
Abstract
Deficiency of glycogen branching enzyme (GBE) causes glycogen storage disease type IV (GSD IV), which is characterized by the accumulation of a less branched, poorly soluble form of glycogen called polyglucosan (PG) in multiple tissues. This study evaluates the efficacy of gene therapy with an adeno-associated viral (AAV) vector in a mouse model of adult form of GSD IV (Gbe1ys/ys ). An AAV serotype 9 (AAV9) vector containing a human GBE expression cassette (AAV-GBE) was intravenously injected into 14-day-old Gbe1ys/ys mice at a dose of 5 × 1011 vector genomes per mouse. Mice were euthanized at 3 and 9 months of age. In the AAV-treated mice at 3 months of age, GBE enzyme activity was highly elevated in heart, which is consistent with the high copy number of the viral vector genome detected. GBE activity also increased significantly in skeletal muscles and the brain, but not in the liver. The glycogen content was reduced to wild-type levels in muscles and significantly reduced in the liver and brain. At 9 months of age, though GBE activity was only significantly elevated in the heart, glycogen levels were significantly reduced in the liver, brain, and skeletal muscles of the AAV-treated mice. In addition, the AAV treatment resulted in an overall decrease in plasma activities of alanine transaminase, aspartate transaminase, and creatine kinase, and a significant increase in fasting plasma glucose concentration at 9 months of age. This suggests an alleviation of damage and improvement of function in the liver and muscles by the AAV treatment. This study demonstrated a long-term benefit of a systemic injection of an AAV-GBE vector in Gbe1ys/ys mice.
Introduction
G
AAV vectors are promising for gene therapy of GSD IV, as the size (2.1 kb) of GBE1 cDNA is small enough to be packaged into the viral vector. AAV serotype 9 (AAV9) has become increasingly attractive because of its high efficiency in delivering genes to major tissues, including the liver and muscles, and its ability to pass the blood–brain barrier to deliver the desired genes to the CNS. 16 –19 Since these are all affected organs and tissues in GSD IV, AAV9-mediated gene therapy is an appealing treatment approach. This study tested the feasibility of AAV9 as a vehicle to deliver human GBE1 cDNA to correct GSD IV in a mouse model for APBD harboring the homozygous Y329S mutation in the Gbe1 gene. 20 These mice are designated as Gbe1ys/ys mice; they are phenotypically very similar to APBD patients, with residual GBE activity and widespread, progressive increase of PG deposition in all tissues, including the CNS. 20 The current study aimed to evaluate long-term efficacy of AAV9-mediated GBE1 gene replacement therapy in Gbe1ys/ys mice.
Materials and Methods
AAV vector construction and preparation
Human GBE cDNA (NCBI Reference Sequence: NM_000158.3) was PCR-amplified from plasmid pSC-hGBE (Origene, Rockville, MD), cloned into an AAV vector containing a CMV enhanced chicken beta-actin hybrid (CB) promoter, 21 and sequenced. The resulting AAV-GBE vector was packaged as AAV2/9 in HEK 293T cells using standard phosphate-mediated transfection, and purified using iodixanol gradient ultracentrifugation. 22 –24 All viral vector stocks were handled according to the Biohazard Safety Level 2 guidelines published by the National Institutes of Health.
Animals and treatment
The GSD IV (Gbe1ys/ys ) mouse colony harboring the Y329S mutation was kindly provided by Drs. Craigen and Akman at Baylor College of Medicine. 20 The AAV-GBE vector was administered intravenously into 14-day-old Gbe1ys/ys mice at a dose of 5 × 1011 vector genomes (vg) per mouse (∼5 × 1013 vg/kg). Four wild-type (Wt; C57BL/6), six AAV-GBE treated, and seven untreated (UT) Gbe1ys/ys mice were sacrificed at 3 months of age after overnight fasting. Fresh tissue specimens were either immediately frozen on dry ice and stored at −80°C until used for biochemical analyses, or fixed in 10% neutral buffered formalin for histological staining. For long-term observation, eight UT and eight AAV-GBE-treated Gbe1ys/ys mice were sacrificed at 9 months of age, their tissues having been processed in the same manner described above. All the mice used in this study were males. All animal procedures were performed in accordance with Duke University Institutional Animal Care and Use Committee–approved guidelines (Duke IACUC protocol registry number: A087-14-04).
AAV copy number determination
DNA was extracted from frozen tissues using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI). Real-time quantitative PCR was performed using SYBR Green (Roche, Basel, Switzerland) and primer pair 5′-CAGATGTGCTGATGGTGGTTTATA-3′ and 5′-CCACGGTGAAATACGATACAAGATC-3′ for human GBE cDNA; mouse beta-actin (primers: 5′-AGAGGGAAATCGTGCGTGAC-3′ and 5′-CAATAGTGATGACCTGGCCGT-3′) was used as an internal control, and the AAV-GBE plasmid DNA was used to generate a standard curve for viral vector copy number calculation.
Tissue lysate preparation for biochemical analyses
Frozen tissues (50–100 mg) were homogenized in ice-cold deionized water (20 mL of water/g tissue) and sonicated three times for 15 s with >30 s intervals using a Misonix XL2020 ultrasonicator (Farmingdale, NY). The homogenate of each tissue was divided into two parts and processed separately: one part was boiled for 5 min and centrifuged, and the supernatant was used for glycogen content measurement by glucosidase digestion as previously described 25 ; the remaining portion was immediately clarified by centrifugation at 4°C, and the supernatant was used for GBE activity assay. For Western blotting, tissues were homogenized in ice-cold RIPA buffer containing proteinase inhibitor cocktail and cleared by centrifugation. Protein concentration was measured using a bicinchoninic acid (BCA) Protein Assay kit (Thermo Scientific, Rockford, IL) according to the manufacturer's guidelines.
Tissue preparation for histopathology analyses
Tissue samples were freshly taken from the cerebrum (cerebral cortex), liver, heart, and skeletal muscles, fixed in 10% neutral buffered formalin, and processed into paraffin blocks. Tissue sections were stained with Periodic Acid-Schiff (PAS) and trichrome stains, and subject to immunohistochemical staining with anti-CD68 (rat monoclonal FA-11; Abcam, Cambridge MA), a marker for macrophages.
GBE activity assay
GBE activity was measured indirectly based on the finding that glucose-1-phosphate (G-1-P) incorporation into glycogen molecules (and concomitant release of inorganic phosphate) is markedly enhanced by the presence of the branching enzyme. 26 The assay (100 μL) contained 20 mM of cacodylate buffer (pH 6.5), 80 mM of G-1-P, 1 mM of AMP, 6 IU/mL of phosphorylase A, and 10–20 μL of homogenate (diluted with deionized water where necessary). The reaction was incubated at 37°C for 1 h and stopped by boiling for 3 min and then centrifuged. Inorganic phosphate content was measured using phosphorous reagent (Pointe Scientific, Ann Arbor, MI). A reaction using deactivated homogenate (boiled 5 min) was conducted in parallel for background reaction subtraction. GBE activity is expressed as nanomoles of phosphate generated per minute per milligram of protein (nmol/min/mg).
Western blotting
Proteins were resolved by SDS-PAGE and blotted to polyvinyl difluoride membrane. The membrane was blocked with milk and incubated with mouse anti-GBE1 (Abcam; ab124023, 1:1000 dilution,) or goat anti GAPDH (Novus Biologicals, Littleton, CO; NB300-320, 1:10,000 dilution), and then with horseradish peroxidase conjugated secondary antibody. The protein was visualized using enhanced chemiluminescence (Amersham, Pittsburgh, PA).
Plasma chemistry testing
Blood was collected into a heparin tube from the inferior vena cava (IVC) for each Gbe1ys/ys mouse at 9 months of age following overnight fasting. After centrifugation, plasma samples were stored at −80°C before being sent to a commercial laboratory (IDEXX Laboratories, Inc. Westbrook, ME) for a panel of biochemical tests, which included alanine transaminase (ALT), aspartate transaminase (AST), alkaline phosphatase (ALP), creatine kinase (CK), glucose, albumin, blood urea nitrogen, and creatinine.
Rotarod performance
A rotarod test was performed to evaluate the functional improvement of muscle strength and motor coordination in the Gbe1ys/ys mice by the AAV treatment. 27 Eight AAV-treated and eight UT Gbe1ys/ys mice were tested monthly from the age of 2 months to 9 months. Wt mice (n = 8) were tested only at 3 months of age.
Statistics
Data are presented as mean ± standard deviation (SD). To analyze data from multiple groups, one-way analysis of variance with Tukey's post hoc test was performed using Prism software (GraphPad, La Jolla, CA). To compare two groups, equal variance, unpaired, two-tailed Student's t-test was performed. A p-value of <0.05 was considered statistically significant.
Results
Disease progression in tissues of Gbe1ys/ys mice
PG aggregates are rarely seen in the muscles of Gbe1ys/ys mice at 1 month of age. However, by 3 months of age, approximately 50% of skeletal muscle cells contain granular PAS-positive glycogen particles, with particle density apparently varying from cell to cell, suggesting different stages of disease progression. By 9 months of age, most myocytes demonstrate mild to severe glycogen accumulation (Fig. 1). Liver glycogen aggregates appear as finely dispersed PAS-positive granules within hepatocytes throughout the liver at 1 month of age. At 3 and 9 months, the glycogen aggregates appear more numerous and densely stained, accompanied by the presence of lymphocytic infiltrates (Fig. 1). In the brain, no PAS-positive materials were found at 1 month of age. At 3 months, widespread fine PG particles were observed, which appeared enlarged at 9 months (Fig. 1). The heart is less affected in this model, as only very few isolated clusters of PAS-positive myocytes were observed at 3 and 9 months of age (not shown).
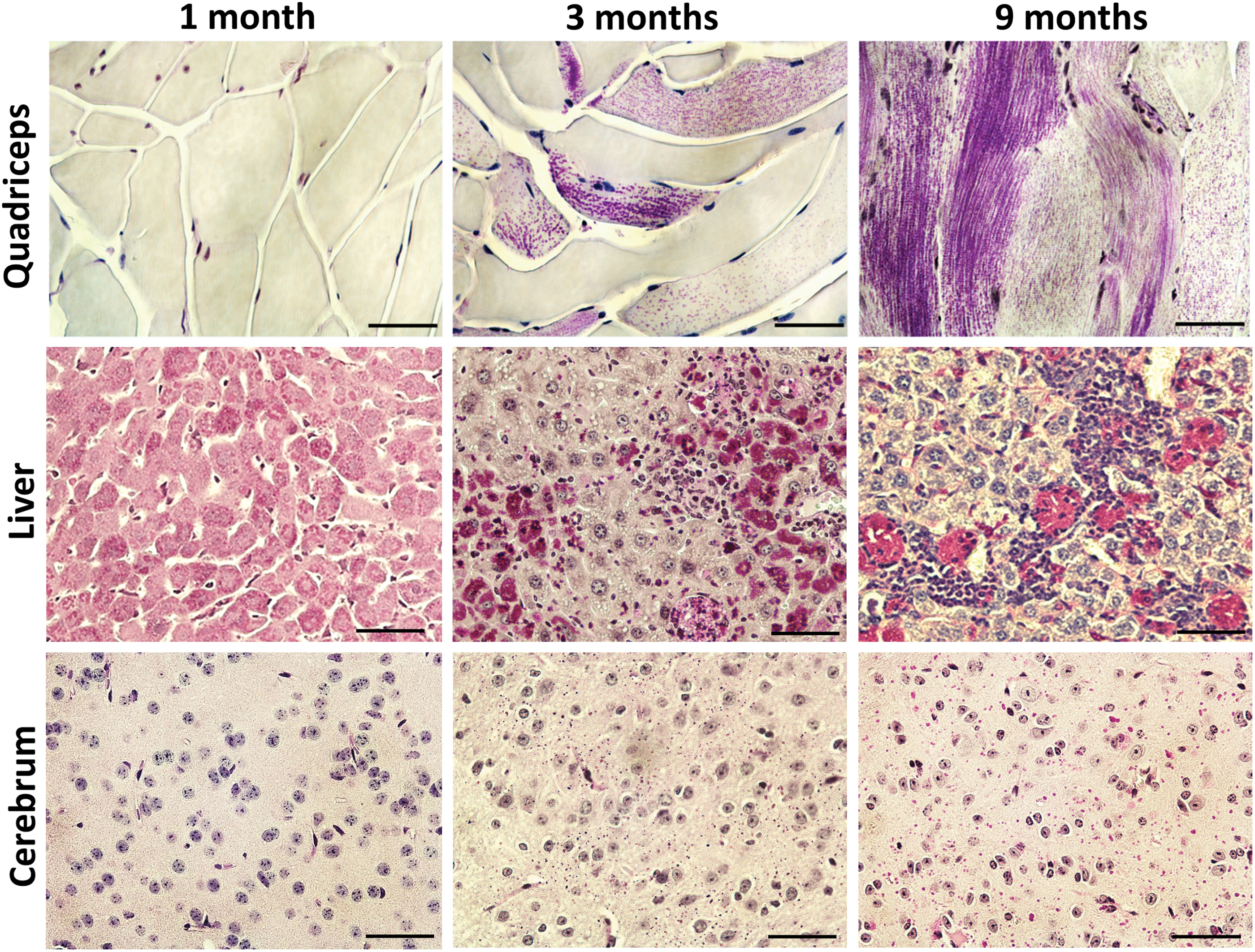
Periodic Acid-Schiff (PAS) staining showing disease progression in the skeletal muscle, liver, and brain of Gbe1ys/ys
mice. No visible PAS-positive particles were found in the quadriceps at 1 month of age. However, significant amounts of PAS-positive cells (purple stain) were observed at 3 months, and most myocytes were affected by glycogen accumulation with varied extents of involvement at 9 months of age. Liver demonstrated fine PAS-positive particles spread throughout all the cells at 1 month of age. By 3 and 9 months of age, the cellular distribution of PAS particles appeared more heterogeneous, along with the influx of lymphocytic infiltrates. PAS-positive particles were absent in the cerebrum (cerebral cortex) at 1 month of age, but emerged at 3 months of age and become larger and more numerous at 9 months. Scale bars: 100 μm. Color images available online at
Quantitation of AAV vector genomes in tissues of Gbe1ys/ys mice
AAV vector DNA in tissues was quantified using real-time PCR (Table 1). Ten weeks after vector administration, an average of 12.29 copies of vector DNA per nucleus (genome) were detected in the heart, 2.22 in the liver, and 0.81 in the brain. In skeletal muscles, 0.31 and 0.73 copies per nucleus were detected in the quadriceps and gastrocnemius, respectively, representing 62–146 copies per muscle cell (assuming an average of 200 nuclei per cell 28 ). In contrast, the AAV vector copy number in 9-month-old mice was <1/10 of that observed in 3-month-old mice in most tissues (Table 1), indicating a gradual loss of viral vectors over time. This waning effect has also been shown in other AAV-treated GSD murine models. 29
Measured AAV copy numbers (copies/nucleus) in tissues a
Mean ± SD. AAV vector was intravenously injected into the Gbe1ys/ys mice at 14 days of age at a dose of 5 × 1011 vg/mouse.
Cerebral cortex.
n = 6.
n = 8.
AAV, adeno-associated virus; ND, not determined.
Expression of GBE in Gbe1ys/ys mice following administration of AAV-GBE vectors
At 3 months of age, residual GBE activities in the major tissues of UT Gbe1ys/ys mice were at 0.65% in the liver, 10.6% in the heart, 29.6% in the brain, 10.7% in the quadriceps, and 18.5% in the gastrocnemius of those in the age-matched Wt mice (Table 2). After AAV treatment, GBE activity was extremely high in the heart (26-folds that of Wt) and moderately higher than Wt levels in skeletal muscles. GBE activity in the brain was increased by 51% (p < 0.001) by the AAV treatment, reaching 45% of the Wt, but surprisingly there was no significant increase of GBE activity in the liver (Table 2). Consistent with the measured enzyme activities, Western blot showed low expression of GBE protein in all tissues of UT Gbe1ys/ys mice compared with Wt, and increased expression in the heart and skeletal muscle by AAV treatment (Fig. 2a). At 9 months of age, significantly increased GBE activity was detected only in the heart of AAV-treated mice, an approximately 71% decrease from that at 3 months of age (Table 2).

Increased expression of glycogen branching enzyme (GBE) and reduced glycogen content in the tissues of Gbe1ys/ys
mice following AAV-GBE administration. (
GBE activity (nmol/min/mg) in mouse tissues at 3 or 9 months of age a
Mean ± SD. AAV vector was intravenously injected at 14 days of age at a dose of 5 × 1011 vg/mouse.
Cerebral cortex.
Wild-type mice, n = 4.
Untreated Gbe1ys/ys mice, n = 7.
AAV-GBE-treated Gbe1ys/ys mice, n = 6.
n = 8 mice for both groups. Data for 3-month-old mice were statistically analyzed with one-way analysis of variance with Tukey's post hoc test; data for 9-month-old mice were analyzed with an unpaired, equal valence, two-tailed t-test.
n.s., not significant (p > 0.05).
Correction of glycogen storage in major tissues by AAV-GBE treatment
Glycogen contents were quantified in mouse tissues using a modified enzymatic method recently developed specifically for GSD IV glycogen measurement. 30 At 3 months of age, glycogen contents in the skeletal muscles (quadriceps and gastrocnemius) of AAV-treated mice were at similar levels of Wt, indicating an effective correction of glycogen storage in skeletal muscles. The glycogen content in the AAV-treated mice was 59% in the fasting liver, and 48% in the brain of those in UT mice (Fig. 2b). At 9 months of age, the glycogen content was significantly reduced in the quadriceps (–86.5%), gastrocnemius (–92.3%), liver (–63.6%), and brain (–22.8%) of AAV-treated mice compared with UT mice (Fig. 2c).
The relatively low level of glycogen in the heart of this mouse model makes it impractical to assess the efficacy of AAV-GBE treatment in the tissue by glycogen content determination. However, PAS staining of heart sections revealed isolated clusters of PAS-positive myocytes in UT Gbe1ys/ys mice, while no visible glycogen particles were found in the AAV-treated hearts of either the 3- or 9-month-old group (not shown).
Consistent with measured glycogen contents, PAS-stained sections from 3-month-old mice revealed no visible PG in the skeletal muscles (quadriceps and diaphragm) of AAV-treated mice (Fig. 3). In addition, AAV treatment markedly improved the overall liver architecture by eliminating focal lymphocytic infiltrates and reducing the number of PAS-positive hepatocytes. PG particles in the AAV-treated cerebrum were apparently less than those in the age-matched UT cerebrum (Fig. 3).
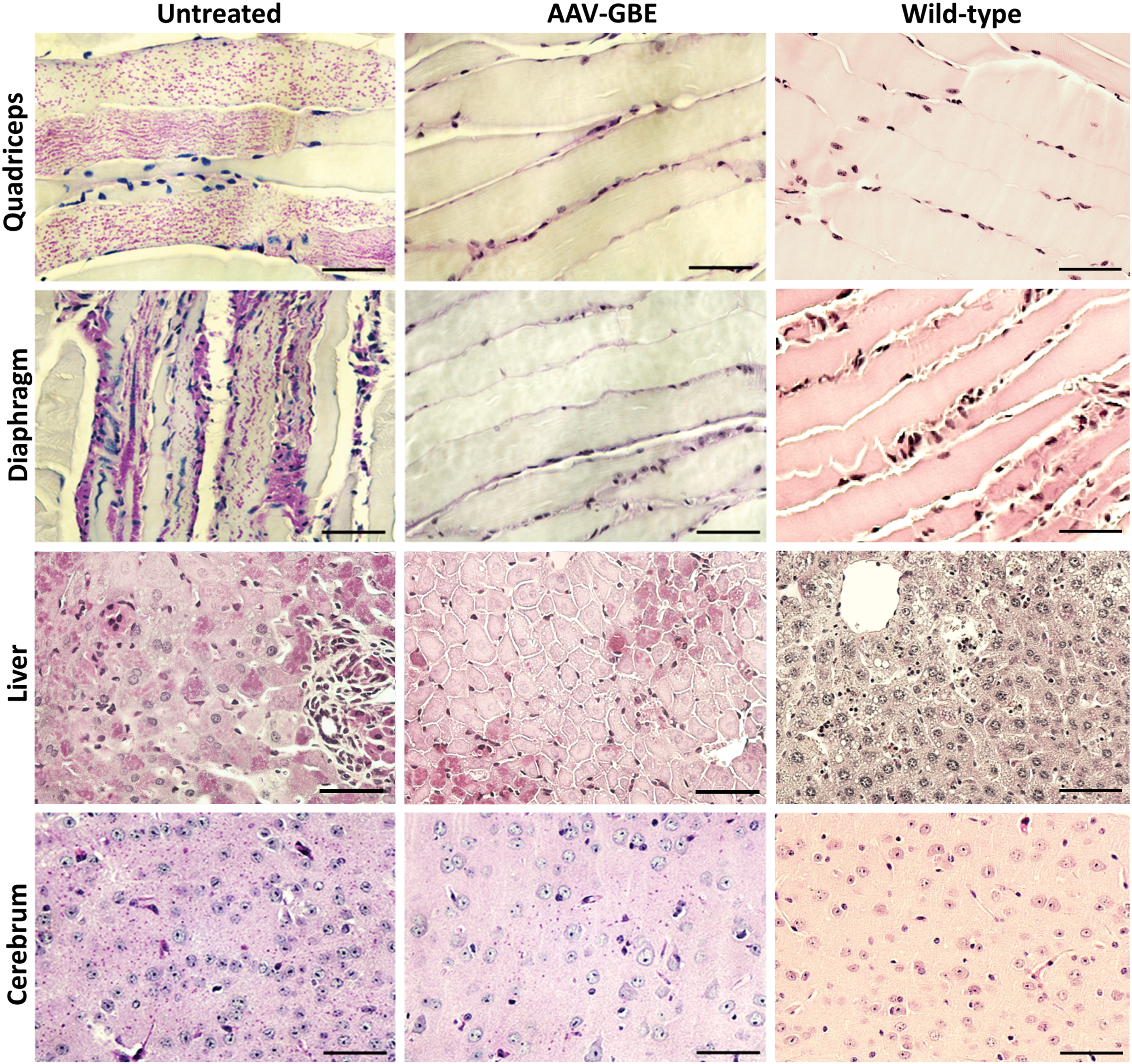
AAV-GBE reduced polyglucosan (PG) accumulation in the tissues of Gbe1ys/ys
mice. Representative PAS staining shows that glycogen deposition (purple stain) was suppressed in the skeletal muscles (quadriceps and diaphragm) and markedly reduced in the liver and cerebrum of 3-month-old AAV-treated mice. Tissues from an age-matched Wt mouse were stained for reference. Scale bars: 100 μm. Color images available online at
Trichrome staining of tissue sections from mice at 9 months of age revealed normal appearance of the liver and muscle of AAV-treated mice (Fig. 4). In UT mice, mild fibrosis was observed (arrows), and lightly stained foamy cells were present in the liver. A significant infiltration of foamy cells between affected myocytes was also seen in the muscle (Fig. 4). These foamy cells were positive for macrophage marker CD68 by immunohistochemistry staining (Supplementary Fig. S1; Supplementary Data are available online at
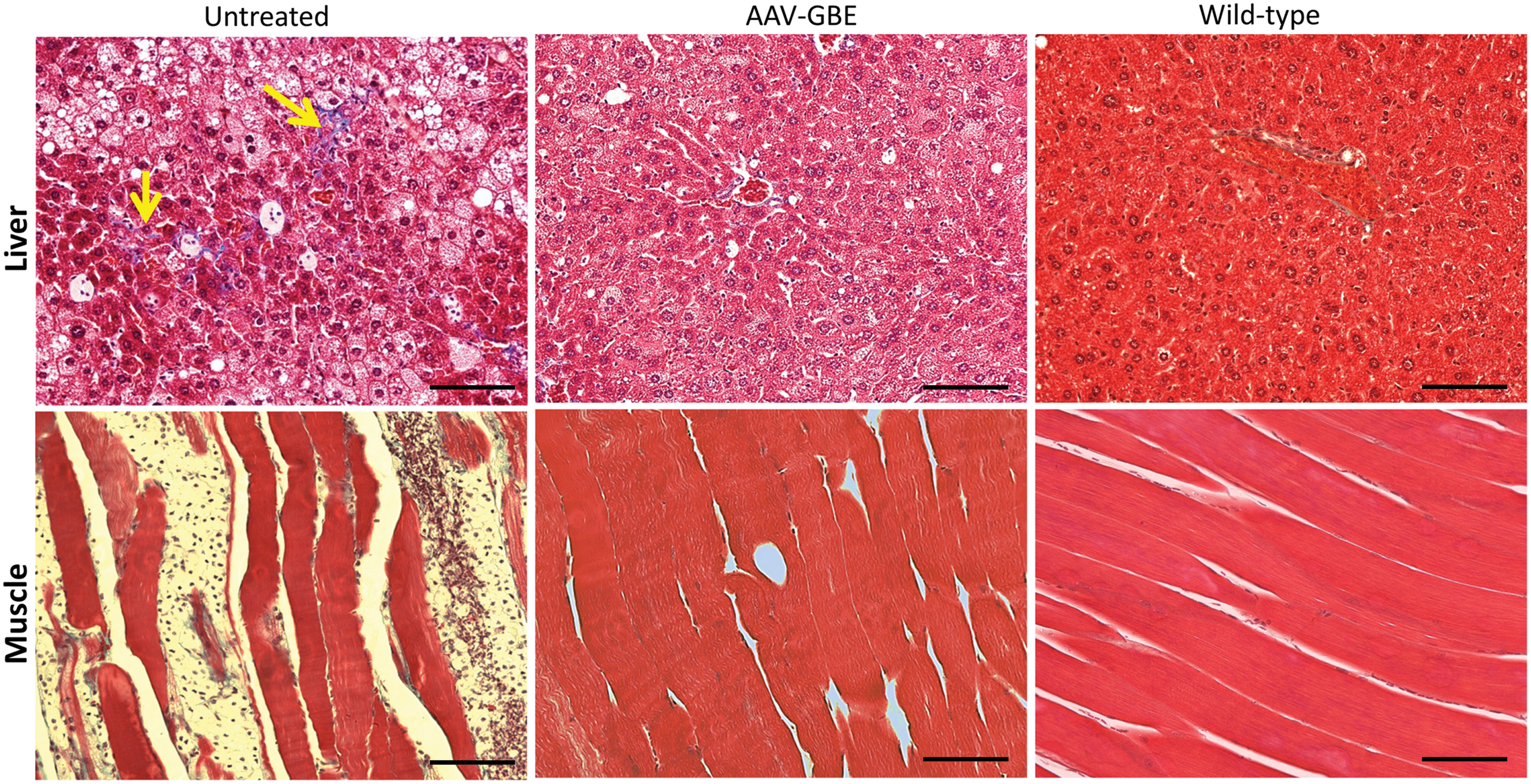
Disruption of tissue structure and infiltration of macrophages in the liver and muscle of UT Gbe1ys/ys
mice at 9 months of age. Trichrome-stained sections of liver and skeletal muscle (quadriceps) from AAV-treated mice appeared normal. In UT mice, mild liver fibrosis is evident (yellow arrows), and larger, foamy cells are present in the hepatic sinusoids. Significant numbers of foamy cells are also present in between myocytes in the skeletal muscle. Tissues from an age-matched Wt mouse were stained for reference. Scale bars: 200 μm. Color images available online at
Effect of long-term AAV-GBE treatment on plasma chemistries
Plasma biochemistry tests were conducted for six UT and six AAV-treated Gbe1ys/ys mice at 9 months of age. All the UT mice had elevated ALT and AST enzyme activities compared with reference values of C57BL/6 Wt controls (normal range: 13–39 IU/L for ALT, 26–79 IU/L for AST), 31 indicating an occurrence of liver damage caused by glycogen deposition. CK activity was within the reference range (24–386 IU/L) 31 for four UT mice but was elevated in two mice (Fig. 5). Other parameters, including ALP, albumin, blood urea nitrogen, and creatinine, were within the reference ranges (data not shown). AAV treatment resulted in an overall trend of decreased plasma enzyme activities of ALT, AST, and CK, suggesting an alleviation of damage in the liver and muscle from the treatment. However the significance of these decreases between the UT- and AAV-treated groups cannot be concluded due to the limited number of mice analyzed. Another interesting finding is that the AAV treatment significantly increased the fasting plasma glucose concentration from 141 ± 10 mg/dL to 174 ± 14 mg/dL (p < 0.001), suggesting functional improvement of the liver.
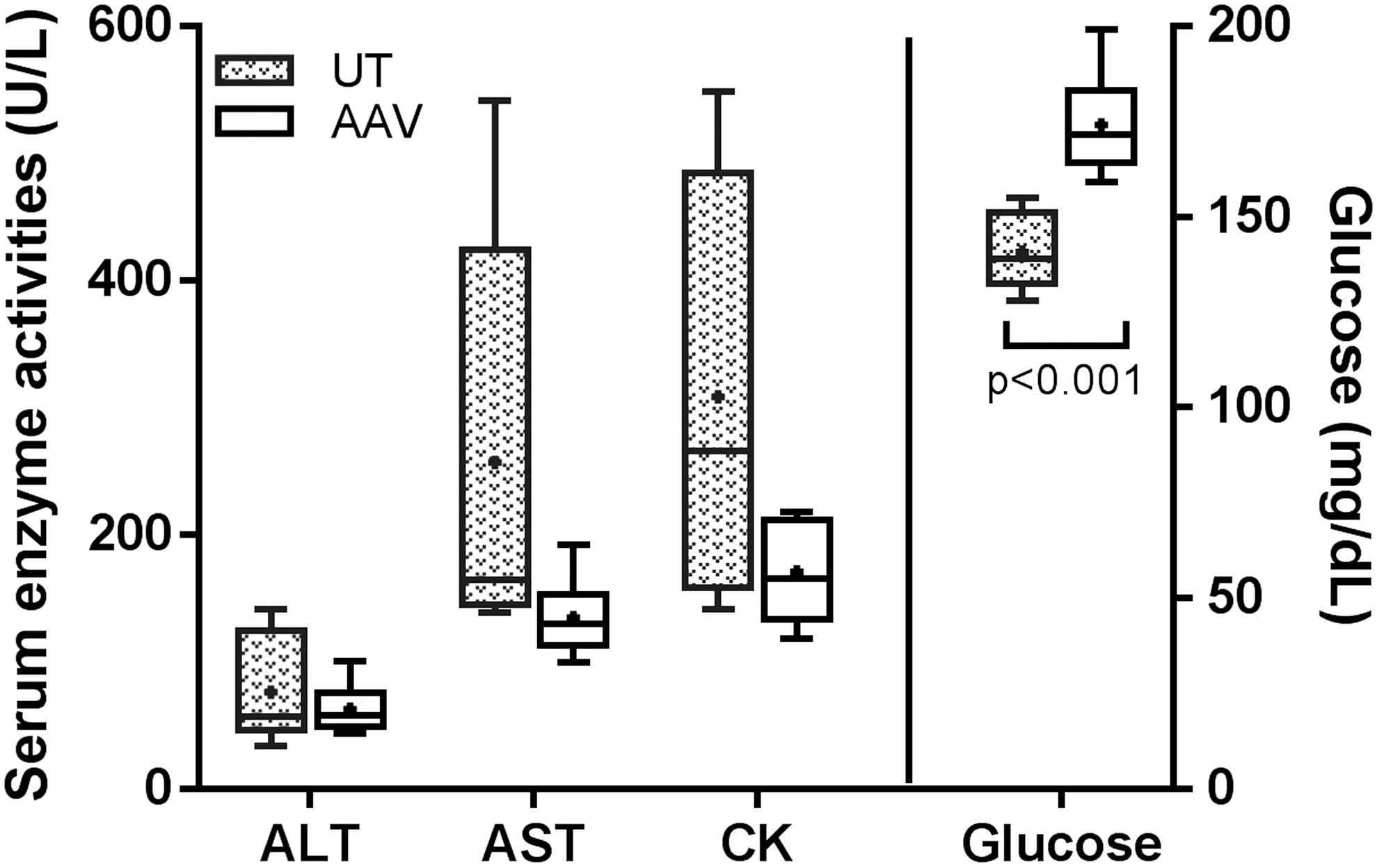
Box and whisker plot for plasma chemistry in Gbe1ys/ys mice at 9 months of age. Fasting plasma from six UT and six AAV-treated Gbe1ys/ys mice were examined. A dot in a box indicates the mean value, a line in a box represents the median value, and whiskers indicate minima and maxima. Note that glucose was plotted to the right y-axis. ALT, alanine transaminase; AST, aspartate transaminase; CK, creatine kinase. Reference values of C57BL/6 Wt controls are: 13–39 IU/L for ALT, 26–79 IU/L for AST, and 24–386 IU/L for CK 29.
Effect of long-term AAV-GBE treatment on rotarod performance
AAV-treatment significantly improved rotarod performance of the Gbe1ys/ys mice to the level of Wt mice at 3 months of age (Supplementary Fig. S2). However, despite significant clearance of glycogen accumulation in the muscle and brain at 9 months of age (Fig. 2c), there was no significant improvement in the rotarod functional test thereafter (Supplementary Fig. S2), suggesting the need for more sensitive functional tests.
Discussion
As with other glycogen storage diseases, adult-onset GSD IV manifests progressive glycogen accumulation in cells. The Gbe1ys/ys mouse model, recently generated by Akman et al., 20 similarly manifests glycogen deposition in the muscles, which shows a punctuated pattern with similar particle sizes but varied particle densities in the affected cells (Fig. 1). Progressive accumulation of PG was observed in the brain of Gbe1ys/ys mice (Fig. 1), in agreement with the fact that functional deterioration of the CNS occurs in elderly patients with APDB. 8 Although progressive liver disease is not typical in human APDB patients carrying the Y329S mutation, 7 there is undoubtedly liver involvement in this Gbe1ys/ys mouse model, as progressive liver damage was observed (Figs. 1 and 4). The heart is not significantly affected in this model, as evidenced by the low glycogen content (Fig. 2b and c) and the histology findings of only the isolated affected cell clusters found at 3 and 9 months of age (not shown). Akman et al. reported a high glycogen level in the heart of adult Gbe1ys/ys mice 20 ; in the present study, high glycogen accumulation was only observed in one of the four mice originally received at the age of 9 months. The measured endogenous GBE activity in the Gbe1ys/ys mice at 3 months of age was below 1% in the liver, between 10% and 20% in the muscles, and approximately 30% in the brain of Wt values (Table 2). It was noticed that the residual GBE activity in the liver of Gbe1ys/ys mice measured in this study (0.65% of Wt) was markedly lower than that reported by Akman et al. (37% of Wt). 20 The cause of this discrepancy is unclear and unlikely to be caused by different assay methods, because the percentages of residual GBE activity over Wt in other tissues were comparable between the two groups. The Western blot tends to support the liver GBE activity observed in this study (Fig. 2a and Table 2).
While GBE activity was not remarkably elevated in the liver (Table 2), the gene therapy effectively reduced the amount of non-degradable glycogen in the liver and protected it from inflammatory cell infiltration and fibrosis (Figs. 2 –4). Usually AAV9 vectors transduce liver much more efficiently than most other tissues in mice do. 17,18,22 Low GBE activity in the liver could not be explained by low transduction efficiency, as the copy number (2.2 copies/nucleus) does not support such a scenario. Rather, it is more likely caused by an inhibitory effect of high glycogen accumulation in the cytoplasm on the GBE expression (e.g., blocking the translocation of AAV vectors to the nucleus and/or the GBE mRNA to ribosomes). Unlike in other tissues where PG is gradually accumulated, Gbe1ys/ys mice have high PG accumulation in the liver at a younger age (Fig. 1).
Akman et al. reported that eventual accumulation of PG in the CNS causes neuropathy in adult Gbe1ys/ys mice, which leads to hind-limb spasticity and stiffness. 20 The ability of AAV serotype 9 to pass the blood–brain barrier and transduce the CNS in mice has been well documented. 19,32 –34 This study demonstrated partial correction of glycogen accumulation in the brain of Gbe1ys/ys mice by the AAV-GBE treatment (Fig. 2). However, despite excellent efficacy of long-term AAV treatment in preventing glycogen deposition in skeletal muscles (Fig. 2c), rotarod performance was significantly improved only at 3 months of age (Supplementary Fig. S2), possibly due to the limited correction of the central and peripheral nervous system dysfunction.
Biochemical measurement of glycogen content is the most accurate method to assess the severity of glycogen storage in tissues. In the past decades, an enzymatic method based on homogenization of tissues in cold water followed by Aspergillus niger amyloglucosidase digestion has become widely used for measuring glycogen content in various GSD tissues. 25,35 –38 When used to quantify glycogen content in GSD IV tissues, however, this method was found to cause significant loss (>90%) of the insoluble glycogen during the preparation of tissue lysates (data not shown). Recently, a modified method was developed that includes an extra boiling step to dissolve the insoluble glycogen, ultimately preserving the glycogen in tissue homogenates from Gbe1ys/ys mice. 30
Blood chemistry tests could provide important clinical information of disease progression and treatment outcome. Elevation of serum aminotransferases AST and ALT is commonly seen in patients with GSD IV, and fasting hypoglycemia is present in some patients with liver cirrhosis. An increase in serum CK activity could be observed in some patients with neuromuscular involvement. 2,3 This study showed that AAV treatment reduced overall plasma ALT, AST, and CK activities, and increased fasting plasma glucose concentration in Gbe1ys/ys mice at 9 months of age, suggesting improvement of the function of the liver and muscle.
In summary, these results show that a single injection of the AAV-GBE vector at a young age effectively prevented glycogen accumulation in skeletal (and cardiac) muscles and, to a lesser extent, in the liver and brain up to 9 months of age. This is the first study showing systemic correction of glycogen storage in a mouse model of GBE deficiency. Although Gbe1ys/ys mice appear to adhere closer to the disease progression of APDB patients, this therapy may serve as a useful therapeutic agent in human patients with all types of GBE deficiency to improve overall muscular, neurologic, hepatic, respiratory, and cardiac functions.
Footnotes
Acknowledgments
We thank Drs. H. Orhan Akman and William Craigen at Baylor College of Medicine for providing the Gbe1ys/ys mouse model. We thank Dr. Dwight Koeberl for his review and discussion of the manuscript. The AAV9 packaging plasmid was provided courtesy of Dr. James M. Wilson at the University of Pennsylvania (Philadelphia, PA). We express our gratitude to the staff of the Duke University Division of Laboratory Animal Resources for their dedicated care of the mice used in this study.
Author Disclosure
The authors declare no competing or financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.