
Abstract
Bispecific antibodies (BsAbs), capable of directing T cells to kill specific cancer cells by transiently binding the two cell types, have emerged as one class of promising cancer immunotherapies. However, their wide clinical application might be hampered by two deficiencies: high cost and inconvenience in drug administration. This study presents concept-proving data that these problems could be bypassed by using an enhanced nonviral DNA vector minicircle (MC) to produce BsAb in vivo. It was found that the anti-CD3/CD20 produced from the minicircle (MC.CD20) could effectively mediate the T-cell killing of multiple CD20-positive human B-cell lymphoma cell lines in vitro. More importantly, it was demonstrated that delivery of 5 μg of MC.CD20 to mouse liver via hydrodynamic injection resulted in both the expression of a therapeutic level of anti-CD3/CD20 throughout the 32-day experiment and effective anticancer activity in a B-cell lymphoma xenograft mouse model. The data suggest that MC encoding the BsAbs may become an attractive cancer immunotherapy modality based on its excellent features of safety, efficacy, and convenience in both preparation and use, and its affordability once the delivery technology matures.
Introduction
B
BsAb has proven to be very powerful in mediating anticancer activity in numerous preclinical and clinical studies. 10,19 For example, it has been shown that administration of blinatumomab, an anti-CD3/CD19 antibody, can result in a complete response in patients with B-cell lymphoma at a dose of 0.015 mg/m2/day or 0.6 ng/mL of serum. 9 Another BsAb, catumaxomab, an anti-CD3/EpCAM antibody, was shown to be effective in treating malignant ascites resulting from abdominal cavity cancers such as gastric cancer and ovarian cancer. 20 Both BsAbs are already being marketed. BsAb comprises only two scFvs connected with a short linker so that it has a small molecular weight of about 55 kDa. The small size has provided it with the advantage of more efficient penetration through the tissue than regular IgG. However, it has also the disadvantage of a short half-life of <2 h in the circulation. 10 This latter feature results in two problems: inconvenience in delivery and high cost of treatment. A pump is needed to deliver the BsAb continuously to maintain an effective therapeutic level during the treatment course, and the high cost correlates with the increased amount of BsAb used. These two problems may prevent BsAbs from wide application in clinics. It was perceived that using nonviral vectors to produce anticancer BsAb could be an excellent solution to these problems. BsAbs produced from cells carrying the genes have been proven to be a feasible way of treating malignancies. 21 The minicircle (MC), a class of enhanced nonviral DNA vectors, 22 –24 is a good candidate for the job. MC comprises almost solely the transgene expression cassette. Free of plasmid backbone DNA, MC DNA vector is devoid of multiple detrimental effects and has excellent transgene expressing profiles. 22 –24 Importantly, technology is available to produce high-quality MC in quantity at only a small fraction of the cost of producing BsAbs. 23,25 Delivery of MC to the appropriate cell type may be able to produce a therapeutic level of BsAb in the circulation for weeks or months. 26
Here, the preliminary results are presented of an anticancer study using immunocompromised NOD-SCID mice inoculated with Raji cells, a human B-cell lymphoma line, as the xenograft cancer model, and MC encoding anti-CD3/CD20 (MC.CD20) as the source of the BsAb. CD20 is a B-cell surface antigen, and anti-CD3/CD20 will erase both normal and malignant B cells. Nevertheless, it has been shown that elimination of normal B cells, either temporally by anti-CD20 × anti-CD3 diabody 11 or permanently by CAR-T armed anti-CD20, 4,27 will result in no serious side effect. The MC.CD20 was infused into the mouse liver using the established hydrodynamic injection technique. Multiple doses of human T lymphocytes were intravenously injected into the mice. Consequently, therapeutic levels of the BsAb were detected throughout the 5-week experiment, and a significant reduction in tumor burden was found that was associated with a significant increase in survival time. The results suggest that MC encoding a BsAb may be an attractive cancer immunotherapy modality once the delivery technology matures.
Materials and Methods
Cell lines and experimental animals
Daudi, Namalwa, Raji, Jurkat, and HEK293T cells were obtained from the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China), and the luciferase-expressing Raji cells were purchased from Lechen Biotechnology Co. Ltd. (Shanghai, China). Daudi, Namalwa, Raji, luciferase-expressing Raji, and Jurkat cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA) and 2 mM of L-glutamine (Gibco) in a humidified atmosphere of 5% CO2 at 37°C. HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and 2 mM of L-glutamine in a humidified atmosphere of 5% CO2 at 37°C. Human mesenchymal stem cells were cultured in DMEM/F-12 at a ratio of 1:1 supplemented with 10% FBS and 2.5 mM of L-glutamine in a humidified atmosphere of 5% CO2 at 37°C. Six- to eight-week-old female NOD-SCID mice were used for all experiments (Charles River Laboratories, Beijing, China). The mice were maintained in an individual ventilation caging system under standardized environmental conditions (20 ± 1°C room temperature, 50 ± 10% relative humidity, 12 h light–dark cycle) and received autoclaved food and bedding and acidified (pH 4.0) drinking water ad libitum. Animal welfare and experimental procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals at the authors' institution.
Production of MC.CD20
MC.CD20 was generated following the protocol of the ZYCY10P3S2T system with modifications. 23 Briefly, a seed culture was prepared using one transformed colony in 20 mL of TB with kanamycin and cultured at 37°C with shaking at 250 rpm until reaching the logarithmic phase (ODA600 = 2.0). Subsequently, an expanded culture was composed by adding 4 mL of the seed culture to 200 mL of TB containing kanamycin in a 500 mL flask. The culture was incubated at 37°C with shaking at 250 rpm for 10 h to the stationary phase. At this point, the MC formation reaction was initiated by adding equal volumes of induction mix comprising 200 mL of LB medium and 40 μL of 20% filtered L-arabinose that was adjusted to pH 7.0 using an appropriate volume of sodium hydroxide. The reaction continued with shaking at 250 rpm at 30°C for an additional 5 h before being processed to isolate the MC DNA from bacterial lysates using a commercially available affinity column (Qiagen, Hilden, Germany). The generated MC DNAs were also adjusted to 1 μg/μL using TE buffer.
In vitro stimulation and expansion of T cells
An established protocol was used to generate T cells from human peripheral blood mononuclear cells (PBMCs). PBMCs were isolated from the whole blood of healthy donors by Ficoll separation. Dynabeads® Human T-Activator CD3/CD28 (Life Technologies AS, Oslo, Norway) was added to the PBMCs as recommended by the manufacturer. To expand the cells, PBMCs were seeded onto a 24-well plate at a density of 1 × 106 cells/well in RPMI-1640 medium containing 25 μL of Dynabeads® (bead-to-cell ratio = 1:1), 100 IU/mL of recombinant human interleukin-2 (R&D Systems, Minneapolis, MN), 2 mM of L-glutamine, and 10% FBS. The stimulation procedure was performed at 37°C and 5% CO2 for different periods of time before being delivered to the mice.
Production of anti-CD3/CD20
HEK293T cells were used to produce anti-CD3/CD20. The cells were cultured in a 10 cm dish until about 70% confluence when transfected with 10 μg of MC DNA mixed with 60 μL of SuperFect® Transfection Reagent (Qiagen). After 3 h of incubation, the transfection medium was replaced with fresh medium, and the cells were continuously incubated at 37°C. The cell culture supernatants were collected 72 h after transfection. The anti-CD3/CD20 was purified by affinity chromatography using a nickel-nitrilotriacetic acid column (Merck Millipore, Billerica, MA), which captures the BsAb through binding to its polyhistidine tag. The purified BsAb was stored at −20°C until use.
Determination of BsAb binding specificity
The CD20-positive B-cell lymphoma lines, including Daudi, Namalwa, Raji, and luciferase-expressing Raji, and the CD3-positive human T cells and Jurkat cells were used for determination of antibody binding specificity. The 500 μL reaction comprising 10 ng of BsAb and 5 × 105 cells was incubated for 30 min on ice. After washing twice with phosphate-buffered saline (PBS), the cells were mixed with 5 μL of His Tag APC-conjugated Antibody (R&D Systems, Minneapolis, MN) and incubated on ice for 30 min. Subsequently, the cells were washed twice with PBS and analyzed by fluorescence cytometry using a FACSCalibur system (BD Biosciences, San Jose, CA). B-lymphoma cell lines and T-cell lines were reacted with APC mouse antihuman CD20 and FITC mouse antihuman CD3 (BD Biosciences) as positive controls, respectively. Data analysis was performed using FlowJo7.6.1 software.
Western blot
The BsAb in the supernatant of the culture and within the cells was determined using Western blot. Cell samples were lysed with radio immunoprecipitation assay lysis buffer (Beyotime, Haimen, China), and the protein concentrations were determined using a BCA kit (ThermoFisher Scientific, Waltham, MA). The whole-cell lysates were subjected to 10% reducing SDS-polyacrylamide gel electrophoresis (PAGE) before transfer onto PVDF membranes (Merck Millipore). The membranes were blocked with 5% skim milk for 1 h at room temperature and incubated with the primary antibody of the mouse anti-flag (Sigma-Aldrich, St. Louis, MO) at 4°C overnight. After washing twice with PBS, the membranes were incubated with HRP-conjugated anti-mouse IgG secondary antibody (Cell Signaling Technology, Danvers, MA) at 4°C for 1 h. The BsAb bands were visualized by using an enhanced chemiluminescent method kit (Cell Signaling Technology). As an internal control for equal protein loading, blots were stripped and probed with antibodies against β-actin (Proteintech, Rosemont, IL).
Mouse cancer xenograft model
Eight-week-old female NOD/SCID mice were conditioned with 20 mg/kg cyclophosphamide (CTX; Sigma-Aldrich) by three daily intraperitoneal injections from day −3 to −1, followed by one intravenous injection of 5 × 105 Raji cells on day 0. On day 2, 5 μg of MC.CD20 in saline with the volume adjusted to 10% of body weight was delivered to the groups of mice using the hydrodynamic injection procedure. Subsequently, groups of mice were given three courses, two doses each and 3 days apart, of human T lymphocytes via tail-vein injection. In each case, the injection volume of the cell suspension was 0.2 mL/mouse. Tumor growth was monitored by bioluminescence imaging. Mice were investigated once per day for health status. Mice that developed hind-limb paralysis were killed by cervical dislocation, and survival time was measured for the evaluation of therapeutic efficacy.
Quantitative bioluminescence
Bioluminescence images were collected on an IVIS Spectrum in vivo imaging system from Caliper Life Sciences (Hopkington, MA) using a cryogenically cooled charge-coupled device camera. Spectrum Living Image 4.0 software (Caliper Life Sciences) was used to acquire and quantitate the bioluminescence imaging data sets. About 10 min before imaging, a single intraperitoneal injection of 150 mg/kg D-luciferin (PerkinElmer, Waltham, MA) in Dulbecco's PBS was administered to each mouse. Subsequently, the animals were induced using isoflurane (Keyuan, China) and positioned within the imaging chamber under anesthesia. Five mice were imaged simultaneously with an exposure time of 2 min. The dorsal images were obtained, and signals were quantified through region of interest (ROI) analysis for each animal. The resulting signal summations were normalized to the ROI area so that all measurements are given in photons/s/cm2/sr. For each image, the normalized background signal from similarly sized ROIs was subtracted.
Pharmacokinetic analysis of anti-CD3/CD20 BsAb
The NOD-SCID mice were injected via hydrodynamic tail vein (HTV) injection with 5 μg of MC.CD20. Whole blood of the animals was obtained by retro-orbital bleeding at 1, 3, 7, 12, 16, and 23 days after HTV injection. Serum was prepared by separation of coagulated whole blood. The anti-CD3/CD20 BsAb serum concentration was quantified by a His Tag Enzyme-Linked Immunosorbent Assay Detection Kit (GenScript, Nanjing, China). The His tag plate in the kit is coated with His-tagged proteins. After addition of 50μL of His tag standards or serum to each well of the His tag plate, 50 μL of anti-His monoclonal antibody was added to all wells. The plate was incubated for 30 min at 25°C and washed four times. After the addition of 100 μL of antibody tracer to all wells and incubation of the plate for 30 min at room temperature, the plate was washed four times and incubated for 10–15 min with 100 μL of TMB substrate. Stop solution was added, and absorbance (450 nm) was read on a Multiskan GO system (ThermoFisher Scientific).
Statistical analysis
All data are expressed as mean ± standard deviation (SD). Each experiment was repeated at least three times. For comparisons of three groups, the values were analyzed by one-way analysis of variance with the Bonferroni's post-test. Linear regression analysis was performed to compare the luminescent signal of Raji and T-cells and MC.CD20. Survival was analyzed by the Kaplan–Meier method and log-rank test. Differences were considered significant when p < 0.05. GraphPad Prism 5 software was used for statistical analysis.
Results
Construction of parental plasmid to produce anti-CD3/CD20
The parental plasmid for production of the MC.CD20 was constructed by inserting the transgene expression cassette into the AgeI and SalI sites of the empty plasmid pMC.BESPX (Fig. 1A and B).
23
The nucleotide sequences of anti-CD3/CD20 are shown in Supplementary Table S1 (Supplementary Data are available online at

Production of minicircle (MC) DNA vector expressing anti-CD3/CD20 bispecific antibodies (BsAb; MC.CD20). (
Fluorescence-activated cell sorter determination of anti-CD3/CD20 binding specificity
Binding of the anti-CD3/CD20 to epitope CD3 was determined by fluorescence-activated cell sorter (FACS) analysis using CD3-positive human T lymphocytes and the Jurkat cell line of T-cell leukemia (Fig. 2A). Similarly, the binding specificity of the BsAb to the CD20 epitope was determined using a number of CD20-positive B-lymphoma cell lines, including the Daudi, Namalwa, and Raji lines. No binding was detectable when the BsAb was incubated with human umbilical mesenchymal stem cells (HuMSC), which express neither CD3 nor the CD20 epitope (Fig. 2B). These observations confirmed the expected dual specific binding activity of the anti-CD3/CD20.

Binding specificity of anti-CD3/CD20 illustrated by fluorescence-activated cell sorter (FACS). (
Anti-CD3/CD20-mediated T-cell killing of lymphoma cells in vitro
Next, the ability of the anti-CD3/CD20 to mediate T-cell killing of the CD20-positive target cells in vitro was tested using the fluorochrome calcein acetomethoxy (AM)-based cytotoxicity assay. The anti-CD3/CD20 was highly active in mediating T-cell killing of the Raji cells at an E:T ratio of 4:1 after incubation at 37°C for 4 h, as evidenced by the heavier loss of the fluorochrome-labeled Raji cells compared with the culture without the BsAb (Fig. 3A). The mechanism underlying the aggregation of the Raji cells in the two cultures with T lymphocytes is not known at present, although the T cell is the obvious mediator.
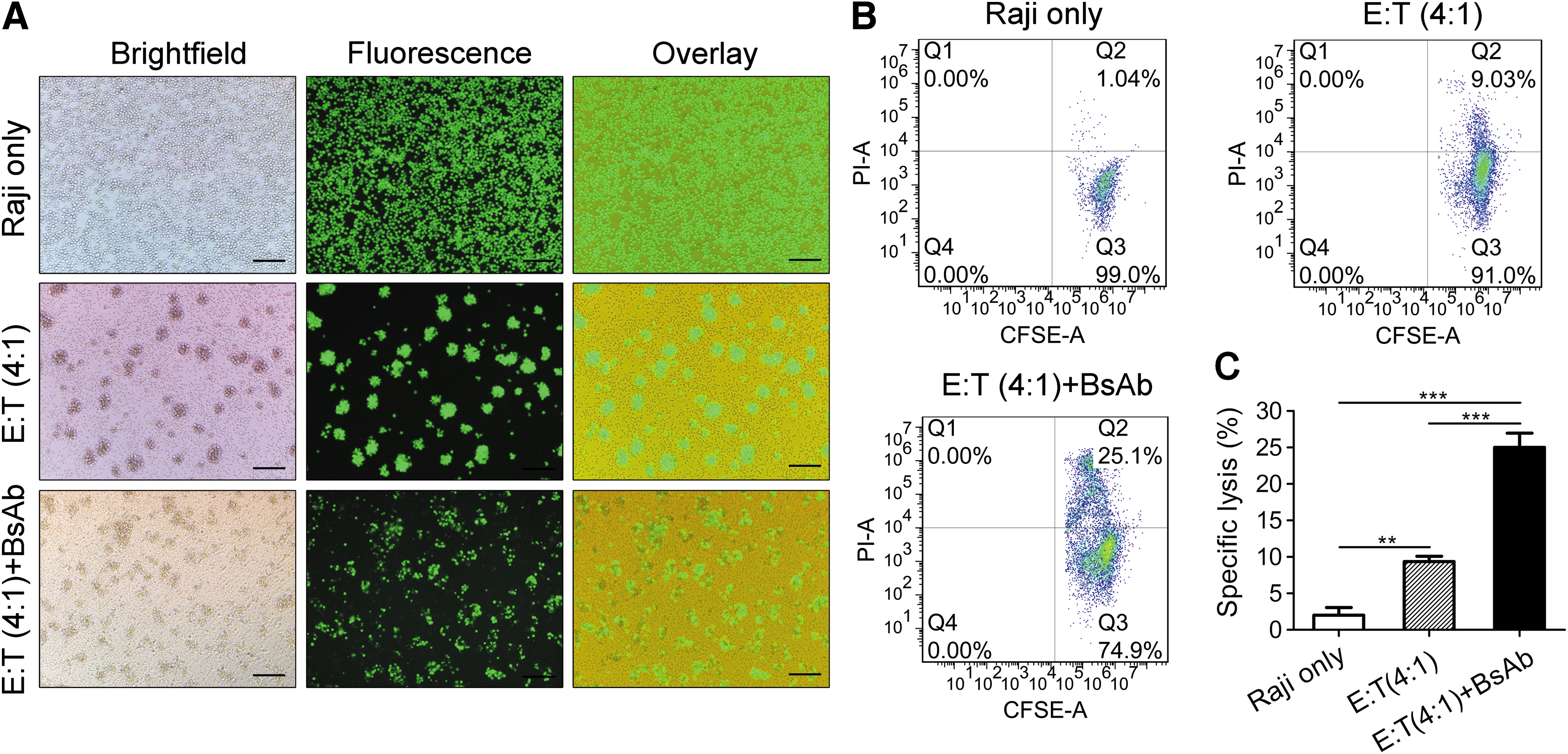
Determination of anti-CD3/CD20-mediated T-cell killing of cancer cells. (
A FACS assay was conducted to quantify the BsAb-mediated T-cell killing of the target cells in the experiments as described above. Here, all of the Raji cells were labeled with the carboxyfluorescein succinimidyl ester (CFDA-SE), whereas the cells killed during the 4 h incubation were labeled with propidium iodide (PI). Consequently, the alive (CFDA-SE alone) and dead cells (CFDA-SE/PI double staining) could be quantified by the FACS assay. Almost no dead cells were detected in the control group comprising Raji cells only. In contrast, the proportion of dead cells in the co-cultures of Raji cells and T cells with or without the anti-CD3/CD20 was 25.0 ± 1.9% and 9.4 ± 0.7%, respectively (Fig. 3B), and was significantly different (Fig. 3C), suggesting that the anti-CD3/CD20 was highly effective in directing the T-cell killing of the B-cell lymphoma cells.
Anticancer effect of MC.CD20 in vivo
In a preliminary experiment, the collected anti-CD3/CD20 serum from murine models was we tested to see if it was functional in vitro. Strong Raji cell-killing activity was found using as little as 0.61 ng of anti-CD3/CD20 from the serum of mice in each 200 μL reaction. The blank serum served as a negative control (Supplementary Fig. S1). Subsequently, a Raji cell xenograft mouse model was used to test whether the anti-CD3/CD20-mediated T-cell killing of the cancer cells in vitro could be translated into an anticancer therapeutic effect in vivo (Fig. 4). Groups of immunocompromised NOD/SCID mice were inoculated with Raji cells carrying an integrated luciferase gene (5 × 105 in each mouse). Two days later, a single dose of 5 μg of MC.CD20 was delivered to the liver of each mouse in selected groups of mice via hydrodynamic injection. Subsequently, groups of mice were intravenously injected with three courses of in vitro-expanded human T lymphocytes, 4 days apart and at two doses each of 1 × 107 cells per dose. Three doses of CTX were administered beforehand to promote the engraftment of the T cells (Fig. 4A). In the mice receiving the MC.CD20, the level of anti-CD3/CD20 as determined by enzyme-linked immunosorbent assay was as high as 6,921 ± 1,753 ng/mL serum (mean ± SD) 24 h after DNA infusions, but dropped sharply to about 1,200 ± 354 ng/mL on day 3 and 112 ± 49 ng/mL on day 7, followed by a period of slower decrease. On day 23, the serum BsAb was 23 ± 10 ng/mL on average for the five surviving mice, although all of them died before the next scheduled BsAb determination time. Conceivably, all five mice had therapeutic levels of BsAb until termination (Fig. 4B). The BsAb expression pattern was similar to that of other reporters expressed from the MC delivered through hydrodynamic injection. 22 The cancer burden of each mouse was determined by periodically measuring the quantum dot of the luciferase fluorescence using a Caliper Life Sciences small animal in vivo imaging system (Fig. 4C). Judging from the intensity of the fluorescence, which was approximately proportional to the number of Raji cells in the mouse, it could be seen that the cancer burden was less in the T-cell group and the least in the T-cell plus MC.CD20 group compared with the Raji cell only group from day 11 to 15 (Fig. 4C). The same order of cancer burdens of the groups is shown in the chart made using the accurately measured quantum number (Fig. 4D). Consistent with the cancer burden, the same order of the survival times of the mice in the groups was revealed in the survival chart (Fig. 4E). Compared with the mice in the untreated and T-cell-treated groups, those in the combined treatment group survived significantly longer (Fig. 4F). Thus, consistent with the in vitro experiment, these data suggested that the anti-CD3/CD20 expressed from MC.CD20 has an anti-human B-cell lymphoma therapeutic effect in vivo.

Anticancer efficacy of MC.CD20 in the mouse cancer xenograft model. (
Discussion
This study demonstrated that when delivered to the liver of the mouse, the MC.CD20 encoding the anti-CD3/CD20 exerted a therapeutic effect in anti-B-cell lymphoma similar to that of the purified anti-CD3/CD2011 or CAR-T facilitated with the same anti-CD20 scFv. 4,27 Consistent with the high efficiency in mediating T-cell killing of cancer cells in vitro, the anti-CD3/CD20 expressed from our MC.CD20 could significantly reduce the cancer burden and prolong the life-span of the mice. Therefore, this study established MC encoding the BsAb (MC.BsAb) as a potential therapeutic modality in cancer immunotherapy.
The effector element of MC.BsAb is the BsAb generated from the MC in vivo. Compared with the purified BsAb, MC.BsAb has many advantages. First, MC is more stable than the BsAb, which is fragile, and it is hard to exclude the possibility of whether BsAb's degraded products would result in unexpected side effects. Second, MC cost much less than BsAb because MC needs only a fraction of the expense of BsAb in every step of production, storage, transportation, and delivery. Third, MC.BsAb is more convenient in clinical applications because a dose of the DNA drug will be able to generate a therapeutic level of BsAb for days, weeks, or longer, whereas BsAb requires a pump for continuous delivery to maintain an effective level due to its short half-life. 10 Finally, MC.BsAb fits into personalized cancer immunotherapy better than BsAb because of the much lower production time and cost to make a MC.BsAb than a BsAb. It has been well documented that each cancer is unique and needs a personalized therapeutic protocol or a unique set of BsAb targets. So time and cost become critical factors in determining clinical potential. 10
Although immunotherapy is very effective in treating hematological malignancies and melanoma, 29 the high cost and long preparation time may prevent the wide application of adoptive cell transfer technologies such as TIL and CAR-T in clinics. 10,30 Furthermore, TIL technology can obtain only one or a limited number of cancer-specific CTL clones from the surgical cancer samples. 31 –33 This may become a major obstacle that prevents its extension from melanoma to other solid cancers. CAR-T technology can solve the problem for a limited number of cancer-specific CTL clones. However, it imposes a safety concern because the viral integration in CAR-T is a mutation event known to have the risk of inducing cancer. 34 The cancer concern may be eliminated by the newly developed technology for making universal CAR-T using the more accurate CRISPER/Cas9-mediated targeting integration technology. 35 However, this new technology, which is associated with a laborious and time-consuming cell culture step, will not significantly improve the ability of CAR-T to compete with MC.BsAb in terms of safety, cost, and flexibility.
Multiple ex vivo technologies have been tried in order to express BsAbs in vivo, for exampledelivering T cells transfected with BsAb mRNA 12 or T cells with integrated BsAb genes. 13 However, these technologies involve cell culture and viral integration, 30 so that will not be able to compete with the MC.BsAb technology in terms of safety, cost, and time frame.
Nevertheless, the MC.CD20 protocol in the present study will require multiple improvements before it can become clinically applicable. For example, a clinically acceptable DNA delivery technology is needed to replace the hydrodynamic injection technique. Initially, this study tried to use the jetPEI transfection reagent, which has already been tested in clinical trials, 36 but it was not possible to reach a therapeutic level of BsAb while simultaneously avoiding toxicity. Moreover, technology has to be developed to allow the MC.BsAb to generate a stable and safe level of BsAb in vivo. In the present study, the rise-and-fall pattern of the serum level of BsAb may be very risky in clinical use in that it may result in the sudden generation of a massive amount of risky cytokines, especially when the cancer load is high.
Footnotes
Acknowledgments
This work was supported by government funds from Shenzhen, China (SFG 2012.566 and SKC 2012.237), the Natural Science Foundation of Guangdong Province of China (2014A030310335), the China Postdoctoral Science Foundation (2015M582446 and 2016M592554), and the Science and Technology Foundation of Shenzhen, China (JCYJ20150521094519466).
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.