
Abstract
This study describes the initial testing of a novel strategy for neutralization of lentiviruses using the fundamental biology of enveloped viruses’ assembly and budding. In the field of gene therapy, viral vector surface proteins have been manipulated in order to redirect host cell specificity by alteration of pseudo-types. This study tested whether known viral pseudo-typing proteins or surface proteins known to be recruited to the human immunodeficiency virus (HIV) envelope could be engineered to carry neutralizing epitopes from another microorganism onto the lentiviral surface. The results identify ICAM1 as a novel vehicle for lentiviral pseudo-typing. Importantly, the study shows that in a model lentiviral system, ICAM1 can be engineered in chimeric form to result in expression of a fragment of the tetanus toxoid on the viral membrane and that these viruses can then be neutralized by human serum antibodies protective against tetanus. This raises the possibility of delivering chimeric antigens as a gene therapy in HIV-infected patients.
Introduction
I
Here, a novel strategy is proposed to circumvent the ability of HIV to evade immune clearance, and data are presented using lentiviral models that support initial proof of concept. This strategy is called Trojan insertion. HIV infection is characterized by continual cycles of immune evasion due to the rapid selection of escape mutations. 10 –13 This strategy involves forcing HIV virions to express on their surface immunogens from other pathogens, to which there is a pre-existing memory response that can rapidly and decisively clear the emerging virus.
Lentiviruses such as HIV are enveloped by the host cell plasma membrane, which coats the virus as it buds from the cell. Some host cell plasma membrane proteins may be actively recruited to sites of HIV budding and can increase infectivity. 14,15 This might be exploited if host cells can be made to express immunogens on their surface in a form that can be incorporated onto budding virions. To this end, chimeric proteins have been designed (Trojans), which consist of a transmembrane domain from proteins known to be incorporated into lentiviral envelope membranes artificially fused to an immunogenic extracellular domain.
The extracellular antigen selected for the model experiments was the tetanus fragment C (TetFrC) antigen. Tetanus toxoid has been historically used with great success for immunization of humans against Clostridium tetani. The serum of immunized humans has been shown to contain anti-toxoid neutralizing antibodies that can be administered as a passive therapy against tetanus infection. 16 In addition, a modified fragment of the tetanus toxin has been shown to be an effective adjuvant fusion molecule to stimulate T-cell responses against human cancers. 17,18
Two potential membrane anchors were tested for fusion to the TetFrC extracellular domain. The first is the vesicular stomatitis virus glycoprotein (VSVg). This is a virus attachment and fusion protein, which confers viral tropism for a wide variety of cell types and has been shown to be successfully incorporated into the surface membrane of lentiviral vectors. 19,20 These properties have been successfully utilized for pseudo-typing many recombinant lentiviral vectors. This protein has been genetically engineered to bear the 52 kDa protein streptavidin in place of its native attachment and fusogenic extracellular domains. Kaikkonen et al. showed that virions expressing streptavidin in this way on their membrane could be targeted to a particular cell type using bridging biotinylated antibodies against cell type–specific markers. 21
The second transmembrane anchor is derived from ICAM1. This protein has been shown to be recruited to the HIV surface via a direct interaction with the HIV protein gag and, as a result, may be present at relatively high levels on free virions. 14,22 ICAM1 has been extensively studied, and the exonic sequences contributing to its transmembrane and cytoplasmic domains are well-defined.
Having designed the chimeric Trojan proteins, this study set out to test whether they can be used to coat lentiviral vectors and whether viruses so coated can be neutralized by a simulated human immune response. The data show that the Trojan proteins can transferred by human lentiviral vector packaging and T-cell lines both transiently and stably. Furthermore, it is shown that lentiviral vectors packaged in the presence of Trojan proteins are measurably infectious and that this infectivity can be neutralized by human anti-tetanus serum antibodies.
Materials and Methods
Chimeric constructs
Chimeric cDNA constructs TV, TI, SV, and SI (Fig. 1) were designed with a four-domain structure consisting of a signal peptide, sequences encoding a 5′ FLAG
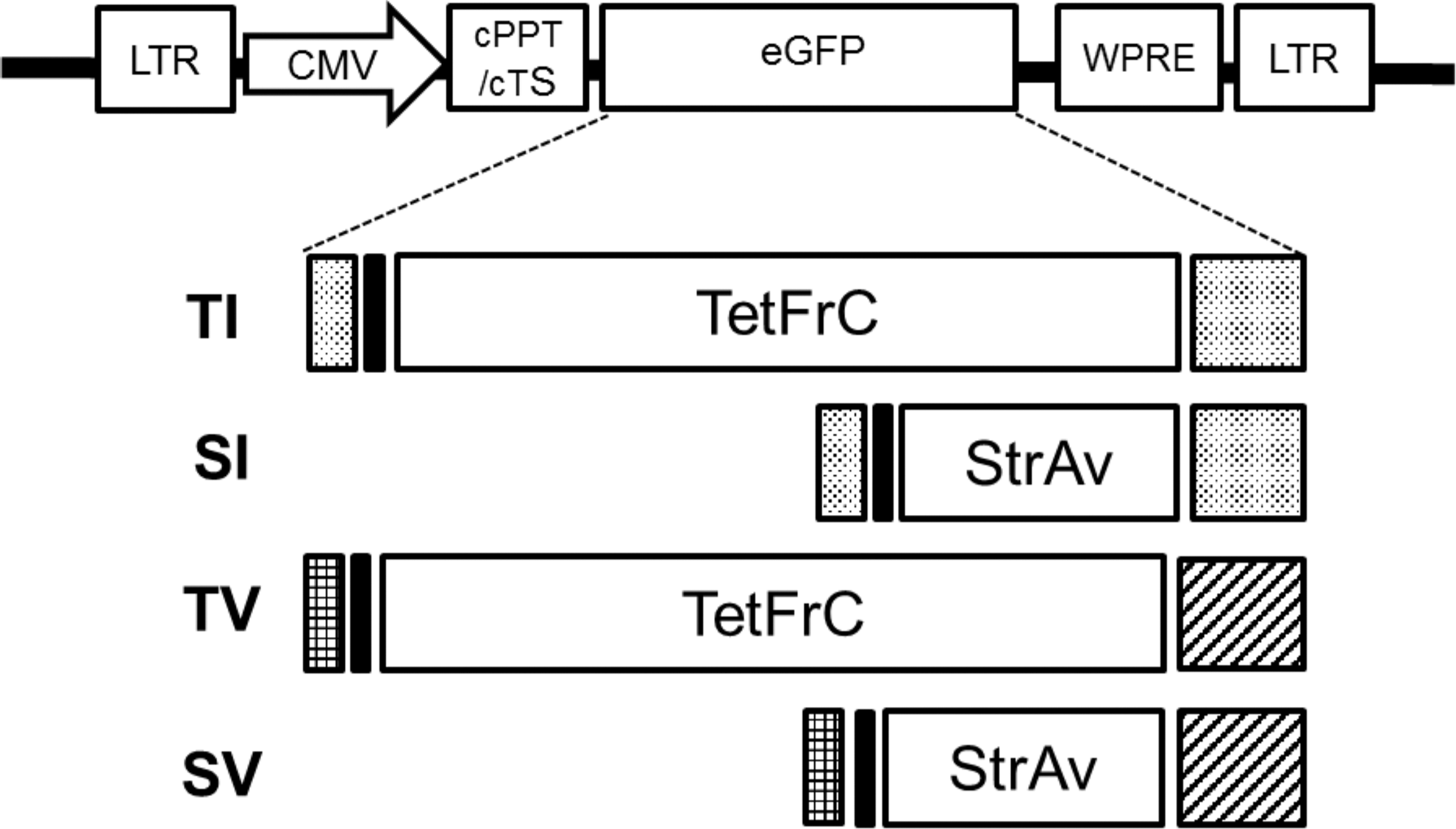
Schematic structure of lentiviral vector and chimeric constructs. Top diagram shows the lentiviral enhanced green fluorescent protein (eGFP) transgene transfer vector pRRLsc_CEW used for control lentivirus production in this study. Below is the domain structure of chimeric transgenes TI, SI, TV, and SV. The TV and SV constructs consist of a gp64 signal peptide (black-checked rectangle, 21 amino acids [aa]), followed by FLAG®-epitope (black rectangle, 9 aa) tagged TetFrC (451 aa) or streptavidin (StrAv, 159 aa) extracellular domain, fused to the transmembrane and cytoplasmic domains (T/C) of vesicular stomatitis virus glycoprotein (Black diagonal-striped rectangle, 72 aa). In the TI and SI constructs, FLAG-tagged tetanus fragment C (TetFrC) or streptavidin extracellular domains are fused to ICAM1 signal peptide (light-gray rectangle, 26 aa) and T/C (light-gray rectangle, 64 aa).
The TetFrC sequence was provided by J. Rice (Southampton University, United Kingdom). 17,23 Sequences for gp64 signal peptide, VSVg transmembrane, and cytoplasmic domain were provided by M. Kaikkonen (University of Kuopio, Finland). 21 ICAM1 signal and transmembrane and cytoplasmic domain sequences were identified from published sequences 24 and NCBI entry CCDS12231.1.
Chimeric cDNA constructs were produced by GeneArt® (Life Technologies). Chimeric cDNA constructs TV and TI were subcloned using EcoRV and NheI restriction enzymes (New England Biolabs) to the pRRLsc_C_W self-inactivating integrating lentiviral vector plasmid with a central polypurine tract/central termination sequence. This vector encodes a lentiviral transfer cassette with expression driven from an hCMV promoter with a Woodchuck hepatitis virus post-transcriptional regulatory element. Resulting plasmids were designated as TV and TI, respectively, and were subjected to Sanger sequencing to confirm chimeric gene sequences. To create an enhanced green fluorescent protein (eGFP)-expressing control virus (LVGFP), the pRRLsc_CEW transfer plasmid was used and has been previously described. 25
Streptavidin cDNA was recovered by FLAG tag primer extension proof-reading PCR amplification of streptavidin sequences from plasmid pCMV-SA-VSV-GED (provided by M. Kaikkonen, University of Kuopio, Finland) with first-round primers forward CAAGGACGATGACGACAAGGACCCCTCCAAGGAC and reverse ATCCCGGGCTGCTGAACGGCGTCGAG and for second-round amplification forward primer ATAGGATCCATGGACTACAAGGACGATGACGACAAG. PCR products were digested with XmaI and BamHI and subcloned into plasmids TV and TI using BamHI and AgeI enzyme sites to create SV and SI, respectively. Sanger sequencing was used to confirm chimeric gene sequences.
Cell lines and culture
Human embryonic kidney (HEK) 293T and human fibrosarcoma HT-1080 cell lines were obtained from ATCC. The PM1 T-cell line was obtained from National Institute for Biological Standards and Control (United Kingdom). All cell lines were cultured in high glucose (4.5 g/L) Dulbecco's modified Eagle's medium (DMEM; PAA) with stable glutamine. Unless otherwise indicated, medium was supplemented with 10% fetal bovine serum (Gibco), 100 IU/mL of penicillin, and 100 μg/mL of streptomycin (Gibco).
Transient expression of chimeric constructs
HEK 293T cells (1 × 106) were grown for 24 h. Cells were then transfected with 5 μg of lentiviral transfer expression cassette plasmids (TV, TI, SV, and SI) using Lipofectamine® (Life Technologies) according to the manufacturer's instructions. On day 1 post transfection, cells were removed from flasks using Trypsin EDTA (Gibco), washed, and returned to new flasks with fresh media. On day 3 post transfection, cells were harvested using 0.5 mM of EDTA in phosphate-buffered saline (PBS; Sigma–Aldrich) and stained with either 5 μg/mL of mouse M2 anti-FLAG antibody (Sigma–Aldrich) or neat mouse anti-TetFrC hybridoma supernatant 31e11 (kindly provided by C. Watts, University of Dundee, United Kingdom). Negative control cells, mock transfected in the absence of plasmid, were stained with 5 μg/mL of isotype control antibody mouse IgG1. The secondary antibody in each case was goat anti-mouse Alexa Fluor® 647 (GaM647; Life Technologies). After staining, cells were fixed with 2% paraformaldehyde, and singlet cells were analyzed by flow cytometry using a FACS Canto II machine (Becton Dickinson). FACS plots and associated measurements were generated using FlowJo software v8.8.6 (Treestar, Inc.).
For further quantitation of chimeric protein expression, 0.2 × 106 293T cells were transfected 24 h post plating with 1.6 μg of transfer cassette plasmid using calcium phosphate, which was to be used in lentiviral packaging. Gene expression was detected at 48 h by antibody staining, as described above. Statistical analysis was performed using GraphPad Prism v6. The levels of gene expression were compared by ordinary one-way analysis of variance (ANOVA) with Tukey's post-hoc test for multiple comparisons.
Lentiviral vector production
Lentiviral vectors were produced by calcium phosphate mediated transfection into HEK-293T cells, using VSVg pseudotype for all vectors. Plasmids used for lentiviral production are as previously described. 26 Cells were transiently transfected with 12.5 μg of packaging plasmid (pMDLg/pRRE), 6.25 μg of pRSV-REV, 7 μg of pMD2.VSV-G, and 25 μg of transfer plasmid.
Viruses were titrated for transducing units/mL by transduction with limiting dilutions and FACS, as previously described, 26 using the HT1080 cell line. Cytoplasmic eGFP gene expression in singlet cell populations was measured directly in the FITC channel. Surface chimeric Trojan protein expression was detected by binding of the M2 anti-FLAG antibody (Sigma–Aldrich), as described above. The percentage of cells expressing surface FLAG epitope above background detected in mock-transduced cells was measured in the APC channel. Mean titers for each lentiviral vector were compared by ordinary one-way ANOVA with Tukey's post-hoc test for multiple comparisons using GraphPad Prism v6.
Lentiviral transduction
To test the production of chimeric proteins in cell lines, 5 × 105 cells were transduced at a multiplicity of infection (MOI) of 1 in DMEM 10% FCS in the presence of 8 μg/mL of polybrene (Sigma–Aldrich). On day 3 post infection, half the cells were analyzed for surface FLAG expression by antibody staining and FACS, as described above. The remaining cells were subjected to clonal dilution (3 cells/mL) and distributed at 200 μL/well to 96-well round-bottom plates. Wells containing growing colonies were expanded until enough cells were available to be sampled for surface FLAG epitope expression by M2 antibody binding and FACS, as described above.
Immunoprecipitation and Western blotting
Lentiviral suspensions were prepared as described above. As a positive control for immunoprecipitation, cell lysates were prepared from 293T cells transiently transfected with TI and 24, as described above. Cells were lysed at 50 × 106 cells/mL in ONYX buffer (20 mM of Tris [pH 7.4], 140 mM of NaCl, 1 mM of EGTA, 1% Triton, 10% glycerol, 50 mM iodoacetamide, and protease inhibitor cocktail [Roche]) according to standard methods.
The protein concentration of viral preparations and cell lysates was analyzed using the Micro BCA kit (Perbio) according to the manufacturer's instructions. Protein (10 μg) from each sample was subjected to immunoprecipitation, as previously described, 27 using human anti-tetanus polyclonal serum IgGs (NIBSC reference antibody TE-3) or isotype human polyclonal IgGs (Sigma–Aldrich). Recovered beads were washed and treated with PNGase F (New England Biolabs). Immunoprecipitated proteins were released from beads during denaturation, as described by the manufacturer, into LDS sample buffer (Life Technologies) with the addition of 50 μM of DTT (Sigma–Aldrich).
After SDS-PAGE and blotting, PVDF membranes were probed with HRP-conjugated M2 anti-FLAG antibody (Sigma–Aldrich), followed by chemiluminescent detection using ECL™ reagent (GE Healthcare). As an additional control, 1 μg of each lentiviral preparation was left unprecipitated, denatured, treated with PNGase F, and then subjected to SDS-PAGE and Western blotting as for the immunoprecipitated proteins.
Neutralization assay
Vector (6.5 × 105 transducing units) was pretreated in 100 μL of PBS for 30 min with 100 μg of human anti-tetanus polyclonal IgG antibody (NIBSC), described above, or with 100 μg of human IgG (hIgG) isotype control antibody (Sigma–Aldrich). Then, 105 HT1080 cells were transduced in standard growth medium for 18 h before vector was removed, and the cells washed with PBS to remove any unbound antibody or viral vector. 48 h post-infection cells were fixed with 2% paraformaldehyde and analyzed by FACS for expression of the relevant transfer cassette, as described above.
The mean percentage of gene expression positive cells above background (from mock-transduced cells) was calculated. Mean gene expression resulting from successful transductions was calculated from a minimum of three transduced wells for each combination of virus and antibodies. Error bars were calculated as mean ± standard deviation (SD). Statistical analysis was performed using GraphPad Prism v6. The effects of the different treatments on transgene gene expression as a proxy for virus infectivity were compared using one-way ANOVA, with Sidak's multiple comparisons post-hoc test comparing the percentage transduction by each treated vector with the corresponding PBS-treated vector control.
Results
Transient expression of chimeric constructs results in surface membrane protein expression
In order to be incorporated into the lentiviral membrane, chimeric proteins must first be incorporated into the host cell membrane. Since the plan was to use 293T cells as packaging cells for lentiviral vector production, first it was important to demonstrate that the novel chimeric proteins could be expressed from the lentiviral transfer expression cassette plasmids onto the surface plasma membrane of these cells.
Initially, the aim was to test whether commercially produced and well-characterized M2 anti-FLAG antibody staining could be used as a marker for TetFrC-chimeric protein expression. To this end, 293T cells were transiently transfected with plasmids TV, TI, SV, or SI, harboring FLAG-tagged TetFrC or streptavidin chimeric protein genes, using Lipofectamine®. On day 3 after transfection, cells were harvested and analyzed for chimeric protein expression (Fig. 2a) by FACS analysis of surface binding of the anti-FLAG epitope antibody M2 (black line plot), or of anti-TetFrC hybridoma supernatant 14e11 (dashed line plot). As a negative control, a sample of each transfectant was stained with an isotype control mouse IgG primary antibody (gray-filled plot).

Transient transfection of lentiviral vector plasmids carrying Trojan constructs.
In TV and TI transfections, surface expression of chimeric proteins was detectable using anti-FLAG epitope antibody M2 in parallel to 14e11 anti-tetanus hybridoma supernatant. Therefore, M2 anti-FLAG antibody staining was used as a marker for TetFrC expression in subsequent experiments.
In SV and SI transfections, surface expression of chimeric proteins was also detectable using anti-FLAG epitope antibody M2. As expected, the anti-tetanus hybridoma supernatant did not bind to the streptavidin extracellular domain-bearing chimeric proteins.
In addition, samples of transfected cells were harvested with trypsin/EDTA instead of EDTA alone, and also with and without fixation to make sure that no epitopes to be detected in later experiments were trypsin or paraformaldehyde sensitive. There was no evidence for a decrease in either anti-TetFrC or anti-FLAG epitope antibody binding with either of these treatments (data not shown).
In order to compare surface expression levels of the different chimeric proteins quantitatively, in experiments independent from the Lipofectamine-mediated transfections, 293T cells were transiently transfected with transfer plasmids using calcium phosphate, which is used in lentiviral packaging, and analyzed for surface expression of chimeric proteins. Plots from representative wells are shown in Fig. 2b. The percentage of positive cells above background and median fluorescence intensities (MFIs) were measured for all wells, and Fig. 2c shows the mean ± SD for each measurement in independent transfections.
The TetFrC-VSVg chimeric plasmid (TV) produced a population of cells with a mean of 48 ± 3% surface FLAG expression, which were detected with a MFI of 714 ± 20 (Fig. 2c). For the TetFrC-ICAM1 chimera (TI), a mean of 77 ± 3% of cells demonstrating anti-FLAG staining above background was observed, and with a MFI of 1306 ± 79 for this population. In the case of the matched streptavidin control chimeras (streptavidin-VSVg, SV, and streptavidin-ICAM1, SI), cells transfected with SV were 60 ± 3% FLAG-positive, with a MFI of 854 ± 50, and 293T cells transfected with SI showed 71 ± 9% surface FLAG-positive cells and a MFI of 1317 ± 468.
Statistical analysis showed that transient transfection with TV (TetFrC-VSVg) resulted in significantly lower mean percentage of FLAG-positive cells than TI and SI but not SV. The greatest significance was seen when comparing transient transfectants of TetFrC-VSVg (TV) with TetFrC-ICAM1 (TI). In addition, SV (streptavidin-VSVg) transfectants had a significantly lower percentage of FLAG-positive cells compared with TI (TetFrC-ICAM1)–transfected cells. MFI comparison did not result in any significant difference between the four different chimeric proteins on the surface of positive cells. These results showed that, with some variation, each chimera could be expected to be expressed on the surface of the cell line to be used for lentiviral vector packaging.
Lentiviral transfer cassettes bearing chimeric constructs can be incorporated into infectious lentiviral vector particles but with variable titers
In order to be able to detect whether lentiviral vectors could incorporate TetFrC chimeras and thus be susceptible to neutralization with anti-TetFrC antibodies, first there was a need to test whether transfer cassette expression in transduced cells could be used to measure infectivity. Transfer plasmids TV, TI, SV, and SI were therefore co-transfected to 293T cells with lentiviral packaging plasmids to create VSVg-pseudotyped lentiviral vectors LVTV, LVTI, LVSV, and LVSI, respectively. On day 2 post transfection, supernatants were harvested and ultra-centrifuged to recover lentiviral particles. Lentiviral preparations were then titrated by transduction of HT1080 fibrosarcoma cells and measurement of transfer cassette expression through binding of anti-FLAG antibody. At least three separate lentiviral preparations were produced for each virus, and putative chimera-bearing viruses were prepared alongside a well-characterized, lentiviral vector expressing cytoplasmic eGFP protein as a positive control for virus manufacture.
The titers (transducing units/mL) produced for each virus are shown in Table 1. Transfer plasmids bearing chimeric constructs TV, TI, SV, and SI were shown to be packaged into lentiviral particles, and detection of chimeric protein expression on target cells through detection of the FLAG epitope could then be used to detect infectivity of lentiviral preparations. Variation in mean titers for viruses LVGFP, LVTV, LVTI, and LVSI did not reach statistical significance. However, for LVSV (streptavidin-ICAM1), the trend was for lower titers, with one batch producing no detectable titer. Therefore, the study proceeded by focusing on the LVTI (TetFrC-ICAM1) and negative control LVSI (streptavidin-ICAM1) pair.
Titer transducing units/mL in HT1080 cells
Chimeric proteins can be stably expressed on HEK293T cells and PM1 T-cells
The lentiviral packaging system used in each virus was integration competent, which means that the transfer expression cassette could spontaneously integrate into the host genome of transduced cells. 28 Therefore, the study tested whether the chimeric constructs could be transferred by infection and stably expressed on human cell lines and, in particular, on a human T-cell line.
It had already been shown that the chimeric proteins were transiently expressed on HEK 293T cells, so 293T cells were transduced with LVTI and LVSI as a control. In addition, the human T-cell line PM1 was transduced, which is a CD4+CCR5+ T-cell line. The two cell lines were transduced with an MOI of 1 of viruses LVTI (encoding TetFrC-ICAM1) and LVSI (encoding streptavidin-ICAM1). On day 3 post infection, a sample of cells transduced with each virus was analyzed for surface chimera expression (Fig. 3a and b). For 293T cells transduced with LVTI (Fig. 3a left), 98% cells were positive for surface FLAG expression, while 59% were positive in cells transduced with LVSI (Fig. 3b right). For PM1 cells transduced with the same viruses, LVTI infection resulted in 67% FLAG-positive cells, and LVSI infection produced 14% positive cells. Attempts to infect PM1 cells with higher MOIs produced cell toxicity (data not shown) and did not increase expression levels.
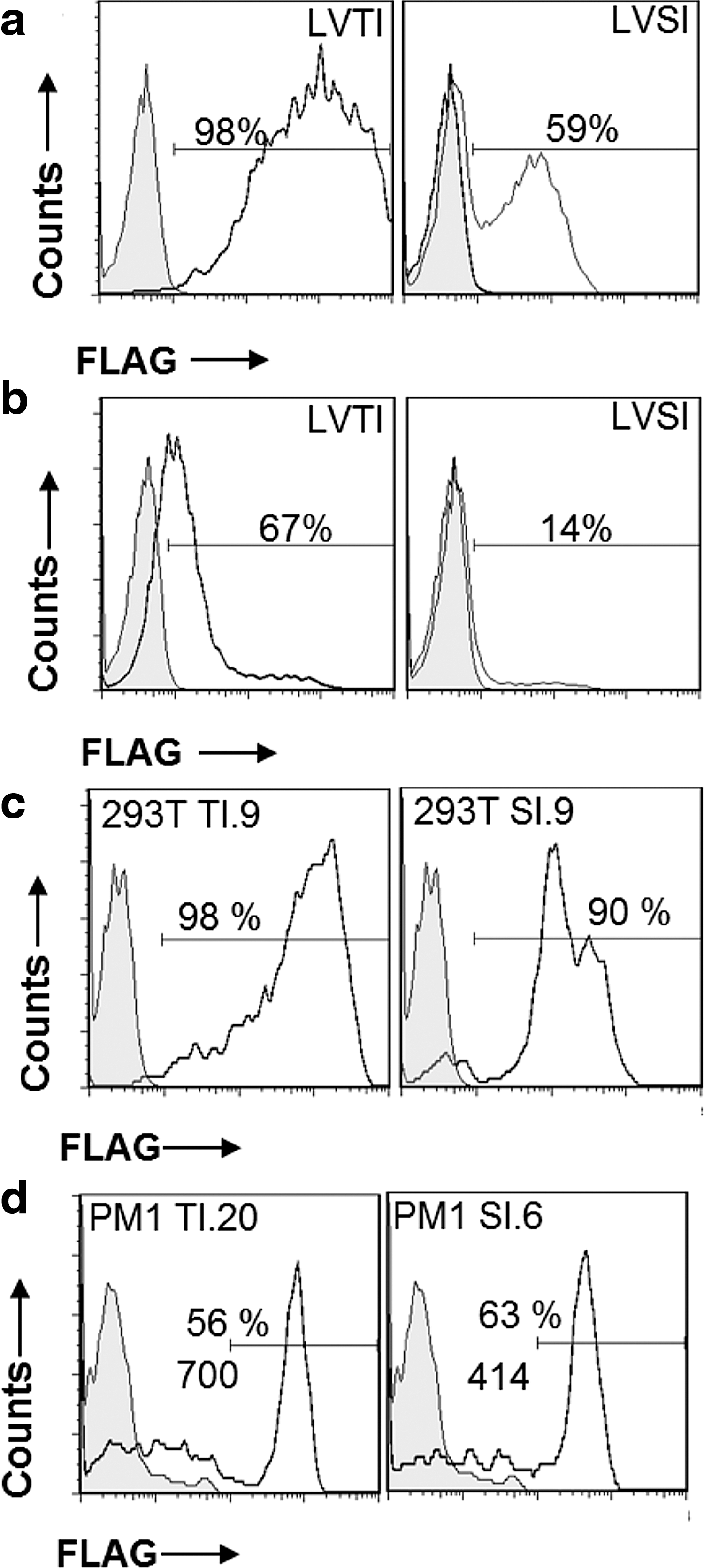
Gene expression from cells infected by lentiviruses bearing chimeric transfer cassettes. (
In order to analyze stable chimeric protein expression on populations derived from single parent cells, cells from the infections described above were diluted to give on average less than one cell seeded per well in 96-well plates. Growing colonies were allowed to expand until enough cells were available for staining with anti-FLAG antibody and FACS analysis; 12–15 colonies for each cell line and infection were analyzed from day 23 post transduction. For 293T cells, there were six positive colonies for LVTI infection and three for LVSI infection. For PM1, there were four positive colonies with LVTI infection and one with LVSI infection.
For each cell line, the highest expressing colony (by percentage FLAG-positive cells above background) for each infection was passaged until 6 weeks post infection and analyzed by FACS for transgene expression (Fig. 3c and d). The LVTI TetFrC-ICAM1 chimeric transgene was expressed on 98% of cells above background on colony 293T TI.9 but with a broad range of fluorescence intensity (Fig. 3c left). The LVSI streptavidin-ICAM1 control chimeric transgene on colony 293T SI.9 was also expressed with a broad range of fluorescence intensity and on 90% of cells above background (Fig. 3c right).
For the selected PM1 colonies, the percentages of cells expressing the transgenes were 56% (PM1 colony TI.20, Fig. 3d left) and 63% (PM1 colony SI.6). Each colony had a discrete peak of higher expressing cells with the MFI of the peak for TetFrC-ICAM1 expression measured at 700 and for streptavidin-ICAM1 at 414. There were also dim and negative cells within each colony, which may represent cells that have downregulated or lost the transgenes. Overall, the results show that the Trojan construct can be transferred to human T-cell lines by infection and expression of the transgenes can be maintained over a 6-week period.
Chimeric proteins in lentiviral preparations can be recognized by immune human anti-tetanus sera
It has been shown that lentiviral vector can be used to confer surface membrane expression of chimeric proteins to target cells. In order to investigate initially whether FLAG-TetFrC chimeric proteins could be detected by TetFrC-specific antibodies in lentiviral preparations, an immunoprecipitation experiment was performed (Fig. 4).
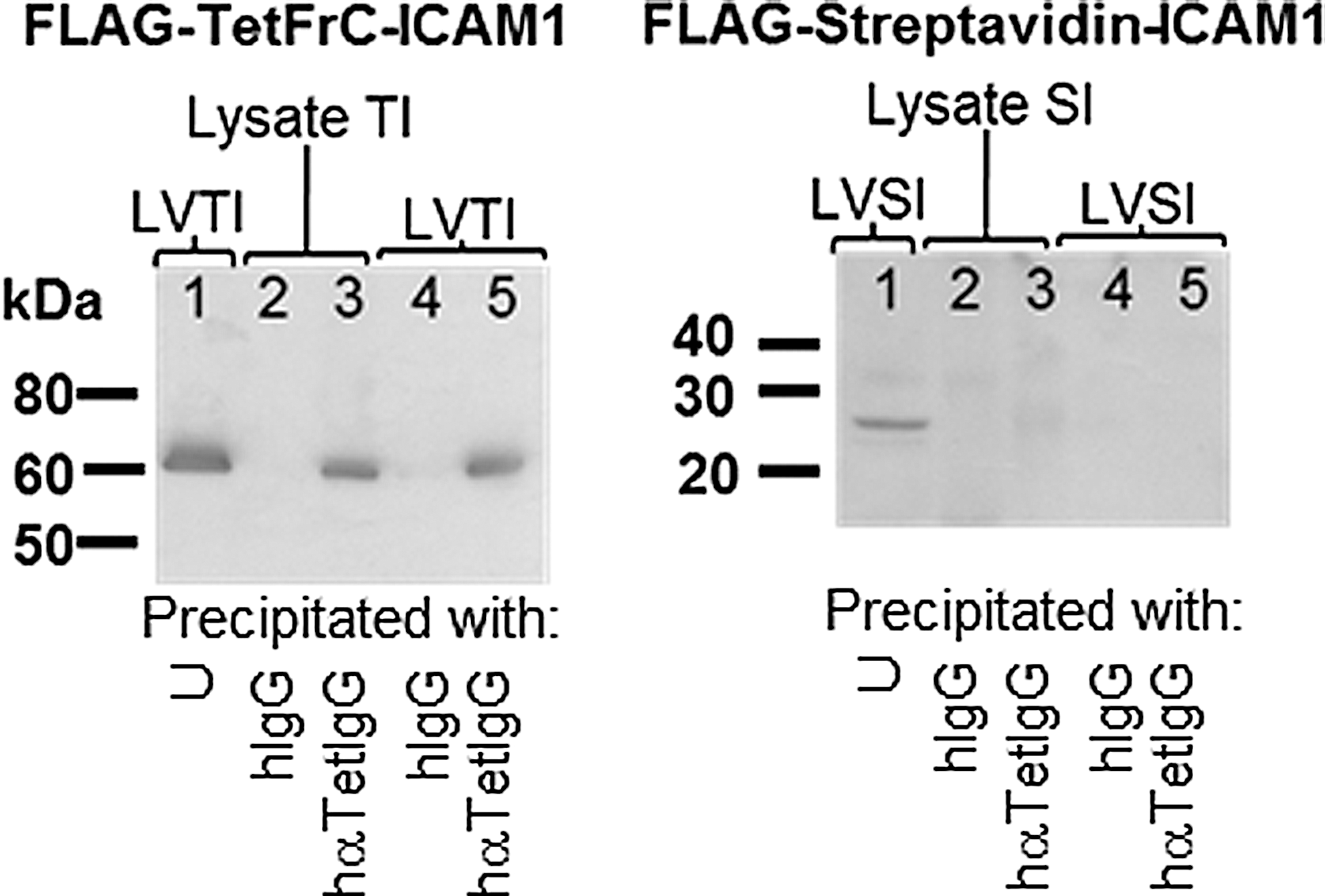
Immunoprecipitation of chimeric proteins in lentiviral preparations with human anti-tetanus antibodies. Lentiviral preparations made using chimeric transfer cassettes were left unprecipitated (U, lane 1), immunoprecipitated with negative control human IgG (hIgG, lane 4) or human anti-tetanus IgG (hαTetIgG, lane 5). For molecular weight controls, lysates were prepared from 293T cells transfected with chimeric constructs, and cell lysates were immunoprecipitated with negative control human IgG (hIgG, lane 2) or human anti-tetanus IgG (hαTetIgG, lane 3). PGNase F treated lysates and immunoprecipitates were separated by SDS-PAGE, and Western blots were probed with M2-HRP antibody.
As a control for the starting sample, equal amounts of protein from each vector were left unprecipitated but otherwise treated as for the immunoprecipitates (lane 1). As a positive control for immunoprecipitation, 293T cells were transiently transfected with each lentiviral chimeric expression vector (data not shown but expression of chimeric proteins demonstrated as for Fig. 1) and lysed. Lysates were immunoprecipitated with protein G-sepharose beads to which negative isotype control polyclonal human IgGs (lane 2) or polyclonal human immune serum anti-tetanus IgGs (lane 3) were stably cross-linked. In parallel, equal amounts of protein from LVTI and SI preparations were incubated with the same beads, namely protein G-sepharose beads conjugated with either human IgGs (lane 4) or human immune serum anti-tetanus IgGs (lane 5). Proteins bound by the antibody-conjugated beads were subject to SDS-PAGE and Western blotting with M2-HRP conjugated antibody probe to detect the FLAG-epitope.
Molecular weight prediction from primary amino acid sequences gave expected average masses of 60 kDa for FLAG-TetFrC-ICAM1 (LVTI) and 27 kDa for FLAG streptavidin-ICAM1 (LVSI) (Expasy Compute pI/MW). In each unprecipitated lentiviral preparation, a band of consistent molecular weight with the appropriate chimeric construct was detected by anti-FLAG antibody showing that each lentiviral vector preparation contained FLAG-tagged proteins consistent in molecular weight with those predicted for the chimeric constructs. When lysates from transfected cells known to be expressing the chimeras were immunoprecipitated with anti-tetanus antibodies from human sera, bands of expected molecular weight were also detected by M2 antibody probe that were not seen in the isotype control lanes.
Finally, when lentiviral preparations themselves were immunoprecipitated, FLAG proteins of the predicted molecular weights for the chimeric constructs were specifically pulled down with human anti-tetanus antibodies but not with isotype control antibodies. These results showed that the chimeric proteins are capable of being specifically bound by human anti-tetanus antibodies and that chimeric proteins are detectable in lentiviral preparations.
Lentiviral vectors with envelope associated chimeric proteins are susceptible to neutralization with anti-tetanus antibodies
Immunoprecipitation showed that chimeric proteins could be bound by anti-tetanus antibodies in lentiviral vector suspensions. However, this did not directly demonstrate that they were associated with the lentiviral envelope membrane due to the possible presence in the lentiviral preparations of exosomes and other cell debris. In order to test this and show that binding of chimeric proteins by anti-tetanus antibodies could produce functional effects, a neutralization assay was performed. Initially, neutralizing antibodies were titrated from 10 to 200 μg on LVGFP and LVTI followed by infection of HT1080 cells (data not shown). With one batch of LVTI, complete neutralization was seen at the lowest dose while the other was maximally neutralized with a dose of 100 μg, and so this dose was selected for subsequent experiments.
In Fig. 5, HT1080 cells were separately transduced with three lentiviral vectors: LVTI (FLAG-TetFrC -ICAM1), whose transfer plasmid expresses surface membrane extracellular FLAG-TetFrC; LVSI (FLAG-streptavidin-ICAM1), a matched negative control virus where TetFrC domain is swapped for streptavidin; and finally LVGFP, whose transfer plasmid expresses cytoplasmic eGFP and would therefore not be expected to bear any surface membrane epitopes for anti-tetanus antibody neutralization. In parallel, HT1080 cells were transduced with lentiviral preparations that had been pre-incubated for 30 min with 100 μg of anti-tetanus antibodies from human sera or in addition, for LVTI viruses, with 100 μg of isotype control human IgGs. Virus infectivity was assessed 48 h post infection by measuring expression of chimeric proteins in target cells by FACS, as described previously.
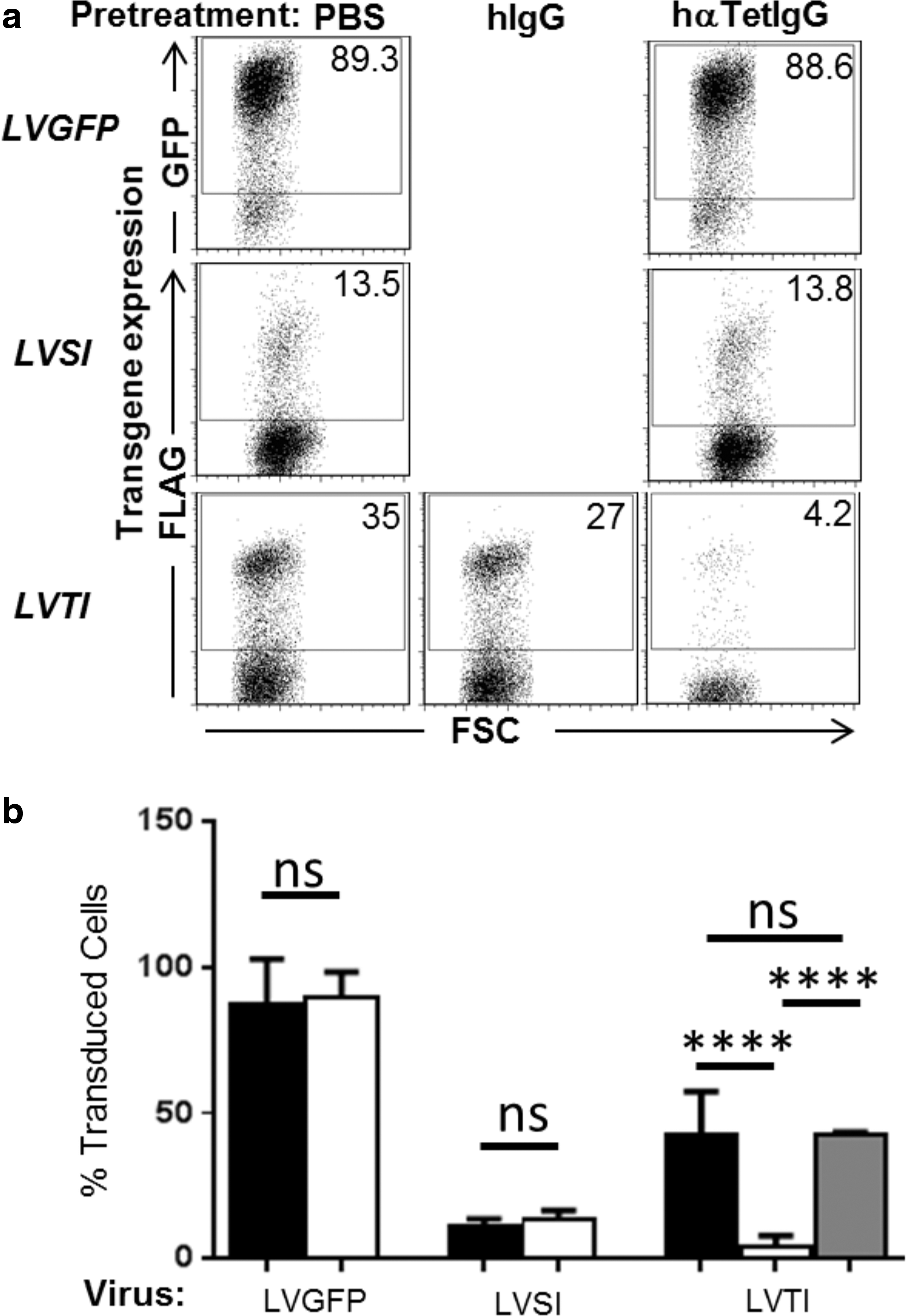
The effect of anti-tetanus antibodies on lentiviruses bearing Trojan chimeric proteins. Lentiviruses were pretreated with phosphate-buffered saline (PBS), isotype control hIgG antibody, or anti-tetanus serum polyclonal IgG antibody. HT1080 cells were then infected for 18 h before virus was removed. Cells were analyzed by FACS 48 h post infection for expression of surface FLAG epitope (or eGFP in the case of LVGFP).
Representative plots from each infection showing gating on positive cells are shown in Fig. 5a. Transduction by untreated LVTI and LVSI was lower than expected based on titrated MOI, but the reasons for this are not yet known. However, treatment of LVTI TetFrC Trojan viruses with human serum anti-tetanus IgG causes a dramatic reduction in transduction, and this is not seen with an isotype control human IgG or when viruses were produced using either an eGFP or streptavidin-ICAM1 expression cassette.
Mean gene expression, as a percentage of FLAG-positive transduced cells, was calculated from a minimum of three wells for each combination of virus and antibodies and is shown in Fig. 5b. Statistical analysis showed a significant (p < 0.0001) decrease in transduced cells after ICAM1-TetFrC bearing viruses were treated with human serum anti-tetanus IgG but not isotype control IgG. As expected, human serum anti-tetanus IgG pretreatment did not produce a significant effect on the mean percentage of cells transduced by LVGFP or LVSI (streptavidin-ICAM1).
Discussion
Altering the host cell range of lentiviral vectors through the introduction of cell-entry glycoproteins from other enveloped viruses is a long-established practice. 19 This study generated chimeric proteins designed to be expressed in the membrane of lentiviral vectors as they bud from host cells. These so-called Trojan proteins are proposed as a means to prevent HIV viruses from avoiding effective immune responses by making HIV virions susceptible to immunity generated by clinically proven vaccination against another pathogen such as tetanus. The results shown are restricted to in vitro models with replication incompetent lentiviruses, but it is hypothesized that Trojan proteins could be delivered as a gene therapy in HIV-infected individuals.
One theoretical application for this is in “shock-and-kill” strategies, which have been proposed as a future method of sterilizing cure for HIV-infected patients on highly active antiretroviral therapy. 7,29 Small molecules are used to reactivate HIV transcription in order to expose viral reservoirs to host immune responses. However, reactivation alone has not been shown to be effective enough for patient immunity to clear the latently infected cells. 7,30 It is proposed that delivery of Trojan genes to latently infected cells under the control of a Tat-responsive promoter would allow these antigens to be used during “shock-and-kill” therapy. It is speculated that activation of HIV transcription by latency reversing agents would cause cell surface expression of Trojan molecules on HIV-producing cells. This would be predicted to have two main sequelae: first, pre-existing anti-tetanus immunity could be used to target and destroy infected cells, but also any emerging HIV virus would be neutralized by serum immunity. Future work will address the success of targeting of Trojan molecules to the surface of lab strain or patient HIV.
The Trojan expression cassette, under the control of an HIV responsive promoter, can be delivered to cells known to harbor latent HIV infection. Delivery of gene therapy as a strategy for HIV treatment has experienced a surge of interest after the “Berlin Patient” report showed that infusion of CCR5-negative cells could provide long-term protection from HIV re-emergence in a HIV-positive individual. 31,32 In addition, the CRISPR/Cas9 system has been tested for HIV co-receptor knockdown to protect cells from infection, as well as a means to target and destroy HIV genomes. 33 –36
Clearly, gene therapy strategies such as the Trojan expression proposed here, as well as the gene-editing strategies discussed above, face challenges of therapeutic delivery. 37 The cellular targets of HIV are well defined, and the key reservoirs for HIV latency have been identified as resting memory T cells and cells of the myeloid lineage, with involvement of central nervous system cells being more controversial. 38 –40 Historically, lentiviral vectors have been posited as ideal vectors for treating HIV and have been shown to infect relevant target cells. 41 In more recent times, lentiviral vectors have been directly tested in HIV blocking strategies using RNAi and gene editing by CRIPSR. 42 –44 Though a VSVg pseudotyped lentiviral vector expression system was used in this in vitro model, technologies to improve the delivery of lentiviral vectors through pseudotyping and cell-type specific retargeting are in development. 20,21,45 Furthermore, the Trojan Chimeras genes could foreseeably be delivered by other gene therapy vectors such as AAV, which have already been tested for use in gene-editing strategies for HIV. 46
With the expression of the Trojan cassette being stimulated in cells containing reactivated HIV, it was predicted that the newly replicated HIV released would be coated with the tetanus antigen. Pretreatment with anti-tetanus vaccination and passive immunization with anti-tetanus human antibodies would be a way to block released virus and potentially clear the latent cellular reservoirs due to expression of tetanus toxoid epitopes.
Initially, four chimeric proteins were constructed and were shown to be expressed transiently on the surface of cells used for lentiviral packaging. The constructs were then used as transfer plasmids in the production of VSVg pseudo-typed lentiviral vectors. Viral titers were variable, and the titers for LVSV containing the FLAG-streptavidin-VSVg chimeric protein were lowest. The reasons for this trend are unknown, as VSVg has been commonly used as a transmembrane carrier for lentiviral pseudo-types. 19,20 It is possible that the chimeric VSVg transmembrane region is competing with the VSVg pseudo-type protein for recruitment to the viral surface resulting in reduced infectivity of the virus. However, the LVTV (FLAG-TetFrC-VSVg) titers were not significantly lower statistically than viruses carrying FLAG-streptavidin-ICAM1 proteins.
Streptavidin on the viral surface might also cause steric hindrance of virus assembly or infectivity; long cytoplasmic tails of pseudo-types from measles viruses were shown to be detrimental to lentiviral titers. 47 In a study investigating the alteration of the lentiviral surface for redirection of infectivity using streptavidin-VSVg and gp64 pseudo-types, Kaikkonen et al. 21 found that ratio of pseudo-type to streptavidin-VSVg plasmids was critical to viral titers. Therefore, it may be that altering the plasmid recipe may be sufficient to improve the low titers seen in the present study.
It was also shown that Trojan lentiviral constructs could be used to transduce human cell lines and lead to surface expression of TetFrC antigen. In the absence of selection, in both cell types and with both viruses, there was TetFrC surface expression in a subset of cells at 2 months post transfection, though longer-term expression was not tested. A broad range of MFI was seen particularly in the 293T wells, but this was not unexpected given the adherent nature of the cells and the dilution method used.
Transduced PM1 wells at 2 months post infection not only showed a narrow peak of expression but also contained some dim and FLAG-negative cells; a gradual loss of expression of chimeric antigens from daughter cells cannot be excluded. Further sorting and screening may identify true stable clones with more restricted ranges of MFI.
The 293T lines so generated have the potential to be used as cell factories for further production of the Trojan-altered viruses, for example with eGFP transgene cassettes. PM1 T-cell lines express the CD4 receptor and co-receptors CCR5 and CXCR4 necessary for infection by macrophage and T-cell trophic (R5 and X4) strains of HIV. PM1 T cells expressing the Trojan antigen can be used to test whether HIV lab strains or primary isolates would be coated with TetFrC protein and therefore be neutralized by anti-tetanus antibodies. 48 Such T-cell lines can also be used to test the potential for HIV mutational escape from the Trojan strategy. 33
The key to generating effective antibodies against HIV envelope protein by vaccination remains elusive. The present results demonstrate that other immunogenic proteins can be delivered to the surface envelope of lentiviruses and that this can make them susceptible to neutralization by antibodies against a different pathogen. In Fig. 5, the equivalent of 2.3 IU/mL of international standard human tetanus immunoglobulin was used for neutralization; units in this antibody are based on in vivo neutralization assays in mice. However, some batches of Trojan virus were completely neutralized with 10-fold less antibody (data not shown). Different amounts of debris in lentiviral vectors prepared by ultracentrifugation without density cushions may be a possible cause of this experimental variation. The amount considered to be protective against tetanus infection in human sera is 0.01 IU/mL. 49 The concentration of antibody needed to neutralize HIV in the context of our proposed Trojan therapy would require further analysis.
The effect on lentiviral titers and stability of altering the viral envelope in this way requires further investigation, since lower transduction than predicted based on original titration was noted, in particular with LVSI, in the neutralization studies. Some error may be inherent to the titration method used, but it is possible that alteration of the envelope may have consequences for the stability of viral vectors during storage and thawing.
It has been demonstrated that lentiviral vector mediated delivery can be used to deliver Trojan proteins for expression on the surface of the T-cell line PM1. Clearly, the effectiveness of lentiviral Trojan delivery to primary patient T cells requires testing. Uncontrolled expression of Trojan proteins on T cells may be undesirable, but lysis of non-HIV-infected cells due to anti-Trojan immunity has the potential to be controlled by making Trojan protein expression dependent on HIV transcription. 50,51 Testing Trojan protein expression in T-cell lines may also reveal whether, in addition to neutralizing cell-free virus, antigen expression on the host cell surface may cause anti-host cell immune responses against tetanus epitopes.
For initial testing, in the in vitro model system, the Trojan expression cassette was under the control of a broadly and constitutively expressed CMV promoter, and therefore the delivery vector particles were also coated with TetFrC antigen. However, pre-existing anti-tetanus immunity in human populations due to vaccination, on which the success of the Trojan strategy relies, has the potential to block therapeutic delivery in its current form. 49 Furthermore, uncontrolled expression of Trojan proteins on non-HIV-infected T cells may result in undesirable cell lysis. Blocking of delivery by anti-tetanus immunity and lysis of non-HIV-infected cells due to anti-Trojan immunity has the potential to be controlled by making Trojan protein expression dependent on HIV transcription. 50,51
To the authors’ knowledge, diverting neutralizing immunity against one pathogen onto another is an entirely novel concept at this time, though suicide gene therapies have been suggested for HIV and other diseases. For example, the conditional expression of a thymidine kinase in T cells has recently been reported to cause cytotoxicity upon ganciclovir treatment in HIV-infected cells, and a similar gene therapy using thymidine kinase-induced drug sensitivity has been tested for prostate cancer. 52,53 In the present experiments, TetFrC was used as a model antigen with known human serum neutralizing antibodies, but it may be possible and desirable to use other or perhaps multiple antigens to reduce the potential for mutational escape and/or reductions in responses due to HIV-mediated damage to immune responses. 54,55
In addition, the ability to display such Trojan proteins on the lentiviral envelope has other possible applications for pseudo-typing of lentiviral vectors. For example, a modified TetFrC has been proposed for use to direct neurotropism of viral vectors. 56 Furthermore, ICAM1 as a transmembrane anchor may offer new ways targeting lentiviral vectors to specific cell types. The expression cassette design offers the possibility for interchangeability of transmembrane and extracellular domains.
Conclusions
Novel chimeric proteins have been generated, designed to coat lentiviruses with antigens from other pathogens to which neutralizing memory immunity is present in vaccinated human populations. It is predicted that altering the HIV particle surface will redirect these immune responses to neutralize HIV. Overall, the foundation data in this paper show that the Trojan chimeric molecules for neutralization of lentiviral particles are functional and merit further investigation.
Footnotes
Acknowledgments
Thanks to J. Rice and F. Stevenson (Southampton University, United Kingdom) for providing sequence information and plasmids for TetFrC, and to M. Kaikkonen and S. Yla-Herttuala (University of Kuopio, Finland) for providing sequences and plasmids for Strep-VSVg. This work was funded by the Bill and Melinda Gates Foundation, Seattle, WA (grant 52116).
Author Disclosure
No competing financial interests exist.