 CAR TC therapy is illustrated in Fig. 2.
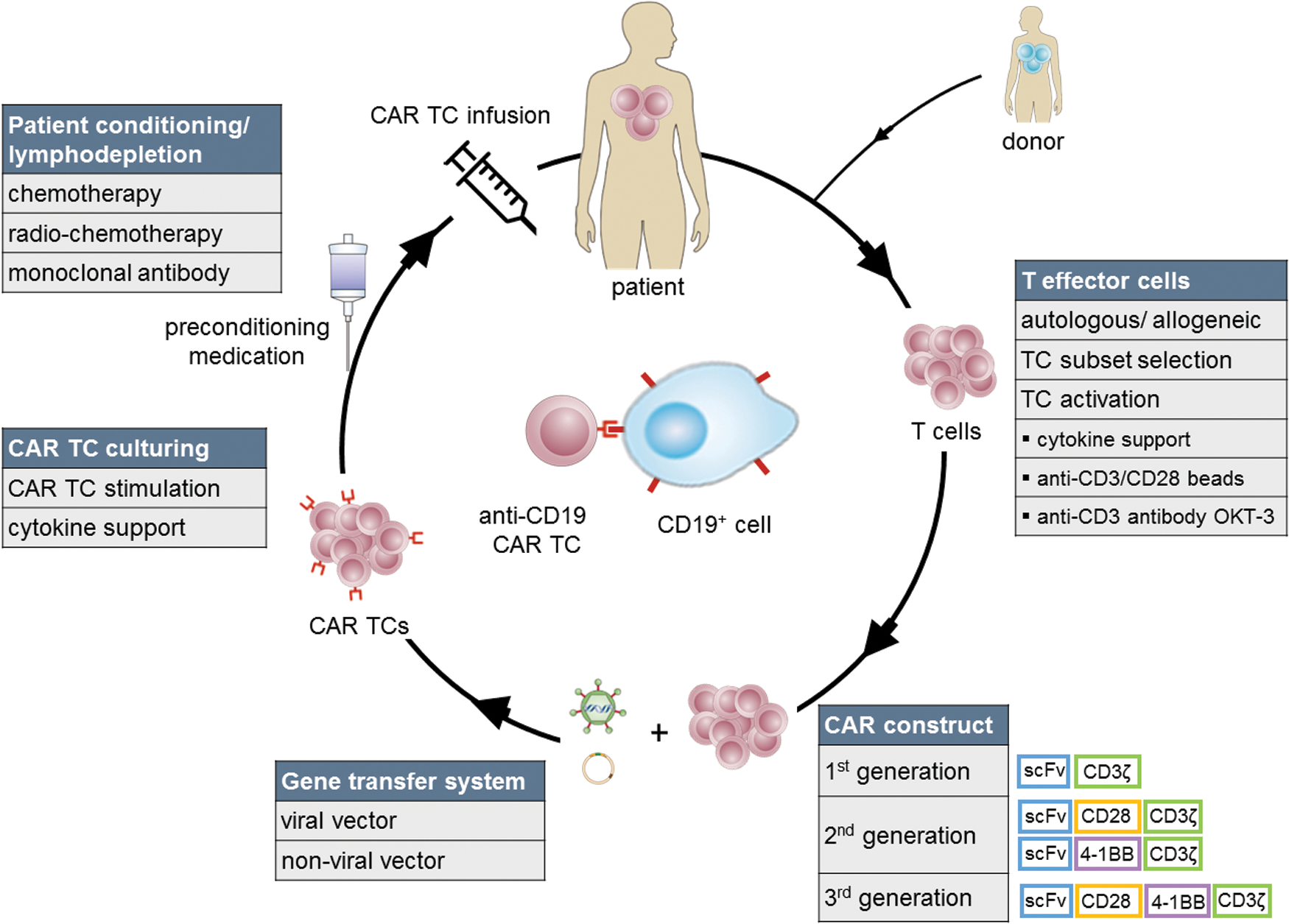
Anti-CD19 CAR T cell therapy. T lymphocytes from the patient (autologous) or a suitable donor (allogeneic) are isolated, enriched, and/or selected for a certain subtype. Before transduction via a viral (retroviral, lentiviral) or nonviral vector (i.e., transposon-based, naked DNA injection) encoding the CAR construct (second- and third-generation clinical CARs most commonly comprise the CD28 and/or 4-1BB costimulatory domains), TCs are activated either via cytokines, coculturing with anti-human CD3/CD28 antibody-coated beads, anti-CD3 monoclonal antibodies, and/or artificial antigen-presenting cells (APCs). Engineered CAR TCs are expanded and transferred into the patient after patient preconditioning by lymphodepleting chemotherapy, radiochemotherapy, or monoclonal antibody treatment. Color images available online at
Summary of clinical trial reports of chimeric antigen receptor-transduced T cells targeting the CD19 antigen
After CAR T cell infusion patients with FL received flu (25 mg/m2) and IL-2 (5 × 105 IU/m2 twice daily × 5) administration in vivo.
Dosage of chemotherapeutic drug not reported.
One patient had received prior allogeneic cord blood transplantation.
Eighteen patients had previously undergone alloSCT.
Pretreatment conditioning according to prior regimens: None (three patients with pancytopenia); other patients: variable regimens including drugs such as flu, cyc, eto, clofarabine, cytarabine, methotrexate, vincristine, Adriamycin (doxorubicin).
One patient died <48 hr after CAR TC infusion.
Four patients had previously undergone alloSCT.
Patients received eight doses of IL-2 in vivo (720,000 IU/kg per dose).
One patient treated in 2012 received a second CAR TC infusion in the trial of 2015.
One patient died of influenza.
One patient died of cardiac arrhythmia.
Eight patients had previously undergone alloSCT.
Three patients had previously undergone alloSCT.
AdV, adenovirus; ALL, acute lymphoblastic leukemia; BCM, Baylor College of Medicine (Houston, TX); BCNU, bis-chloroethylnitrosourea; benda, bendamustine; CLL, chronic lymphocytic leukemia; C-MOAD, cyclophosphamide, mitoxantrone, vindesine, cytarabine, dexamethasone; CMV, cytomegalovirus; COH, City of Hope Medical Center (Duarte, CA); CPGH, Chinese PLA General Hospital (Beijing, China); CR, complete remission; cyc, cyclophosphamide; DLBCL, diffuse large B-cell lymphoma; EBV, Epstein–Barr virus; eto, etoposide; FL, follicular lymphoma; flu, fludarabine; LCL, lymphoblastoid cell line; LG, low grade; MCL, mantle cell lymphoma; MSKCC, Memorial Sloan Kettering Cancer Center (New York, NY); N/A, not assessed; NCI, National Cancer Institute (Bethesda, MD); NE, not evaluable; NHL, non-Hodgkin lymphoma; NR, no response; OKT3, Muromonab-CD3; PD, progressive disease; pento, pentostatin; PMBCL, primary mediastinal B cell lymphoma; PR, partial remission; rituxi, rituximab; SD, stable disease; SMZL, splenic marginal zone lymphoma; TBI, total body irradiation; U Penn, University of Pennsylvania (Philadelphia, PA); VST, virus-specific T lymphocytes.
Car T Cells and the Role of Costimulation
Early clinical CAR TC trials were disappointing as only limited expansion and in vivo persistence of transduced TCs were noted. 14 Physiologically full TC activation and proliferation induction requires at least three signals: TCR engagement (first signal), costimulation provided by antigen-presenting cells (APCs) (second signal), and cytokine release (third signal). 15 Lacking expansion and persistence of initial CAR TCs was attributed mainly to restricted CAR TC activation during interaction with target cells given that malignant cells have shown deficiency not only for expression of activating costimulatory receptors but also for secretion of helper cytokines. 16 In addition, CAR TCs failed to interact with regular APCs because no specificity of the CAR receptor against APC-expressed antigens exists. CAR TC products lacking any costimulation and first-generation CARs comprising only the single CD3ζ stimulatory domain from the CD3ζ chain of a TCR were shown to be insufficient in clinical application: they were lost in vivo less than 7 days after transfusion into patients with lymphoma. 17
To overcome limited CAR TC persistence, and to mediate superior activation, proliferation, and in vivo persistence to TCs compared with first-generation CARs, 18 second-generation CARs were developed. These CARs incorporate a costimulatory molecule, such as CD28, CD27, DAP-10, 4-1BB (CD137), OX40 (CD134), or inducible TC costimulator (ICOS) adjacent to the CD3ζ signal within their intracellular signaling domain (Fig. 1). CD28 and 4-1BB have been used most commonly in clinical trials and have yielded remarkable clinical responses. Nonetheless, even incorporation of a single costimulatory molecule might not be sufficient for full CAR TC activation. 16,19 Consequently, third-generation CARs including two costimulatory molecules within CAR constructs have been generated. 20 It remains to be elucidated whether third-generation CARs are superior to second-generation CARs in ongoing trials. 21 Moreover, fourth-generation CAR TCs redirected for universal cytokine killing (TRUCKs) that express cytokines or costimulatory ligands in order to enhance other effectors of the immune system have been developed. 22,23
CAR TC activation and proliferation can be further supported by providing optimal cell culturing conditions to transduced lymphocytes. Magnetic beads coated with CD3 and CD28 antibodies not only provide a cross-linking TCR signal but also mediate CD28-based costimulation already ex vivo. 24 Alternatively, artificial APCs 25,26 or coculturing with various cytokines including interleukin (IL)-15 or IL-12 27,28 have been included in various TC expansion protocols to augment CAR TC activity.
Mediating Car Expression on T Cells
For CAR expression, TCs are provided with genetic information via gene transfer vectors based on either nonviral or viral systems. Viral vectors comprise the intrinsic ability to gain access to cells by means of infection and exploit the host's cellular machinery for replication. Gammaretroviral vectors, based mainly on murine leukemia virus (MLV), 29 and lentiviral vectors, based mainly on the human immunodeficiency virus (HIV) type 1, 30 have evolved the preferred gene transfer systems in CAR TC trials. No genotoxicity for gene transfer into differentiated cells including TCs has been reported, and to date no retrovirus-related transformational event has been observed in more than 500 cumulative follow-up years of patients treated with engineered TCs. 31,32 Hence, gammaretroviruses continue to be widely and safely used vectors for clinical TC gene therapy. 31,33,34 In addition, alternative nonviral vectors based on transposons have been developed. 35
Clinical Efficacy of Anti-CD19 Car T Cells for All, Cll, and Indolent Lymphoma
At present, reports of 17 clinical CAR TC trials comprising approximately 182 patients with B cell malignancies and targeting the CD19 antigen have been published (note that 17 patients have been enrolled in more than one trial report; hence, the effective number of treated patients is 165). The majority of trials used second-generation CAR vectors in the context of phase I clinical studies and have been performed at four research institutions: Baylor College of Medicine (BCM, Houston, TX), Memorial Sloan Kettering Cancer Center (MSKCC, New York, NY), University of Pennsylvania (UPenn, Philadelphia, PA), and the National Cancer Institute (NCI, Bethesda, MD). Whereas UPenn uses a 4-1BB costimulatory endodomain within their CAR construct, BCM, MSKCC, and NCI have focused on CD28 as costimulatory element (Fig. 1B). Patient number and targeted malignancy, CAR vector construct design and manufacturing details, as well as clinical outcomes are summarized in Table 1. The vast majority of trials focused on ALL, enrolling a total of approximately 93 patients, whereas 46 and 43 patients have been treated for CLL and lymphoma, respectively. In addition, more than 58 anti-CD19 CAR TC clinical trials are ongoing (Table 2). Although all trials share basic elements, study protocols differ significantly considering disease entity, age of involved patients, vector design, origin of TCs, CAR TC manufacturing process, preconditioning chemotherapy, cytokine support, or infused CAR TC dosage (Table 1).
Ongoing clinical anti-CD19 chimeric antigen receptor-transduced T cell trials registered in 2016
τ, truncated; A, trial active, not recruiting; Ad, adenovirus; ALL, acute lymphoblastic leukemia, BCM, Baylor College of Medicine; C, trial completed; CAR, chimeric antigen receptor; CLL, chronic lymphocytic leukemia; CMV, cytomegalovirus; COHM, City of Hope Medical Center; CPGH, Chinese PLA General Hospital; EBV, Epstein–Barr virus; EGFR, epidermal growth factor receptor; FHCRC, Fred Hutchinson Cancer Research Center; gen, generation; HCL, hairy cell leukemia; IL, interleukin; LVV, lentiviral vector; MSKCC, Memorial Sloan Kettering Cancer Center; N, not yet recruiting; N/A, not available; NCI, National Cancer Institute; NOS, not otherwise specified; R, recruiting; RVV, retroviral vector; S, trial suspended; SCH, Seattle Children's Hospital; SIN, self-inactivating; SSPH, Shenzen Second People's Hospital; TAA, tumor associated antigen; TC, T cell; TCM, central memory T cell; TN/MEM, naive memory T cell; UCL, University College, London; UPenn, University of Pennsylvania; VST, virus-specific T cell; WM, Waldenström macroglobulinemia.
Note: Search term: CD19 CAR (completed May 25, 2016).
Source:
CAR T cell trials targeting ALL
CAR TCs in patients with ALL have displayed remarkable success: One of the first trials of ALL CAR TC treatment was published at the MSKCC by Brentjens and colleagues, who reported complete remission (CR) in an adult patient with relapsed leukemia. After cyclophosphamide preconditioning, autologous TCs transduced retrovirally with a second-generation scFv.CD19.CD28.CD3ζ CAR construct had been infused into the patient. 16 Using the same vector, long-term molecular CR (mCR) was observed for all five patients who had been included in a subsequent study (the patient in the Brentjens and colleagues 2011 trial was also involved). 36 A further update was published in 2014 by Davila and colleagues, who extended the patient cohort by enrolling an additional 11 patients with refractory, high-risk, BCR-ABL-positive (four patients) or post-alloSCT (four patients) relapsed ALL. An overall 88% CR rate including a total of 75% mCRs were reported. Seven patients (44%) were subsequently eligible for alloSCT without any evidence of relapse up to 2 years after alloSCT. CAR TC persistence in the bone marrow up to 8 weeks after CAR TC infusion was observed. 37
Grupp and colleagues at UPenn treated two children with TCs that had been lentivirally transduced with a scFv.CD19.4-1BB.CD3ζ CAR construct. Whereas one patient treated with autologous CAR TCs without preconditioning therapy displayed mCR for up to 9 months, the second patient treated with transduced TCs derived from a previous allogeneic umbilical cord donor and pretreated with cyclophosphamide and etoposide preconditioning therapy relapsed 2 months after CAR TC infusion with CD19-negative leukemia. 38 This was not only the first report of B cell antigen escape after anti-CD19 CAR TC treatment, but also the first trial observing long-term engraftment and persistence of CAR TCs up to 1 year. Prolonged CAR TC persistence was attributed mainly to inclusion of the 4-1BB costimulatory molecule within the CAR construct 38 (see Number and Subtypes of Infused CAR T Cells, below).
In contrast to previous studies using donor-derived TCs for CAR TC production that reported no evidence for GvHD, 39,40 Dai and colleagues from the Chinese General Hospital of Beijing treated nine patients with ALL with CAR TCs transduced with a scFv.CD19.4-1BB.CD3ζ CAR construct and reported for the first time the occurrence of GvHD in two of three patients who had received CAR TCs of donor origin. The GvHD occurrence was attributed to a higher dose of infused CAR TCs as well as to mixed chimerism in affected patients, resulting in decreased tolerance to donor TC infusions. The third patient had received donor-derived but autologously collected CAR TCs and did not show any sign of GvHD. 41
Overall, nine trials of anti-CD19 CAR TC treatment for ALL in 93 patients have reported responses in 76 patients (81%), including 73 (78%) CRs.
CAR T cell trials targeting CLL
Some of the first studies targeting CLL via anti-CD19 CAR TCs were performed at UPenn. The first trial treated three patients with advanced, chemotherapy-resistant CLL with an scFv.CD19.4-1BB.3ζ CAR construct that had been lentivirally transferred into autologous TCs. Patients had been pretreated with various preconditioning regimens (Table 1). Two patients achieved CR and the third patient had a partial response (PR) lasting more than 8 months. Notably, Kalos and colleagues calculated that each CAR TC expanded more than 1000-fold in vivo and destroyed at least 1000 leukemia cells in the patients. Furthermore, CAR TCs were shown to have maintained effector function up to 169 days after infusion and to have trafficked and expanded within the patient's bone marrow. 42,43 The most recent report of CAR TCs at UPenn, performed again by Porter and colleagues, included 14 patients with CLL who had been pretreated with various chemotherapy regimens before CAR TC administration. Durable CR was reported in four patients, of whom none experienced progressive disease. Of the remaining 10 patients, four had a PR and six showed no response to treatment, and all 10 patients died of disease progression or disease-related complications 1–10 months after CAR TC therapy. Interestingly, this trial determined no significant correlation between clinical response and patient age, sex, number of previous therapies, disease stage at enrollment, in vitro anti-CD19 activity, or infused CAR TC dose. However, the degree of CAR TC expansion in vivo as well as duration of CAR TC persistence and B cell aplasia correlated with clinical outcome. CAR TC functional persistence in patients who had obtained CR was observed beyond 4 years. 44
So far, a total of eight CAR TC trials enrolling 46 patients with CLL have observed responses in 23 patients (50%), including 12 (26%) CRs (note that six patients with ALL have been enrolled in more than one trial; Table 1).
CAR T cell trials targeting lymphoma
The first clinical trial of CAR TCs in lymphoma was published by Jensen and colleagues from the City of Hope National Medical Center (Duarte, CA) in 2010 and used a first-generation scFvCD19.3ζ-CAR construct. TCs were transfected via a nonviral vector system and infused into four nonpretreated patients with lymphoma. Although clinical response was evident, all patients progressed rapidly after CAR TC administration and showed only poor CAR TC levels in the peripheral blood, probably due to immunoreactivity against infused CAR TCs. 17 Savoldo and colleagues from the BCM performed a comparative study: six patients with lymphoma who had received no prior lymphodepletion were treated either with a first-generation (scFv.CD19.CD3ζ) or a second-generation (scFv.CD19.CD28.CD3ζ) CAR construct. Although only modest clinical results were reported in two patients displaying transient SD (stable disease), this trial proved proliferative and persistence advantage of second-generation CARs in a clinical setting. 18
The NCI has conducted the majority of CAR TC studies targeting lymphoma to date and has used a CD28-containing CAR vector (scFvCD19.CD28.CD3ζ) for this purpose. In 2010, a case report of a patient with advanced lymphoma described a PR for up to 36 weeks. The patient had been pretreated with cyclophosphamide and fludarabine, and infusion of autologous retrovirally transduced CAR TCs was followed by intravenous IL-2 application. 19 After relapse this patient was retreated again 1 year later with CAR TCs and achieved another PR that lasted for more than 26 months. 45 Applying the same protocol, an additional three patients were treated in 2012. Except for one patient who died of influenza, patients obtained stable remissions lasting up to 12 months. 46 In subsequent trials, the study protocol was modified and exogenous IL-2 infusion after CAR TC infusion was eliminated: Six enrolled patients who had progressed after alloSCT and DLI and had been treated with CAR TCs without prior lymphodepleting therapy all displayed a clinical response (one PR, five SD). 47 Significant clinical response was, furthermore, reported for the largest NCI lymphoma trial. Of 11 enrolled patients, one patient died, most probably of cardiac arrhythmia, and one patient was lost to follow-up. The nine remaining patients, representing 81% of treated patients, had a clinical response: five CRs, three PRs, and one SD. 48 One additional patient with diffuse large B-cell lymphoma (DLBCL) progressed despite CAR TC treatment after cyclophosphamide/fludarabine pretreatment chemotherapy. 49 Mixed results concerning 10 treated patients receiving CAR TCs of donor origin have been reported. Whereas one patient progressed with his disease, seven patients displayed SD and two patients achieved CR and PR, respectively. 40
To date, eight anti-CD19 CAR TC lymphoma trials involving 43 patients have been published and reported a response in 17 patients (39.5%), including eight CRs (19%).
Conditioning Regimens
Depletion of recipient lymphocytes before CAR TC infusion via conditioning chemotherapy, chemoradiotherapy, and monoclonal antibodies have been associated with enhanced engraftment, persistence, and efficacy of CAR TCs. 50 The positive conditioning effect has been related to three mechanisms: First, tight homeostatic regulation maintains the number of TCs at a constant level. Hence, lymphodepletion reduces resident cell populations and creates space for infused cells to expand. Second, conditioning reduces the number of regulatory TCs that restrict CAR TC expansion by secretion of inhibitory cytokines such as IL-10 or transforming growth factor (TGF)-β and consumption of proliferation-enhancing IL-2. Third, lymphodepletion has been shown to stimulate stromal cells to produce TC growth cytokines IL-7 and IL-15, both associated with enhanced expansion of infused cells. 51,52 In fact, the finding that patients without any preconditioning treatment displayed poorer clinical responses 16,18,41 compared with patients receiving lymphodepleting chemotherapy before CAR TC infusion 16 has been confirmed in a meta-analysis. 53
As for ALL, conditioning consisting of either cyclophosphamide alone 16,36,37 or cyclophosphamide combined with fludarabine 48,49,54 has yielded a comparable clinical outcome. In patients treated for CLL, however, cyclophosphamide alone 16 has turned out to be less effective than other chemotherapeutic conditioning regimens combining different agents. 42 For lymphoma, in turn, a more intense conditioning regimen seems necessary for significant response in treated patients, as high-dose cyclophosphamide 49 has been demonstrated to mediate superior effects on CAR TC activity compared with low-dose cyclophosphamide. 19,46 However, the optimal conditioning regimen before CAR TC administration remains to be identified given that some trials have also reported CRs in patients who had not received any lymphodepleting pretreatment. 38 –40,47 It should also be kept in mind that commonly used conditioning chemotherapeutics such as cyclophosphamide or fludarabine endow direct activity against B cells and per se might contribute to clinical responses in CAR TC therapy.
Number and Subtypes of Transfused Car T Cells
Although the first intention-to-treat clinical anti-CD19 CAR TC trial defined the maximum tolerated CAR TC dose as 1 × 106 CAR TCs/kg body weight, 49 the optimal number of infused CAR TCs for maximized clinical efficacy has not yet been assessed for several reasons: First, TCs are dynamic “living drugs” that undergo expansion or repression and are subjected to tight regulatory mechanisms in vivo. Second, interindividual variation may result in significant differences in CAR TC products in terms of quality and quantity. Third, several reports have indicated that no correlation of transfused CAR TC number and clinical outcome exists, and that peak blood levels of CAR TCs could not be predicted by the dose of administered CAR TCs. 16,19,40,42,43,45 Hence, factors other than infused TC quantity seem responsible for CAR TC expansion and efficacy in vivo. Attention has consequently turned to identification of the optimal TC starting population. Ideally, infused engineered TCs should not only be able to traffic to distant tumor sites to mediate antitumor responses, but should also expand and adopt preferentially a memory-associated phenotype to persist long-term. For this purpose, terminally differentiated effector memory TCs (TEM) were initially considered an ideal TC starting population. However, TEM have shown only poor plasticity and proliferative capacity. 55 Hence, to date more naive cells such as central memory TCs (TCM) or stem cell memory TCs (TSCM) have captured attention given that they have been shown to provide more suitable characteristics of plasticity, long-term expansion, and persistence. 56 To obtain starting populations of TCM or TSCM cell-sorting procedures have been developed, 57 although it must be mentioned that the CAR TC manufacturing process itself requires long periods of time and multiple rounds of ex vivo expansion of lymphocyte replication to render sufficient TC numbers of lymphocytes, thereby resulting in a senescent, anergic TC product. Whereas proliferation and low cytokine levels of these exhausted TCs are reduced, high apoptosis rates and high levels of inhibitory receptor expression including PD-1 (programmed cell death protein 1) and TIM-3 (T cell immunoglobulin and mucin-domain containing protein 3) have been observed. 58 Interestingly, the CD28 domain within CAR constructs has been associated with increased cell exhaustion and promotion of a terminally differentiated CAR TC phenotype. 4-1BB, on the other hand, has been shown to improve cell exhaustion. 58 These finding confer a reasonable explanation for observed prolonged in vivo persistence of 4-1BB-containing CAR TCs in clinical trials. 38,42,44
Exhausted TCs have been related not only to the type of costimulatory CAR molecule used, but also to the disease of origin, provided the starting lymphocyte subset for CAR TC manufacturing has been demonstrated to accelerate cell exhaustion. For example, in CLL, TCs with more differentiated phenotypes prevail, 59 hence rendering a starting TC population in an already pseudo-exhausted state with functional characteristics of terminally differentiated lymphocytes.
Adverse Events and Toxicity Related to Anti-CD19 Car T Cells
Except for the trial conducted by Cruz and colleagues, 39 all reports of anti-CD19 CAR TC trials have reported toxicities. Most toxicities were mild and reversible, including fever, chills, hypotension, dyspnea, headaches, fatigue, and neurologic alterations. However, severe toxicities including renal failure, marked hypotension, seizures, altered mental status, cardiac arrest, and acute respiratory distress requiring mechanical ventilation, pressor support, or reanimation have also been observed. Moreover, although in the majority of cases not directly related to CAR TC treatment, a low number of patients (three of 165) died after CAR TC administration. 16,60 In general, CAR TC-related toxicity can be categorized as direct on-tumor-off-target toxicity, indirect cytokine-associated toxicity, and neurotoxicity. Other undesired CAR TC effects include immunogenic reactions and/or immune escape.
On-tumor-off-target toxicity
On-tumor-off-target toxicity of CAR TCs relates to the fact that a tumor antigen targeted by CAR TCs might not be exclusively expressed by malignant tissue. In fact, infusion of more than 1010 CAR TCs targeting HER-2 after lymphodepletion in a patient with metastatic colon cancer resulted in pulmonary toxicity and the death of the patient 4 days after CAR TC administration. 61 The fatal outcome was most likely due to HER2 expression in the pulmonary endothelium. Because CD19 expression is strictly limited to B cells, 9 severe cross-reactivity of anti-CD19 CAR TCs seems unlikely. Isolated aplasia of malignant as well as nonmalignant B cells, however, represents a common and expected consequence of anti-CD19 CAR TC treatment, thus serving as a biomarker for an ongoing anti-CD19 on-target CAR TC effect. It remains to be elucidated whether long-term B cell depletion mimicking a condition similar to Bruton agammaglobulinemia (with increased risk of opportunistic infections) might have further consequences in affected patients. Although commonly treatable with immunoglobulin infusions and/or administration of antibiotics, a patient with B cell aplasia died 21 months after CAR TC treatment from ecthyma gangrenosum after a Pseudomonas wound infection. 44
Cytokine-related toxicity
Cytokine release syndrome (CRS), a systemic non-antigen-specific inflammatory process, represents a consequence of on-target CAR TC activity and elimination of malignant cells and has been suggested as a probably essential effect of anti-CD19 CAR TC therapy. 62 It is associated with increased cytokine serum levels and symptoms that range from mild and self-limiting (fever, chills, myalgia, hypoxia, mild hypotension, and/or neurologic changes) requiring approximately 15 days of intrahospital care to severe courses (renal insufficiency, acute respiratory failure, severe hypotension, cardiac dysfunction, coagulopathy, and/or marked cytopenia) requiring intensive care monitoring and in-hospital treatment for approximately 57 days. 49 Whereas mild CRS has been reported for almost 100% of patients, severe CRS occurs in about 30% of treated patients. Onset has been shown to be variable in timing, starting from 24 hr 16,41,54 up to 3 weeks 38 after CAR TC infusion. Of note, an earlier beginning of symptoms after CAR TC administration has been associated with more severe courses of CRS. 54 In general, the severity of CRS has been shown to correlate with high TC expansion levels, clinical response, and elevated serum levels of IL-6, interferon (IFN)-γ, ferritin, C-reactive protein (CRP), soluble IL-2 receptor (sIL-2R), and coagulopathy markers including D-dimers and fibrinogen. 44,49,54 Interestingly, for ALL an extensive disease burden has been shown to be associated with higher severity of CRS. 49,54 However, no such correlation has been reported for CLL. Considering the frequency of CRS in anti-CD19 clinical trials, the definition of CRS has long remained unspecific and difficult to distinguish from other postimmunotherapy conditions such as macrophage-activation syndrome (MAS) characterized by hyperferritinemia, hepatosplenomegaly, fever, bilirubinemia, and elevated triglycerides because both syndromes share partly similar features and CRS itself activates macrophages. 38 Only more recently have criteria for CRS and severe CRS been established: According to Davila and colleagues, definition of severe CRS requires the fulfillment of three criteria including (1) persistent fever, (2) elevation of selected cytokines, and (3) additional clinical manifestations of toxicity. 37 Furthermore, CRP has been shown to correlate with IL-6 concentration and CRS severity 37,44,49 and has therefore been suggested as a useful biomarker for prediction of severe CRS. Elevated CRP levels (exceeding a threshold of 20 mg/dl) predicted with a sensitivity of 86% the risk of developing severe CRS. 37 In addition, ferritin has been proposed as a useful biomarker for CRS activity. 44 Like the definition of CRS criteria, the management of CRS has been unspecific and mostly supportive, initially applying high-dose steroids to affected patients. Although CRS symptoms showed rapid improvement, application of steroids limited the expansion and persistence of CAR TCs. All three patients enrolled in the trial by Davila and colleagues and who had achieved CR and were subsequently treated for CRS with steroids relapsed, whereas no change in clinical response in patients who remained steroid-untreated was observed. 37 These findings were confirmed also by Lee and colleagues 49 and more recently in the trial, conducted by Dai and colleagues, that also reported progressive malignancy in two patients who had been treated for CRS and GvHD with steroids. 41 In contrast, humanized monoclonal anti-IL-6 receptor antibody tocilizumab targets CRS without affecting CAR TC efficacy. Initially used by Grupp and colleagues, 38 tocilizumab has mediated rapid drop in cytokine levels, rapid defervescence, stabilization of blood pressure, as well as weaning from mechanical ventilation and has therefore been proposed as the first-line treatment for severe CRS. 37,41,49,54 Nonetheless, it must be noted that it is still a matter of discussion whether blocking the homeostatic cytokine IL-6 compromises CAR TC efficacy.
Neurotoxicity
Neurologic side effects have been reported in approximately 26–50% of treated patients. They display variable courses of encephalopathy, confusion, delirium, seizures, myoclonus, tremor, gait disturbances, paralysis, apraxia, aphasia, or visual hallucinations. Symptoms mimic in some ways the toxic side effects associated with the anti-CD19 anti-CD3 monoclonal antibody blinatumomab. Except for the report by Lee and colleagues, who observed an abnormal magnetic resonance imaging (MRI) result, 49 no correlation of neurotoxicity and neuroimaging or electroencephalographic (EEG) alterations has been reported by others. 37,44 Symptoms resolved spontaneously and no apparent sequelae in affected patients were noticed. Analysis of cerebrospinal fluid (CSF) has identified anti-CD19 CAR TCs within the CNS in some of the patients. Maude and colleagues did not determine correlation between the presence of CAR TCs within the CNS and neurotoxicity and did not report CAR-associated encephalopathy in patients with CNS leukemia, 54 and Davila and colleagues detected CAR TCs in some but not all patients with neurologic symptoms after CAR TC infusion. 37 On the other hand, Lee and colleagues observed higher levels of CAR TC within the CSF of patients displaying neurologic symptoms. 49 Interestingly, tocilizumab had no effect on neurotoxicity, 48 and the severity of neurologic symptoms was not shown to correlate with the severity of CRS. 54 More recently, Dai and colleagues included two patients with CNS leukemia within their study protocol and observed efficient migration of CAR TCs into the CSF and CNS leukemia remission in both patients. 41 Given these contrary observations, further studies are needed to assess the mechanism behind CAR TC neurotoxicity. Furthermore, it remains unclear whether neurotoxicity is due to inflammation or to a direct TC-mediated effect of CAR TCs within the CNS of affected patients.
Immunogenicity
scFv domains within CARs have been derived mostly from murine monoclonal antibodies (Fig. 1B) and therefore represent a source of immunogenicity in CAR TC therapy given their potential to induce allergic responses or rejection of the infused CAR TC product. Although to date immunogenic reactions or formation of anti-murine antibodies in treated patients have not been reported, tight monitoring of anti-CAR antibody production and the development of humanized scFv CARs remain important issues for the safety of CAR TC products in humans.
Immune escape
Antigen loss under selective pressure is one of the most important mechanisms of tumor escape. Several trials have reported CD19-negative relapse in approximately 10% of patients with ALL undergoing anti-CD19 CAR TC treatment. 38,49,54 This tumor escape strategy cannot be prevented by secondary CAR TC infusions or the use of enhanced CAR constructs with optimized costimulation. However, simultaneously targeting more than one tumor antigen might prevent tumor escape. For example, dual-targeted CAR approaches directed additionally against CD20 in lymphoma, 17 CD22 in ALL, 21 or CD23 in CLL 63 could enhance clinical efficacy and protect from CD19-negative tumor relapse. In addition, tumor-mediated immunosuppression is targeted by novel CAR constructs. Checkpoint inhibitors such as cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) or PD-1 negatively regulate TCs. Therefore, blocking of CTLA-4 or PD-1 via concomitant administration of antibodies could prolong in vivo persistence and provide a selective advantage to CAR TCs. 64 Moreover, incorporation of a safety switch such as inducible caspase 9 within a CAR TC enables controlled elimination of the engineered TCs in case of toxicity 65 (see Sadelain 66 for further review).
Conclusions and Perspectives
After two decades of preclinical tests in vitro and in animal models, CAR TC therapy has reached clinical application. Clinical phase I and II trials as well as ongoing studies have been performed, yielding astonishing clinical responses and sparking new hope for patients with CD19-positive diseases that have already undergone multiple prior treatments including cytostatic drugs, monoclonal antibodies, and even SCT. The global health business is currently investing an enormous amount of money into this novel technology, still endowed with open questions: 1. The vector systems are still “under construction,” that is, the added value of more than one costimulatory molecular is currently under evaluation. 2. The most appropriate preconditioning regimen still needs to be defined. Whereas some investigators favor defined lymphodepleting protocols, others argue that conditioning should be chosen in a more personalized manner. 3. Side effects of CAR TC therapy such as CRS or neurotoxicity are still not fully understood, and proposed criteria and prediction markers have yet to prove their usefulness in the clinical routine. 4. The factors responsible for efficient CAR TC function that contribute to successful clinical outcome are still a vaguely specified issue. Further trials are needed to be able to generate highly effective yet personalized anti-CD19 lymphocyte weapons. 5. The appropriate number of transfused CAR TCs remains to be defined. Although correlation between applied cell dose and clinical outcome might not exist, the mechanisms and kinetics of CAR TCs in patients require further investigation. It seems that, in particular, quality and TC subset selection, not quantity of TCs, might be of decisive importance. 6. Although to date CAR TCs in the clinical setting represent personalized, patient-specific therapies associated with individualized resource-intensive manufacturing processes, more universal approaches in CAR TC development, that is, TALEN (transcription activator-like effector nuclease)-modified, genome-edited CAR TCs,
67
in the future might offer “off-the-shelf” CAR TC therapy the possibility for standardized and even automated CAR TC production. 7. Interestingly, the majority of CAR TC trials so far have been conducted in the United States and China, whereas European countries are strikingly underrepresented. This might be due to regulatory hurdles and complex organization among the various European countries resulting in less effective translation of CAR TC research from preclinical to clinical phase I/II trials in Europe than in the United States, where only one regulatory authority defines upcoming questions. To make European academic institutions and pharmaceutical industries more competitive in a global world, the European landscape urgently needs streamlining of regulatory processes from the local authorities up to the Committee for Advanced Therapies (CAT) at the supranational level. 8. Depending on the results of ongoing trials, clinical application of CAR TC therapy will prove whether this treatment will replace SCT or will constitute an additional option for patients primarily ineligible for SCT, turning them into eligible patients. So far, CAR TC therapy has been used mainly to bridge the time from chemotherapy to alloSCT. Given that anti-CD19 CAR TCs target tumor cells but not stem cells it is unclear at this moment whether or not, and to which extent, CAR TC therapy might replace SCT in the allogeneic and/or autologous setting.
In summary, CD19-targeting CAR TC therapy holds much promise, particularly for patients with CD19-positive leukemia and lymphoma at relapse. The approach is currently being tested in phase I to III trials and will certainly find its way into the clinic. Many players in the field, from academia to pharmaceutical and biotechnology companies, are involved in this novel therapeutic field. In the future, the clinical perspective of CAR TC therapy must be defined between monoclonal antibodies, small molecules, and well-established SCT.
Footnotes
Author Disclosure
P.D. is a consultant (Novartis). P.W.: Honoraria and membership on Advisory Boards of Sanofi-Aventis. Membership on Advisory Boards and Travel Grants from Hexal AG. All of the other authors declare no conflict of interest.