
Abstract
Hematopoietic stem and progenitor cells (HSPCs) have great therapeutic potential because of their ability to both self-renew and differentiate. It has been proposed that, given their unique properties, a small number of genetically modified HSPCs could accomplish lifelong, corrective reconstitution of the entire hematopoietic system in patients with various hematologic disorders. Scientists have demonstrated that gene addition therapies—targeted to HSPCs and using integrating retroviral vectors—possess clear clinical benefits in multiple diseases, among them immunodeficiencies, storage disorders, and hemoglobinopathies. Scientists attempting to develop clinically relevant gene therapy protocols have, however, encountered a number of unexpected hurdles because of their incomplete knowledge of target cells, genomic control, and gene transfer technologies. Targeted gene-editing technologies using engineered nucleases such as ZFN, TALEN, and/or CRISPR/Cas9 RGEN show great clinical promise, allowing for the site-specific correction of disease-causing mutations—a process with important applications in autosomal dominant or dominant-negative genetic disorders. The relative simplicity of the CRISPR/Cas9 system, in particular, has sparked an exponential increase in the scientific community's interest in and use of these gene-editing technologies. In this minireview, we discuss the specific applications of gene-editing technologies in human HSPCs, as informed by prior experience with gene addition strategies. HSPCs are desirable but challenging targets; the specific mechanisms these cells evolved to protect themselves from DNA damage render them potentially more susceptible to oncogenesis, especially given their ability to self-renew and their long-term proliferative potential. We further review scientists' experience with gene-editing technologies to date, focusing on strategies to move these techniques toward implementation in safe and effective clinical trials.
Introduction
T
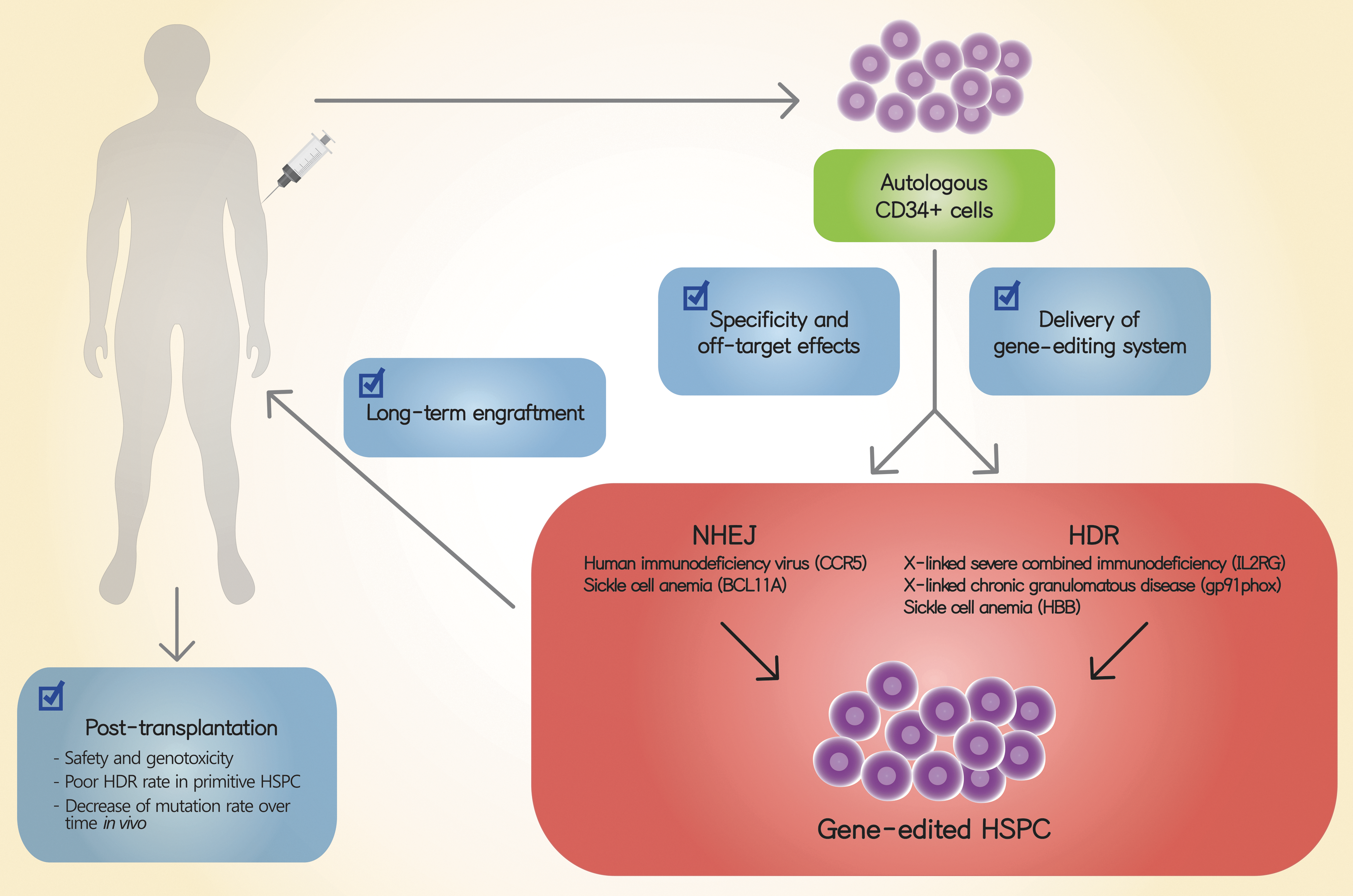
Strategy for therapeutic gene editing in human hematopoietic stem and progenitor cells (HSPCs). Autologous human CD34+ HSPCs are isolated from the bone marrow or mobilized peripheral blood of a patient affected with a broad spectrum of congenital or hereditary diseases including hematologic, immunologic, and metabolic disorders. After culture ex vivo with cell proliferation-stimulating cytokines, ZFN, TALEN, or CRISPR/CAS9 RGENs are delivered into human HSPCs to correct or knock out specific genes linked to various diseases including HIV, sickle cell disease (SCD), X-linked severe combined immunodeficiency (SCID-X1), and X-linked chronic granulomatous disease (X-CGD). Gene-edited HSPCs are then infused back into the patient, generally after administration of a conditioning regimen meant to facilitate modified cell engraftment. To achieve clinically relevant rates of successful gene editing, investigators must maximize the specificity and minimize the off-target effects of gene-editing tools while continuing to optimize the method used to deliver these gene-editing systems into cells. Some of the major concerns investigators must address include potential threats posed to patient safety, low HDR rates in edited cells, and a low mutation rate over time posttransplantation. HDR, homology-directed repair; NHEJ, nonhomologous end joining.
Genome-Editing Technologies
In the last two decades, programmable nucleases able to induce double-strand DNA breaks (DSBs) in a site-specific fashion have been discovered or artificially synthesized. Today, three classes of DNA-editing nucleases—zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) systems, also known as RNA-guided engineered nucleases (RGENs)—dominate the field. All three nucleases operate in a similar manner, cleaving a target region of DNA to allow for endogenous homology-directed repair (HDR) or nonhomologous end joining (NHEJ), thus enabling a large range of genetic modifications including gene disruption, gene insertion and correction (with codelivery of a homologous donor template), and chromosomal rearrangement. 13
TALENs and ZFNs comprise amalgamations of the nuclease domain of the FokI restriction enzyme with the TALE or ZF DNA-binding domains, respectively. The TALE DNA-binding domain, found in TALE proteins native to plant pathogenic bacteria, is made up of 33- to 35-amino acid repeats individually able to recognize a single DNA base pair. Each repeat derives its specificity from the repeat variable di-residues (RVDs) contained within its region. 14 Designer ZF domains are engineered using proprietary algorithms encompassing knowledge of the DNA-binding specificities of hundreds of endogenous cellular ZF proteins. 15 These domains consist of roughly 30 amino acid residues, arranged in a ββα configuration, with the amino acid responsible for DNA specificity located within the α-helical domain. This configuration lends each ZF the ability to bind, on average, three DNA bases. It is possible to engineer synthetic TALENs and ZFNs capable of recognizing a wide range of DNA sequences. However, given that the FokI nuclease domain functions as a dimer, two TALEN and ZFN monomers are required to initiate DNA cleavage, potentially augmenting both specificity and off-target effects due to a propensity for dimer mismatches, 16 while also necessarily increasing the size and complexity of the gene product or transgene being introduced into target cells. 17
The CRISPR/Cas9 system, first reported in January 2013 18 –21 and thereafter rapidly supplanting TALENs and ZFNs in many laboratories, originates from the RNA-guided DNA cleavage procedure developed by bacteria and archaea to defend against the foreign DNA of invading plasmids or phages. Under this system, bacteria and archaea are able to co-opt short sequences (∼20 bp) from the DNA of the invading organism (known as protospacers) and introduce these fragments into their own genome, producing a CRISPR. These CRISPR domains are then transcribed and processed, resulting in target-sensitive CRISPR RNA (crRNA) and invariable target-independent trans-activating crRNA (tracrRNA). Finally, crRNA and tracrRNA complex with the Cas9 protein, initially derived from the Streptococcus pyogenes (Sp) bacterial species, to create an active DNA endonuclease able to cleave a 23-bp DNA target composed of the protospacer and the protospacer-adjacent motif (PAM), typically a 5′-NGG-3′ sequence. 16 It has been shown that crRNA and tracrRNA can be united to form a simplified, chimeric single-chain guide RNA (sgRNA). 22 Thus the CRISPR/Cas9 RGEN, comprising Cas9 protein and sgRNA, can be easily prepared by selecting an approximately 20-bp target DNA sequence of interest adjacent to a PAM motif and engineering a vector or vectors to allow expression of an appropriate sgRNA and Cas9. The simplicity and flexibility of this approach, requiring only the design of specific targeted sgRNAs for use with a single endonuclease, confers some advantages over TALEN and ZFN editing, both of which require complex protein engineering for each new target site. 16 In addition, CRISPR/Cas9 RGEN complexes are active as monomers, permitting the simultaneous modification of multiple target genes without the exacerbation of off-target effects inherent in TALEN- or ZFN-mediated multiplex genome editing.
Potential for Clinical Translation
To date, investigators have performed laboratory studies highlighting the therapeutic potential of gene editing with TALENs, ZFNs, or CRISPR/Cas9 for a variety of diseases (Table 1). These studies constitute the first steps toward establishing clinical protocols. Two target genomic loci—chemokine coreceptor 5 (CCR5) and BCL11A—have received the most attention as practical early clinical targets because of the potential therapeutic benefit derived from simple knockout of these loci via NHEJ, without need for correction via HDR. A patient with both HIV-1 infection and leukemia underwent allogeneic transplantation with donor HSPCs naturally homozygous for a deletion in the CCR5 gene and subsequently became HIV negative, thus stimulating interest in a CCR5-knockout approach to eradicate the disease. 23 Gene knockout of the CCR5 locus in autologous HSPCs holds promise as a less toxic and more generally available approach to HIV cure, and is the most frequently studied locus in the gene-editing field to date. Identification of the BCL11A transcription factor as critical in shutting off expression of fetal γ-globin during development has stimulated investigators to attempt NHEJ-mediated knockout of either the BCL11A gene itself, or of the BCL11A erythroid-specific enhancer, in order to reactivate fetal γ-globin in autologous HSPCs as a potential treatment for sickle cell anemia. 24 The application of gene editing to treat other human diseases via actual gene correction is much more challenging, and must be weighed against simpler lentiviral vector-mediated gene addition therapies for diseases such as SCID-X1 and chronic granulomatous disease (CGD).
Summary of gene-editing methods, efficacy, target, and disease type in hematopoietic stem and progenitor cells
AAV, adeno-associated virus; AML, acute myeloid leukemia; BM, bone marrow; CB, cord blood; CRISPR, clustered regularly interspaced short palindromic repeats; ddPCR, droplet digital PCR; ESCs, embryonic stem cells; FACS, fluorescence-activated cell sorting; GCD, genomic cleavage detection; GFP, green fluorescent protein; gRNA, guide RNA; HDR, homology-directed repair; HEKn, human epidermal keratinocytes, neonatal; HSPCs, hematopoietic stem and progenitor cells; IDLV, integration defective lentiviral vector; indel, insertion or deletion of bases; iPSCs, induced pluripotent stem cells; MLL, mixed-lineage leukemia; mPB, mobilized peripheral blood; N/A, not applicable; NGS, next-generation sequencing; NHEJ, nonhomologous end joining; PB, peripheral blood; RFLP, restriction fragment length polymorphism; RNP, ribonucleoprotein; SCD, sickle cell disease; SCID-X1, X-linked severe combined immunodeficiency; sgRNA, single-chain guide RNA; SNP, single-nucleotide polymorphism; ssDNA, single-stranded DNA; TALEN, transcription activator-like effector nuclease; TIDE, tracking of indels by decomposition; X-CGD, X-linked chronic granulomatous disease; ZFN, zinc finger nuclease.
Optimization of Efficacy, Toxicity, and Delivery of Gene-Editing Components in HSPCs
Viral delivery
The efficient and safe delivery of gene-editing systems into HSPCs constitutes a major roadblock facing the implementation of gene-editing technologies in laboratory and particularly clinical settings. Viral vectors co-opt cell-entry and DNA-integration machinery, developed by the parental viruses to be both highly efficient and nontoxic to target cells. 25 Lentiviruses have previously been used to successfully deliver both sgRNA and Cas9 coding sequences into murine HSPCs, thereby generating loss-of-function mutations in various genes implicated in human acute myeloid leukemia (AML). 26 However, simultaneous delivery of both sgRNA and Cas9 into human HSPCs via an all-in-one integrating lentiviral vector has proven more challenging. Our preliminary observations indicate that sustained expression of SpCas9 in human HSPCs via an integrating lentiviral vector results in significant cytotoxicity. 27 Moreover, sustained expression of site-specific endonucleases is key in determining the extent of potentially detrimental off-target effects, 28 and integrating vector delivery would result in permanent expression in both HSPCs and their progeny, an unintended result not likely to be clinically acceptable.
The integration-defective lentiviral vector (IDLV) system is an attractive alternative because it enables efficient delivery to primary human cells with minimal accompanying vector integration, due to the integrase mutations. 29 Promisingly, transduction of human cord blood (CB)-derived CD34+ cells with IDLV constructs expressing targeted ZFN and donor DNA template for gene correction was shown to sufficiently stimulate gene correction with detectable ZFN activity for the first 3–4 days after transduction. 30,31 However, human primary HSPCs had the lowest gene-editing efficiency of the various cell types studied, possibly due to stress caused by the simultaneous delivery of three different IDLVs: two distinct IDLVs to deliver the ZFN monomers, and a third IDLV to deliver the donor sequence. 30 In our experiments, even several days of Cas9 expression from an IDLV resulted in significant toxicity to human HSPCs, although at a reduced level compared with the cytotoxicity engendered by integrating lentiviral Cas9 gene delivery.
Several other nonintegrating viral delivery systems have been used to transport nucleases and/or gene correction cassettes. Certain adenoviral serotypes can transduce human HPSCs and deliver large transgene cassettes without detectable genomic integration. HSPCs were evaluated for gene disruption after transduction with an adenoviral vector encoding a CCR5-specific pair of ZFNs. 32 Gene disruption of this locus was high (26.4–31.3%) when HSPCs were pretreated with protein kinase C (PKC) activators before transduction with the adenoviral vectors; however, this approach resulted in low cell viability and a reduction in human cell engraftment in a humanized mouse model. 32 A second study used a clever microRNA approach to suppress expression of TALENs or ZFNs in the adenovirus producer cells (given the toxicity associated with high levels of nuclease), permitting high-titer production of vector particles that were shown to efficiently knock out several target loci in human HSPCs. 33 There is some concern that residual adenovector particles may be highly immunogenic and thus not ideal for clinical use. Because of their low levels of integration, immune stimulation, and pathogenicity, recombinant adeno-associated viral vectors (rAAVs) comprise a third nonintegrating viral delivery method for gene editing. 34 However, their relatively small packaging capacity (rAAV can deliver just 4.7 kb) limits their ability to encode and transport large nucleases such as TALENs or RGENs. A Cas9 ortholog (Staphylococcus aureus Cas9, measuring 3.2 kb) small enough to be delivered—along with sgRNA—via rAAV has been thoroughly characterized, 35,36 necessitating further study of the potential uses of rAAV to deliver gene-editing components to human HSPCs. AAV6 has already been used to successfully deliver a DNA donor cassette for targeted gene addition to CB CD34+ cells, in combination with mRNA encoding the ZFN pair (see below). 37
DNA transfection
Unlike traditional gene therapy, genome editing does not require sustained expression of the editing machinery. A “hit-and-run” strategy can be used, whereby transient expression of the nuclease complex serves to permanently modify the genome. DNA transfection is the most widely reported method currently used to deliver coding sequences. In one study, transfection of TALEN pairs resulted in endogenous activation of mixed-lineage leukemia (MLL) oncogenes in human CB-derived HSPCs and leukemia in immunodeficient mice; however, activation of leukemia survival pathways in successfully edited cells may have overcome the toxicity caused by DNA transfection. 38 In another, transfection of Cas9-2A-GFP and gRNA-encoding plasmids effectively ablated B2M and CCR5 in mobilized human HSPCs. Disappointingly, engraftment of these cells in immunodeficient mice was minimal, suggesting that the transfection process may have been toxic to the most primitive HSPCs. 39 One promising study reported retained engraftment ability and 8% homozygous knockout of the CCR5 locus after transfection of ZFN plasmid DNA into human HSPCs. 40 Overall, despite its simple application, the introduction of exogenous DNA into HSPCs via electroporation remains far from ideal because of its associated potential for random recombination with the genome, DNA-related cytotoxicity, cyclic GMP-AMP synthase activation, and the disruption/activation of endogenous genes.
RNA transfection
Transfection of mRNA or mRNA analogs generated through in vitro transcription was developed as an alternative to DNA delivery and has emerged as the preferred method for ex vivo gene editing of human HSPCs. 41 In one study, electroporation of mRNA encoding a CCR5-targeted ZFN pair resulted in more than 50% indels (insertions, deletions, and mutations) in mobilized CD34+ cells, with retention of engraftment ability in immunodeficient mice. 37,42 –44 In the sole gene knockout study carried out in a clinically relevant large animal model, Peterson and colleagues delivered CCR5-targeting ZFN mRNA into pigtailed macaque HSPCs, obtaining up to approximately 64% edited cells ex vivo. After transplantation of modified cells back into the donor monkey, edited cells persisted in vivo, constituting 40% of circulating cells early posttransplantation and stabilizing at between approximately 3 and 5% in the blood 6 months later. Engraftment with edited HSPCs resulted in normal differentiation to all lineages and produced progeny cells capable of traveling to secondary lymphoid tissues. 44 Chang and colleagues achieved efficient disruption of the BCL11A locus in marrow CD34+ cells via transfection of targeted ZFN mRNA, with editing of both BCL11A alleles in up to 80% of burst-forming unit erythroid (BFU-E) and half of these with knockout/knockout alleles. This resulted in significant upregulation of hemoglobin F to levels likely high enough to inhibit hemoglobin S polymerization, thus supporting the notion of a novel sickle cell treatment based on nuclease-mediated knockout of BCL11A in human HSPCs. 45
Other Approaches
In addition to transfection of standard in vitro-transcribed mRNA, several other approaches for component delivery are under development. In one study, introduction of unmodified sgRNA and Cas9 mRNA gave rise to a modest 7% gene-editing efficiency in K562, whereas chemically modified sgRNA with improved stability yielded gene-editing efficiencies of 60–80% in the same cell line. 46 Investigators have also mixed translated Cas9 protein with sgRNA in vitro to create a ribonucleoprotein (RNP) complex, and then transfected this complex into target cells. This approach has been successful in generating indels. 46,47 The improved efficiency of Cas9 RNP in human HSPCs may be due to the ability of Cas9 protein to protect sgRNAs from degradation. 46
Transfection of nuclease-encoding mRNA—and, in the case of CRISPR/Cas9 approaches, sgRNA and Cas9 mRNA together or RNP alone—is often more efficient and definitely less toxic than DNA electroporation or viral vector delivery in terms of indel generation and gene knockout. Gene correction, however, requires the additional delivery of a corrective DNA cassette, which cannot be transported as an RNA or protein moiety. Thus, a number of groups have combined mRNA transfection of nucleases (and sgRNA for CRISPR approaches) with DNA electroporation, IDLV/AAV viral vectors, or DNA oligonucleotides in order to deliver a corrective HDR cassette. Genovese and colleagues delivered IL2RG-targeting ZFN mRNA and an IDLV-encoded corrective donor DNA template encoding green fluorescent protein (GFP) into human HSPCs from healthy donors and one patient afflicted with SCID-X1, yielding approximately 5% GFP+ cells. When transplanted into mice, modified cells engrafted and retained the ability to differentiate into progeny of multiple lineages. At 12 weeks, GFP+ cells made up approximately 2% of human cells in murine bone marrow (BM). 42 The same investigators delivered IL2RG-specific ZFN and donor DNA into mouse HSPCs, achieving a gene-editing efficacy of 6% in vitro, but rates of in vivo gene modification sank to almost undetectable levels long-term. 48 Wang and coworkers transfected human HSPCs with AAVS1 or CCR5-targeting ZFN mRNA before transduction with AAV6 carrying a GFP-encoding donor DNA, achieving insertion of the GFP cassette at the AAVS1 or CCR5 locus at rates of 17–43%. 37 This approach was particularly encouraging because it resulted in no observed loss of HPSC engrafting ability in immunodeficient mice, and the most primitive subpopulation of CD34+CD90+CD133+ cells was edited with the same efficiency as the overall CD34+ cell population. De Ravin and colleagues used AAVS1-targeting ZFN mRNA along with AAV6-delivered donor DNA to deliver a wild-type gp91phox transgene into the AAVS1 safe harbor locus of human HSPCs from patients with X-linked CGD (X-CGD). The gp91phox transgene was appropriately targeted in approximately 15% of CD34+ cells, and resulted in augmented NADPH oxidase activity in neutrophils differentiated from gene-edited patient cells. Furthermore, when modified HSPCs were transplanted into NSG mice, approximately 4–11% of human cells in the marrow expressed gp91phox long-term. This study was the first to demonstrate that codelivery of nucleases and donor DNA can lead to clinically relevant levels of targeted integration into AAVS1. The result produced by De Ravin and colleagues has potentially broad applications in the field of genome editing as it relates to treatment of monogenic disorders, given that this approach would eliminate the need for individual design and testing of new designer nucleases or sgRNA for each specific disease-inducing mutation. 43 Hoban and coworkers compared the effectiveness of introducing a human β-globin-targeted cassette with a sickle cell mutation via either transfection with a double-stranded DNA oligonucleotide or transduction with an IDLV, combined with mRNA encoding the ZFN pair, and achieved an editing efficiency of 30–40% for ZFN plus oligos and approximately 18% for ZFN plus IDLV in CD34+ cells in vitro. However, only 0.85 or 0.21% of ZFN plus oligos, or ZFN plus IDLV-treated engrafted human cells, respectively, had the sickle cell mutation in immunodeficient mice at 16 weeks posttransplantation. They also demonstrated that marrow CD34+ cells from patients with sickle cell disease, once edited with ZFN mRNA and donor oligo-corrective DNA, led to the production of wild-type hemoglobin tetramers in vitro. 49 Another laboratory reported slightly better results when they combined electroporation of a hemoglobin gene-targeted RNP with single-stranded DNA homologous to the same locus. 50 These findings lend credence to the concept of autologous transplantation with gene-edited HSPCs as a potential treatment for sickle cell disease, if efficiency can be improved.
Specificity and Off-Target Effects
Genome-editing strategies typically comprise either target integration at safe genomic harbors, such as AAVS1, or disease-specific allelic disruption/correction in an attempt to minimize collateral genomic damage like that associated with random insertion-related oncogene activation. Nonetheless, gene-editing tools can also generate unintended, permanent, deleterious changes in the genome including genomic instability, chromosomal translocation, chromosome loss, and aneuploidy. 51 NHEJ and HDR can also occur at off-target sites, often at loci homologous to the intended nuclease target. If either type of off-target mutation occurs in long-term repopulating HSPCs and alters genes or genomic loci important for cell survival, self-renewal, or proliferation, either cell death or aberrant cell expansion can occur, followed by the acquisition of secondary or tertiary mutations. Thus, the specificity and off-target effects of gene-editing systems must be key considerations in their development, particularly in terms of the potential clinical applications of these methods in human HSPCs.
Investigators' modification of the FokI domain—an alteration ensuring that heterodimerization on DNA binding is necessary to form a catalytically active nuclease complex—dramatically augmented the specificity of ZFNs and TALENs. Specifically, ZFN editing efficiency improved after an amino acid substitution at the dimer interface mediating recognition of the target site, 52 while TALEN efficiency increased after the development of an expanded set of RVD for the TAL effector recognition domain. 53 Many groups have attempted to increase the specificity of CRISPR/Cas9 editing because this system's relatively short Watson–Crick base-pairing makes it likely to cause more unintended, off-target editing than either ZFNs or TALENs. 54 Somewhat paradoxically, the use of truncated gRNAs, with shorter regions of target complementarity (17–19 bp), yielded a decrease in undesired mutagenesis 55 ; however, this truncation may also have lowered the absolute efficiency of on-target genome editing. 56 Modification of the Cas9 component so that two gRNA/Cas9 complexes are necessary to cleave DNA—achieved either via conversion to a nickase enzyme or through fusion of a “dead” Cas9 with FokI—has been shown to greatly decrease off-target editing. 57,58 Most recently, investigators developed a CRISPR/Cpf1 system that recognizes thymidine-rich PAM sequences instead of the guanosine-rich sequences recognized by spCas9. 59,60 At a genome-wide level, Cpf1 resulted in reduced levels of off-target effects compared with Cas9, possibly because Cpf1, a single-RNA-guided nuclease, does not require a tracrRNA and therefore produces cohesive DSBs. 59 Although various strategies exist to improve the specificities of genome-editing tools, few have been applied to human HSPCs to date, necessitating further investigation.
To assess the specificities of various genome-editing tools, several groups have attempted to characterize the off-target profile of these tools in HSPCs. Heckl and colleagues performed targeted sequencing of the top five off-target sites for each sgRNA used in their CRISPR/Cas9 study and reported no off-target mutations in edited cells. 26 Mandal and colleagues confirmed on-target sites (n = 5) and predicted off-target sites (n = 126) for CRISPR/Cas9 editing in human HSPCs, using capture sequencing. CCR5-targeting sgRNA generated one- or two-base indels in the highly homologous CCR2 gene, but statistical evaluation of all captured off-target sites yielded only a single site with unintentional editing (1 of 126; 0.6%), suggesting minimal off-target mutagenesis. 39 Li and colleagues analyzed the top 23 predicted off-target sites for their CCR5-specific ZFN and detected off-target modification at the CCR2 locus, a gene highly homologous to CCR5, at a frequency only 1 log lower than modification rates at the targeted CCR5 site. The loss of CCR2 in mice did not produce a deleterious effect 61 but, rather, led to a beneficial phenotype due to the combined effect of CCR2 and CCR5 in preventing HIV entry. 62 Hoban and colleagues evaluated the specificity of ZFN targeting β-globin, and discovered off-target cleavage only in the highly homologous δ-globin gene, which is known to be functionally dispensable. 49 Genovese and colleagues deep-sequenced both the intended ZFN IL2RG target site and 12 genomic loci bearing homology to the intended site, previously identified by genome-wide screening performed in K562 cells. 63 The top two off-target sites possessed minimal indels in both in vitro (at 0.17–0.7%) and in vivo samples (at 0.02%). 42
Most of the off-target studies in HSPCs to date have evaluated mutations at potential off-target sites as determined by computational prediction or screening in various cell lines. This approach, it is important to note, might overlook certain off-target sites. Whole-genome sequencing could provide a complete catalog of off-target sites; however, this method is impractical because of the high cost of the analysis and the existence of naturally occurring background mutations, possibly caused by ex vivo expansion. 64,65 The number and the pattern of off-target effects can vary widely among different target sequences within different cell types; thus, additional methods both for making off-target predictions and for evaluating off-target effects in HSPCs are needed.
Conclusions and Future Perspectives
Given that HSPC transplantation replaces some or all of the patient's bone marrow with donor cells, this procedure has the potential to treat a broad spectrum of congenital or hereditary diseases including hematologic, immunologic, and metabolic disorders. Because it is often difficult to find an HLA-matched donor, autologous transplantation with gene therapy has been proposed as an alternative treatment for patients with these diseases. Advances in genome-editing technologies have the potential to revolutionize HSPC gene therapy by the avoidance of adverse effects such as insertional mutagenesis and insufficient or dysregulated corrective transgene expression. However, the field of genome editing in HSPCs remains in its infancy, and many questions and concerns regarding this new technology are yet to be addressed and explored.
There are several hurdles preventing rapid or widespread clinical applications of HSPC gene editing. Primitive long-term repopulating HSPCs are more sensitive to DSBs than committed progenitors given their high level of p53, 66 thus decreasing HDR rates and increasing the toxicity associated with nuclease activity in these cells. Primitive HSPC sensitivity to DSBs is likely a protective mechanism, evolved to protect long-lived and proliferative stem cells from DNA damage and transformation, and we should perhaps be cautious in trying to circumvent it. Although it is not yet clear how HSPCs choose between NHEJ and HDR, the particularly poor HDR rate observed in primitive HSPCs is probably due to quiescence, or slow cycling, of these cells. 42 Shortening the length of exposure to nuclease activity may help eliminate this issue, but the benefits inherent in such a step must be balanced against the need for efficient editing. Several studies, in addition to our unpublished observations, have suggested that levels of genome editing and cell viability are inversely correlated. Furthermore, cell viability is closely correlated with engraftment rate, and the most primitive HSPCs, those responsible for engraftment, are likely also the most sensitive to nuclease toxicity. It is clearly necessary to optimize the delivery method used to transport gene-editing tools into HSPCs to avoid additional damage to engrafting cells. One possible route to optimization is first to enrich for edited HSPCs, for instance via flow-sorting of cells with the CCR5 deletion or knock-in of a selectable marker gene, followed by in vitro expansion to ensure a safe dose of engrafting cells before transplantation. However, convincing evidence for the significant expansion of actual long-term engrafting HSPCs in vitro is currently lacking, and the mutational burden associated with in vitro expansion could itself be oncogenic. 67
Almost every study reporting on genome editing of HSPCs to date has studied edited cells in vitro, or after transplantation into immunodeficient mice. It has been reported that repopulating cells in the NOD/SCID mouse contribute only to short-term repopulation and do not differentiate into all lineages. 68 –70 Only one long-term multilineage engraftment study has been conducted in nonhuman primates. 44 In this study, the levels of CCR5 disruption dropped drastically from 40% in vivo early posttransplantation to only 3–5% at 6 months after transplantation. Furthermore, even in the murine studies, gene-editing efficiencies dropped significantly over time posttransplantation. This result suggests that the “real” primitive long-term repopulating HSCs have either failed to undergo genome editing or have become so damaged postediting that they have lost the ability to self-renew or have been destroyed by the immune system and/or genome integrity-protective pathways. It is clear that further mechanistic studies must be performed to determine the underlying reason(s) for the low genome-editing efficacy consistently observed long-term posttransplantation.
Although we have listed many challenges and noted some of the significant basic and translational questions that remain to be answered, it must be said that the rapidly advancing field of genome editing has tremendous potential as applied to HSPCs and could eventually lead to life-changing treatments able to cure diseases currently beyond the reach of conventional gene addition therapies. A clinical trial relying on ZFN-mediated deletion of CCR5 in T cells from patients with HIV has already been reported, 71 in addition to a study that used TALEN-mediated knockout of endogenous T cell receptors (TCRs) to manufacture “off-the-shelf” chimeric antigen receptor (CAR) T cells, 72 and at least one trial targeting CCR5 in CD34+ cells from patients with HIV is ongoing. While investigators develop and carry out further clinical trials, gene-editing technologies targeting HSPCs will continue to aid scientists investigating hematopoiesis, stem cell biology, and leukemogenesis.
Footnotes
Acknowledgment
The authors are supported by funding through the intramural research program of the National Heart, Lung, and Blood Institute.
Author Disclosure
The authors have no relevant financial interests to declare.