
Abstract
We previously developed a mini-intronic plasmid (MIP) expression system in which the essential bacterial elements for plasmid replication and selection are placed within an engineered intron contained within a universal 5′ UTR noncoding exon. Like minicircle DNA plasmids (devoid of bacterial backbone sequences), MIP plasmids overcome transcriptional silencing of the transgene. However, in addition MIP plasmids increase transgene expression by 2 and often >10 times higher than minicircle vectors in vivo and in vitro. Based on these findings, we examined the effects of the MIP intronic sequences in a recombinant adeno-associated virus (AAV) vector system. Recombinant AAV vectors containing an intron with a bacterial replication origin and bacterial selectable marker increased transgene expression by 40 to 100 times in vivo when compared with conventional AAV vectors. Therefore, inclusion of this noncoding exon/intron sequence upstream of the coding region can substantially enhance AAV-mediated gene expression in vivo.
Introduction
T
In quiescent tissues, minicircle DNA vectors devoid of the bacterial backbone can provide 10 or more times higher sustained levels of transgene expression compared to that achieved with a canonical plasmid containing the same expression cassette. 1 –6 The reason for this is that transcriptional silencing of the transgene occurs when >1 kb of spacer DNA exists between the 5′ end of promoter and 3′ end of polyadenylation (polyA) site. 7 Based on these findings, we constructed a mini-intronic plasmid (MIP) in which the components required for plasmid propagation in bacteria (bacterial replication origin and selectable marker) are positioned within an intron of a noncoding exon contained within the expression cassette. 8 Because there is virtually no spacer outside of the expression sequence, MIP vectors are not subject to transgene silencing and in addition provide 2–10 times or higher levels of transgene expression compared with minicircle vectors in vivo and in vitro.
The detailed mechanism of MIP-generated enhanced expression over minicircle DNAs and plasmid DNAs is not clear. It has been previously reported that introns may increase gene expression through enhancing mRNA export from the nucleus to the cytoplasm. 9 Splicing of promoter proximal introns has been shown to provide enhanced transgene expression. 10 Additionally many recombinant genes are expressed inefficiently when their introns are removed. 11 –13
With the success of MIP vector, we then tested the potential of MIP intron (bacterial replication origin and selectable marker sequences) in promoting transgene expression from adeno-associated virus (AAV) vectors. Enhancing the expression abilities of AAV vectors will significantly lower the dose of the vector needed to achieve therapeutic levels of expression, reducing the cost and potential for vector mediated immune responses directed against the vectors' capsids in transduced cells. 14,15
Materials and Methods
Materials
A 0.68 kb truncated pUC origin (Nature Technology Corporation) was used to replace the 1.0 kb pUC origin in optimized intron with pUC origin and RNA-OUT (OIPR) to generate the 1 kb OIPR (Supplementary Fig. S1a, b; Supplementary Data are available online at
Vector construction
pRHB (pRSV.hAAT.bpA), MC.RHB, and MIP.RHB were previously described.
2,8
RSV.hAAT.bpA and RSV.OIPR.hAAT.bpA fragments were PCR amplified from pRHB and MIP.RHB, respectively, with PCR primers 5′ ATATAT
MIP.RHB.R6K and MIP.RHB.ColE2 were generated through replacing the intronic pUC origin in RSV promoter hAAT MIP vector JM158 (MIP.RHB) with the R6K or ColE2 miniorigins. Briefly, the JM158 vector was PCR amplified using the primers 5′ AGGCGG
pRHB.R6K and pRHB.ColE2 were produced through replacing the pRHB bacterial backbone with R6K-RNA-OUT or ColE2-RNA-OUT sequences. The RSV promoter- hAAT -bovine growth hormone polyA (bpA) region from MC.RHB was PCR amplified using primers 5′ AGGCGA
ApoE.HCR promoter R6K and ColE2 origin MIP vectors were constructed by replacing the OIPR with the R6K intron, containing R6K origin-RNA-OUT as follows. The JM179 ApoE.HCR promoter human factor 9 (hFIX) transgene OIPR MIP vector was PCR amplified to remove the intronic pUC origin and digested with SacI and SacII to create a JM179 backbone acceptor PCR fragment using primers 5′ AGGCGG
The recombinant AAV vectors AAV.MIP.RHB, AAV.RHB, AAV.R6K.RHB, AAV.ColE.RHB, AAV.ApoE.HCR.MIP.hFIX, AAV.ApoE.HCR.R6K.hFIX, AAV.ApoE.HCR.hFIX, AAV.PGK.hAAT, AAV.PGK.MIP.hAAT, AAV.PGK.R6K.hAAT, and AAV.PGK.ColE2.hAAT were generated through inserting the expression cassette sequences (promoter-coding gene-polyA signal) into the recombinant AAV production vector p388-AC-For. 8
Animal studies
Six to eight week old female C57BL/6J mice purchased from Jackson Laboratory were used for DNA or recombinant AAV (rAAV) vector infusion. To ensure the same molar amount of DNA was injected into each animal, 3.63 μg/kb DNA was used for various sized constructs. Each DNA construct was diluted into 1.8 mL 0.9% NaCl for each animal and was delivered through hydrodynamic tail vein injection. For rAAV vector infusion, each animal received 1.0 × 1011 VG diluted in 200 μL 1 × PBS through tail vein injection. Five animals were tested for each DNA or rAAV construct in each tested experimental group. After infusion, blood samples were collected periodically by a retro-orbital technique. The serum hAAT and the plasma hFIX levels were quantified by enzyme-linked immunosorbent assay (ELISA) measurements.
AAV production. All AAV vectors were packaged as previously described using a Ca3(PO4)2 transfection protocol followed by CsCl gradient purification. 19 All AAV productions were prepared using HEK293 cells and were titrated by quantitative dot blot as described previously. 19
ELISA
ELISA for hAAT and hFIX was performed as previously described 8 with the following antibodies: goat anti-human alpha-1 antitrypsin primary antibody at 1:1000 (Strategic Biosolutions S0311G000-S4), goat pAB to alpha-1 antitrypsin (HRP) (Abcam ab7635), mouse anti-human factor 9 (F9) immunoglobulin G (IgG) primary antibody at 1:1000 (Sigma F2645), and polyclonal goat anti-human F9 peroxidase–conjugated IgG secondary antibody at 1:4200 (Enzyme Research GAFIX-APHRP).
Quantitative PCR analysis
Fifty nanograms of XbaI digested genomic DNA from each sample was used as the template for quantitative PCR. Two animal samples from each injection group were selected and two 20 μL reactions were performed for each animal sample. Various copy numbers (50 copies to 5 × 108 copies) of XbaI digested standard vector DNA along with 50 ng noninfused control genomic DNA per reaction was used to make the copy number standard curve. Forward primer 5′ AAGGCAAATGGGAGAGACCT 3′ and reverse primer 5′ TACCCAGCTGGACAGCTTCT 3′ oligos were used to amplify a 150 bp fragment from the hAAT cDNA region. Forward primer 5′ TTGCTGACAGGATGCAGAAG 3′ and reverse primer 5′ TGATCCACATCTGCTGGAAG 3′ oligos were used to amplify a 150 bp fragment from β-actin as loading control. The tested transgene signal was then normalized to the β-actin signal. The mass of a single diploid copy of mouse genome is 5.88 pg; thus, 50 ng genomic DNA contains 85,034 copies of diploid genome (5 × 105 pg/5.88 pg). The average transgene copy number in 50 ng genomic DNA from each group was then divided by 85,034 to achieve the transgene construct copy number in each cell. All calculations were based on methods described by AppliedBiosystems 20 and by using the University of Rhode Island internet tool. 21 Quantitative PCR was performed by using Corbett Research RG6000 PCR machine.
Results
Incorporation of OIPR (optimized intron with pUC and RNA-OUT) into recombinant AAV construct enhances transgene expression
Our previous study demonstrated that adding the 1.3 kb mini-intronic sequence to a 5′ non-coding exon called, OIPR (Fig. 1a; Supplementary Fig. S1a) significantly enhanced transgene expression levels 2- to 10-fold compared with canonical minicircle vectors, and 100- to 1000-fold compared with canonical plasmids in vivo. The OIPR is made up of the optimized pUC replication origin and the prokaryotic RNA-OUT antibiotic-free 16,22 selectable marker sequences and flanked by synthesized strong exonic splicing enhancers, 23 G triplets, 24 a tract of 11 continuous polypyrimidines, 25 and a consensus branch point sequence to ensure proper and efficient splicing. 8 We wanted to evaluate if the OIPR sequences enhanced expression from a viral vector. To do this we incorporated the OIPR intron into an AAV vector expression cassette. The first expression cassette contained a human α1-antitrypsin (hAAT) coding sequence driven by Rous sarcoma virus (RSV) promoter and terminated by bovine growth hormone polyA (bpA), AAV.RHB (Fig. 1a). The recombinant OIPR AAV construct, AAV.MIP.RHB, was constructed by inserting an OIPR intron into AAV.RHB before the start codon of hAAT. The AAV.MIP.RHB and AAV.RHB expression cassettes were packaged into rAAV8 vectors. These vectors were infused into the livers of 6–8 week old C57BL/J female mice by tail vein injection, respectively, and the levels of serum hAAT transgene product were measured at various time points (Fig. 1b). Consistent with our previous findings, inserting the OIPR intron subsequently improved transgene expression in vivo. As indicated in Fig. 1b, animals infused with rAAV vector (AAV.MIP.RHB) expressed hAAT transgene at 40-fold higher levels than the other rAAV vector (AAV.RHB).
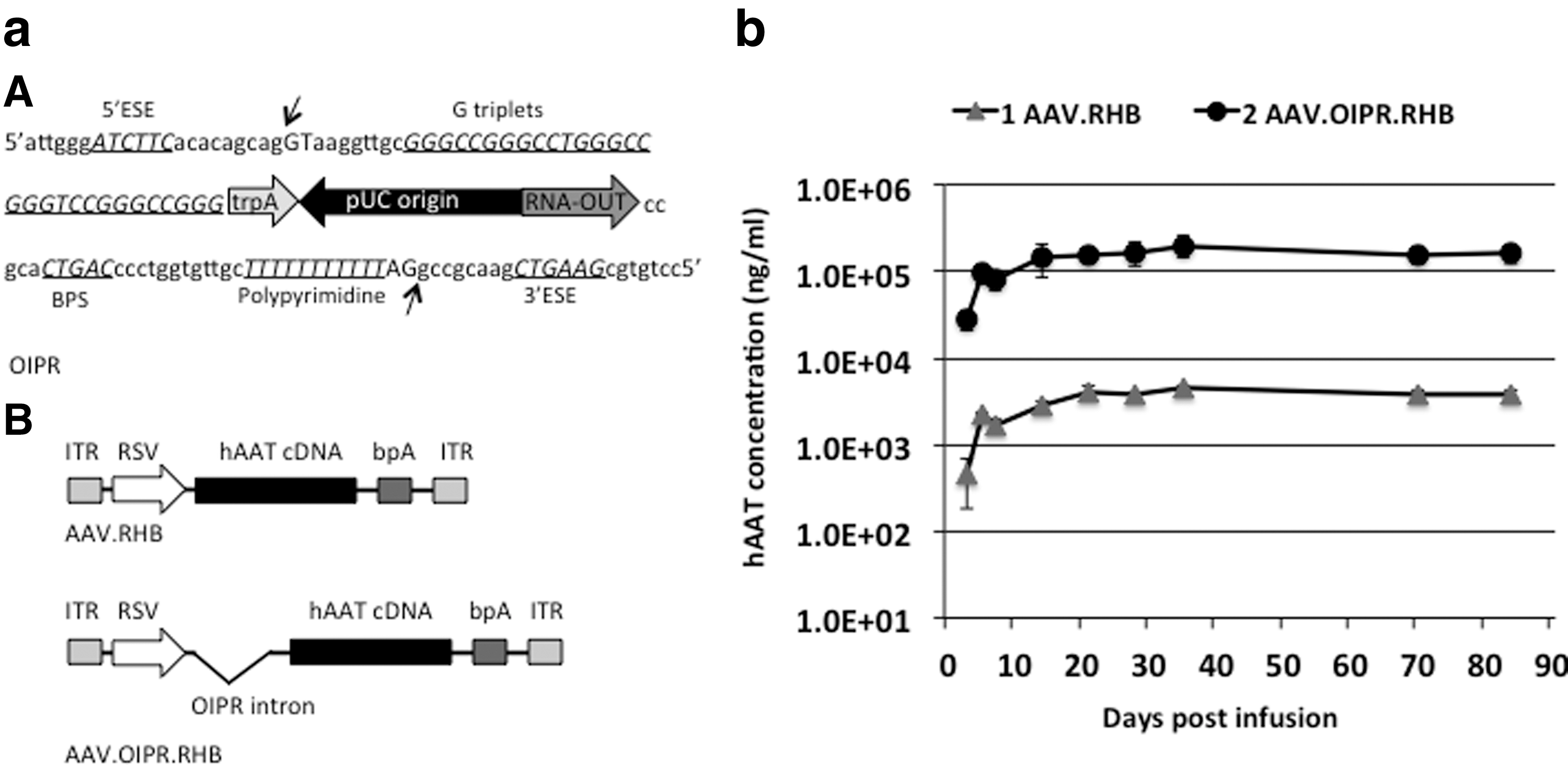
RSV-hAAT expression, recombinant adeno-associated virus (AAV) vectors, and transgene expression in mice.
OIPR introns that lack either pUC replication origin or RNA-OUT selectable marker were not sufficient to improve transgene expression
The rAAV vectors have a packaging limit of ∼4.4 kb foreign DNA, and because the OIPR intron is about 1.3 kb, only 3.1 kb of additional DNA can be incorporated into the recombinant AAV.MIP construct. Therefore, we attempted to further reduce the intron size.
The OIPR intron was originally designed for MIP plasmid to relocate the bacterial replication origin and selectable marker from the backbone region to the expression cassette region for MIP plasmid vector propagation. However, for the recombinant AAV construct, prokaryotic bacterial replication and selection sequences are not required. We therefore removed either pUC replication origin or RNA-OUT selectable marker sequences from the OIPR intron and the remaining intron was tested for their ability to enhance transgene expression in vivo. As shown in Fig. 2a, pUC replication origin was removed from the original OIPR intron in MIP vector MIP.RHB.R, and RNA-OUT selectable marker sequences were removed from OIPR intron in MIP vector MIP.RHB.P. These two minimized MIP constructs along with a canonical plasmid pRHB (silencing control), a canonical minicircle MC.RHB (nonsilencing control), and original MIP vector MIP.RHB (transgene enhancement control) were infused into the livers of mice, respectively, and the levels of serum hAAT transgene product were measured at various time points (Fig. 2a). Intact OIPR intron was able to enhance transgene expression compared with minicircle vector; however, the OIPR intron that lacks either pUC replication origin or RNA-OUT selectable marker was only able to maintain a similar transgene expression level as minicircle (Fig. 2a).
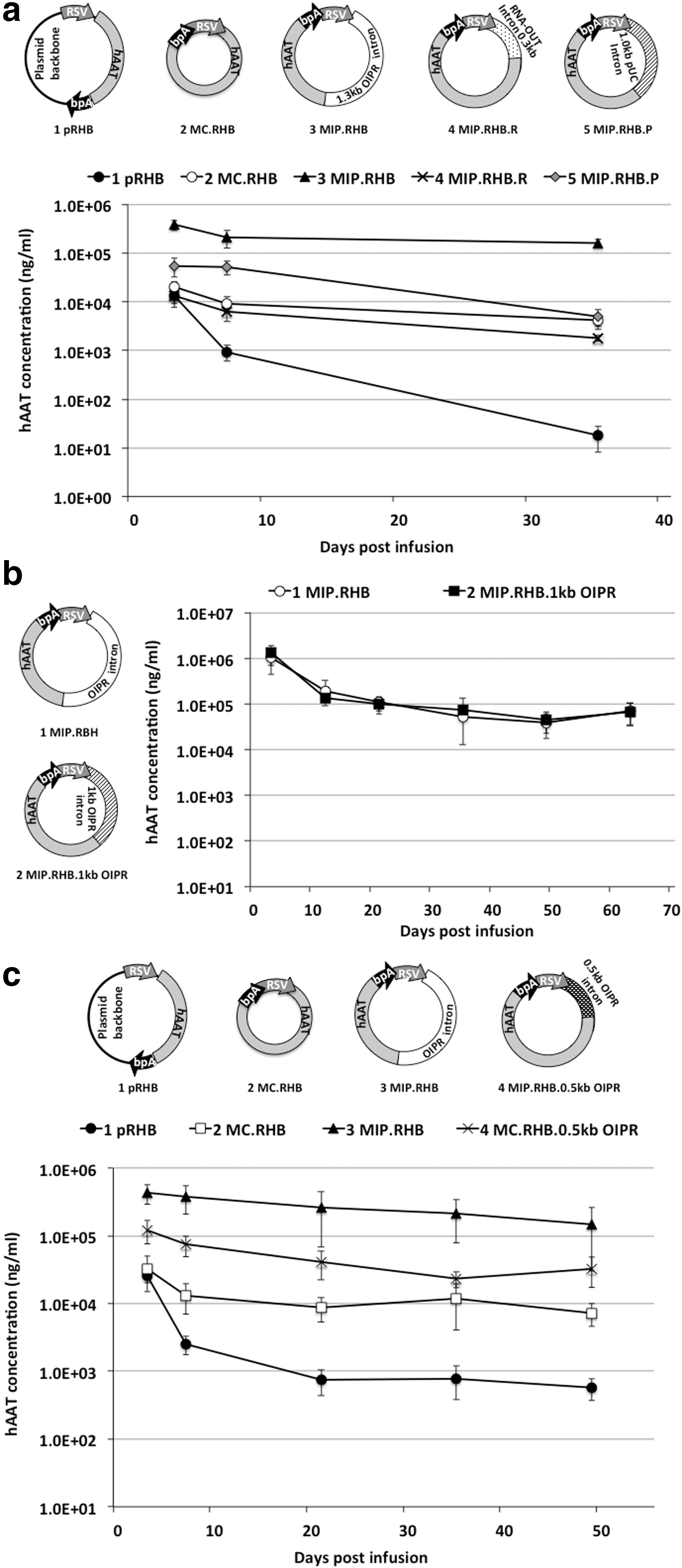
RSV-hAAT expression constructs and transgene expression in mice.
This suggested that enhancement of the expression required both the replication origin and RNA-OUT selectable marker sequences. Instead of removing the pUC or RNA-OUT sequences, we reduced the size of pUC sequences and made two shortened OIPR introns, 1 kb OIPR and 0.5 kb OIPR (Supplementary Fig. S1b, c). As shown in Fig. 2b, animals infused with MIP vector with 1 kb OIPR (MIP.RHB.1kb OIPR) expressed the same enhanced hAAT levels as animals infused with original MIP vector (MIP.RHB). The MIP vector with a further shortened OIPR intron (MIP.RHB.0.5kb OIPR) had an intermediate enhancement of transgene expression compared with minicircle vector (MC.RHB) (Fig. 2c).
R6K and ColE2 miniorigins were able to enhance transgene expression in vivo when used as MIP introns
In addition to the pUC replication origins, we tested other shortened bacterial replication origins for their ability to improve transgene expression when engineered into an intron. Two minimized bacterial replication origins, R6K and ColE2 (developed by Nature Technology Corporation), 17 were tested. Plasmids containing these miniorigins as plasmid backbone (Nanoplasmid™ vectors) expressed enhanced transgene levels, indicating that these miniorigins might have the same function in improving gene expression. 17 The R6K miniorigin is modified from a R6K gamma replication origin (Supplementary Fig. S2a), and the ColE2 miniorigin is modified from a ColE2-P9 replication origin (Supplementary Fig. S2c). The combined size of R6K miniorigin and RNA-OUT selectable marker is 456 bp, and the combined size of ColE2 miniorigin and RNA-OUT selectable marker is only 271 bp. The minimized size of these miniorigins makes them ideal to use as enhancing introns in recombinant AAV constructs.
These miniorigins were tested for expression enhancement in MIP vectors in vivo. As shown in Fig. 3a, R6K-RNA-OUT or ColE2-RNA-OUT sequences were introduced to replace the original OIPR intron, in plasmid MIP.RHB.R6K or MIP.RHB.ColE2, respectively, to create R6K intron and ColE2 intron (Supplementary Fig. S2b, d). In comparison, the R6K-RNA-OUT and ColE2-RNA-OUT were also used in a canonical plasmid backbone vector, pRHB.R6K and pRHB.ColE2 (Fig. 3a). These DNA vectors along with a canonical plasmid pRHB (silencing control) and a minicircle vector MC.RHB (non-silencing control) were infused into the livers of mice by hydrodynamic tail vein injection, and the levels of serum hAAT transgene product were measured at various time points (Fig. 3a). As previously described, R6K and ColE2 miniorigins were able to enhance transgene expression when placed outside of expression cassette (Nanoplasmid™ vectors). 17 The miniorigins themselves when used to replace conventional plasmid backbone (pRHB.R6K and pRHB.ColE2) were able to enhance transgene expression compared with conventional plasmid pRHB. However, placement of these sequences into an intron resulted in even greater transgene expression (Fig. 3a). This was demonstrated when the miniorigins with RNA-OUT sequences were used as MIP introns—the resulting MIP vectors (MIP.RHB.R6K and MIP.RHB.ColE2) were able to further enhance transgene expression. Nevertheless, none of these vectors were as robust as the original 1.3 kb MIP intronic vector (MIP.RHB) (Fig. 3b).

R6K and ColE2 miniorigins enhanced transgene expression.
Interestingly, R6K-RNA-OUT and 0.5 kb OIPR introns were similar in length (443 bp for 0.5 kb OIPR, Supplementary Fig. S1c; 545 bp for R6K intron, Supplementary Fig. S2b), and as shown in Fig. 3c, R6K-RNA-OUT and 0.5 kb OIPR showed similar enhancement of transgene expression when used as MIP introns (MIP.RHB.R6K, MIP.RHB.0.5kb OIPR).
R6K and ColE2 miniorigins were able to enhance transgene expression from AAV when used as 5′ promoter proximal introns
Because of their small size, the R6K and ColE2 miniorigins make them attractive candidates to use as introns in recombinant AAV constructs to enhance transgene expression, and hence we elected to test their functionality in rAAV vectors. Figure 4a shows the schematic drawings of recombinant AAV constructs with miniorigin introns (AAV.R6K.RHB and AAV.ColE2.RHB), the recombinant AAV construct with OIPR intron (AAV.MIP.RHB), and the canonical AAV construct (AAV.RHB). The packaged rAAV8 vectors of these constructs were infused into mice and serum hAAT followed over time. As expected, R6K and ColE2 miniorigins with RNA-OUT sequences significantly enhanced transgene expression when used as 5′ promoter proximal introns (Fig. 4a). The most encouraging finding from this experiment is that the 545 bp intron (R6K intron; Supplementary Fig. S2b) was able to enhance the levels of transgene expression by ∼40-fold compared with canonical rAAV vectors.
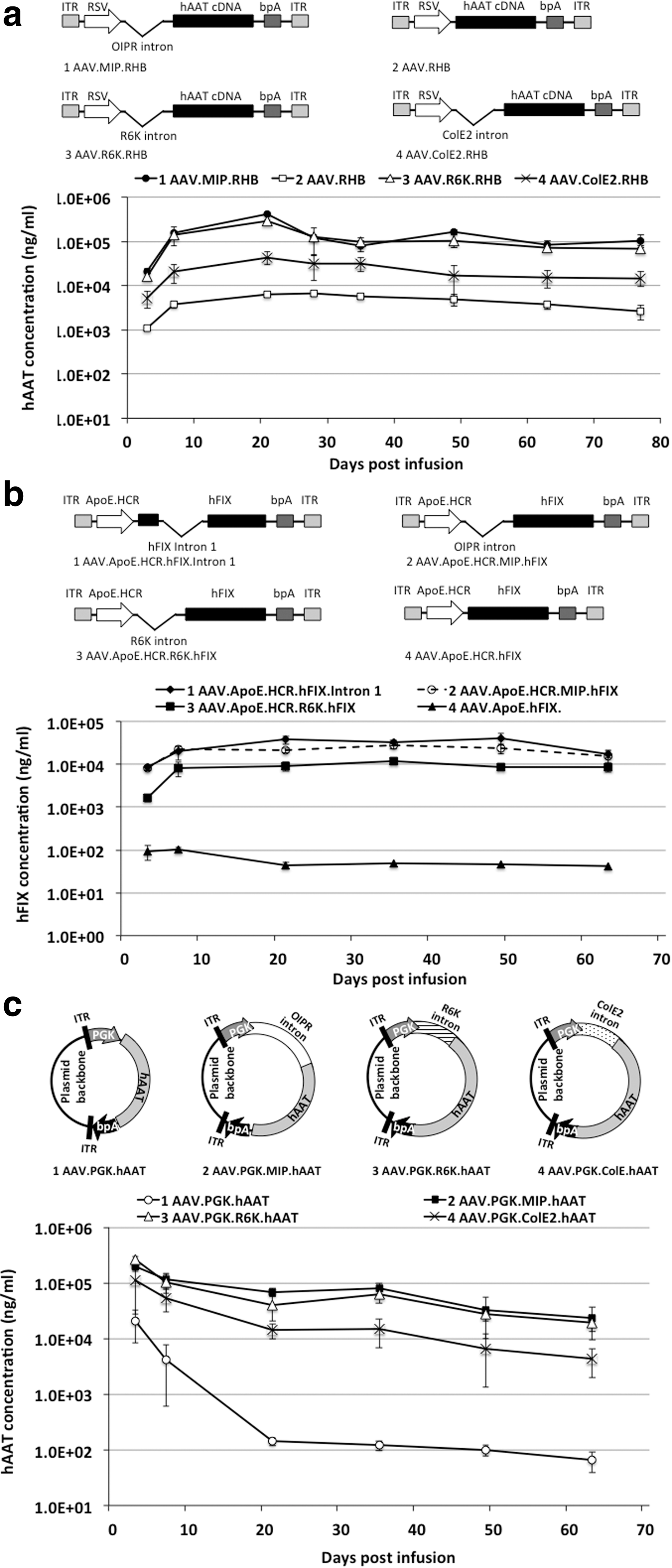
R6K and ColE2 miniorigins were able to enhance transgene expression from AAV when used as 5′ promoter proximal introns.
To avoid any potential bias toward specific reporter gene and promoter combinations, additional promoters (ApoE.HCR promoter and phosphoglycerate kinase [PGK] promoter) and transgene hFIX were tested. To compare the expression enhancement abilities of the R6K-RNA-OUT and OIPR introns to an alternative intron, these introns were compared with a widely used version of the hFIX endogenous intron 1. This 1.4 kb intron has been proven to robustly enhance hFIX expression in vivo 26 and has been incorporated into clinical trial AAV vectors. 27 As shown in Fig. 4b, the R6K-RNA-OUT intron and the OIPR intron when incorporated as a 5′ promoter proximal intron in rAAV8 vectors (AAV.ApoE.HCR.R6K.hFIX and AAV.ApoE.HCR.MIP.hFIX) were able to enhance hFIX expression to a similar level as the hFIX intron 1 containing vector (AAV.ApoE.HCR.hFIX.Intron 1). In addition, this represented ∼100-fold enhancement when compared with conventional rAAV8 (AAV.ApoE.HCR-hFIX). The smaller size of R6K-RNA-OUT intron and the flexibility of inserting both R6K-RNA-OUT intron and OIPR intron into the 5′ promoter proximal location give these introns advantages to be used with a broader range of transgenes.
Similar results were obtained when the PGK promoter was used. As indicated in Fig. 4c, OIPR, R6K-RNA-OUT, and ColE2-RNA-OUT introns were incorporated into recombinant AAV constructs to make AAV.PGK.MIP.hAAT, AAV.PGK.R6K.hAAT, and AAV.PGK.ColE2.hAAT. As a first test, the recombinant AAV plasmid constructs were infused into mice through hydrodynamic tail vein injection. Consistent with previous findings, adding any of these intron to the 5′ promoter proximal location enhanced the transgene expression, with OIPR and R6K-RNA-OUT introns giving the highest enhancement, while ColE2 gave moderate enhancement when compared with the conventional recombinant AAV plasmid construct (AAV.PGK.hAAT) (Fig. 4c).
The vector copy number is not affected when incorporating the miniorigin introns
In our previous studies we have shown that enhanced expression from minicircle or MIP DNA vectors were not related to a difference in DNA copy number; we wanted to establish that the copy number of the rAAV vector–derived DNAs were also not a factor.
To quantify copy number of each rAAV vector construct per diploid genome, liver samples of animals infused with different rAAVs (Fig. 4a) were extracted at 77 days after infusion. By quantitative PCR, we were able to establish that the rAAVs with miniorigin introns were maintained at the same levels as conventional rAAV, ranging from 0.12 to 0.30 copies per diploid genome 11 weeks after infusion (Fig. 5). However, rAAV with OIPR intron (AAV.MIP.RHB) was maintained at about 0.8 copies per diploid genome at the same time point. Since both AAV.MIP.RHB and AAV.R6K.RHB expressed transgene at similar levels (Fig. 4a), this indicates that rAAV with R6K miniorigin (AAV.R6K.RHB) expresses transgene at >2-fold higher per genome copy than rAAV with OIPR intron.
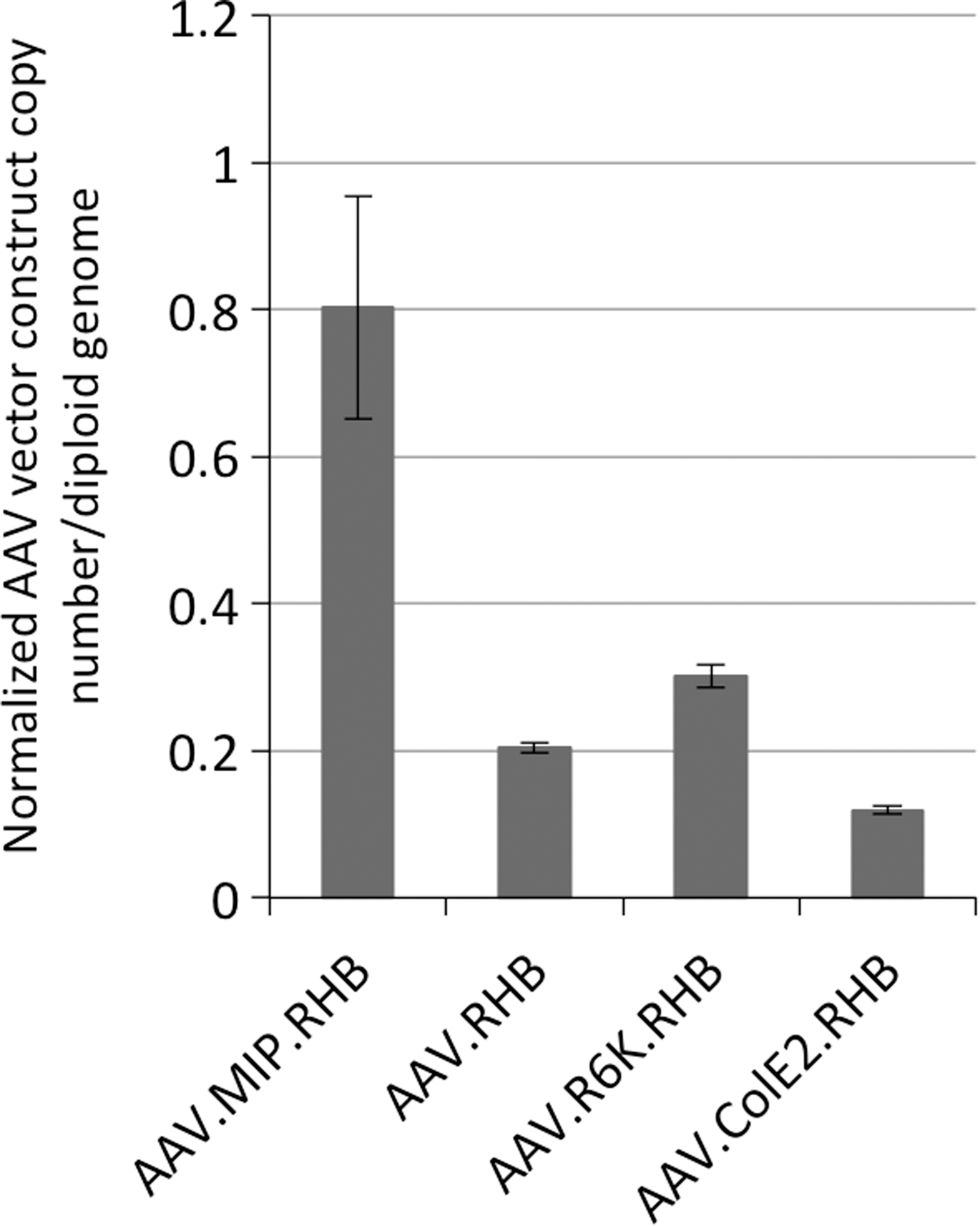
Incorporating of the miniorigin introns does not change rAAV vector copy number in vivo. Liver DNA samples of animals infused with different rAAVs from experiment shown in Fig. 4a were extracted at 77 days after injection. Copy number of each vector per diploid genome in these liver samples was determined by quantitative real-time PCR. Two duplicate samples from each animal and two technical repeats for each sample were tested. The standard deviation was determined by using four quantitaive PCR reactions of animal samples from each infusion group.
The insertion of introns containing bacterial replication origin and RNA-OUT sequences has been proved to significantly improve transgene expression in vivo. These rAAVs can be easily prepared through standard rAAV production methods, and the yields are similar to conventional rAAV vectors (Supplementary Fig. S3a, b).
Discussion
Our studies on MIP vectors and AAV constructs with introns have demonstrated that adding an intron just downstream of the promoter in a noncoding first exon containing a bacterial replication origin and RNA-OUT selectable marker significantly enhances transgene expression from a recombinant AAV vector.
MIP DNA plasmid vectors by design require a bacterial replication origin and selectable marker in the intronic sequences to be able to propagate the extrachromosomal circular DNA vector in bacteria. In contrast, for recombinant AAV constructs, it is not necessary to maintain the bacterial replication origin and selectable marker sequences in the intron. To our surprise, when either bacterial replication origin or RNA-OUT selectable marker was removed from an intron, they lost the capability to enhance transgene expression. The 1.3 kb OIPR that maintains full pUC origin sequences enhances transgene expression more robustly than the 0.5 kb OIPR. However, the length differences of these introns are irrelevant to their capacities to enhance transgene expression. The 1 kb OIPR intron (Fig. 2b) is able to enhance transgene expression, while the 1 kb intron in MIP.RHB.P (Fig. 2a) is insufficient to maintain transgene expression. In our previous study on developing MIP vector, we have demonstrated that replacing the OIPR intron sequences with other sequences (hAAT endogenous intron 2) completely abolished the transgene expression enhancing abilities. 8 Removing pUC origin or RNA-OUT sequences from OIPR introns may potentially affect the splicing efficiency of these introns. If the splicing efficiency is affected, it is more likely to see the transgene products significantly reduced or abolished. As shown in Fig. 2a, the truncated introns (MIP.RHB.R and MIP.RHB.P) were not capable of enhancing transgene expression to the levels observed in MIP.RHB vector. However, they were still able to sustain high levels of transgene expression compared with minicircle vector MC.RHB. This indicates that the splicing activity itself is likely still intact in this case and has no influence in transgene expression strength. However, splicing out the OIPR intron enhances transgene expression. All these facts strongly suggest that the transgene expression enhancing effects are sequence specific and both the bacterial replication origin, and RNA-OUT sequences contribute to these effects. Removing portions of each resulted in a reduced effect and at this time suggests there is not one specific region in each segment that was responsible for enhanced transgene expression. Further studies are needed to uncover the underlying mechanisms of this phenomenon.
The presence of AUG sequences within the 5′ UTR region of expression cassettes has been demonstrated to reduce gene expression. 28 While the OIPR intron contains 8 copies of AUG sequences, the R6K-RNA-OUT intron has 10, and the ColE2-RNA-OUT intron has 5 copies of such sequences, all of these motifs are intronic with no AUG motifs in the 5′ UTR. Additionally, removing 3 copies of intron AUG sequences from OIPR by deleting RNA-OUT sequences (MIP.RHB.P) depleted the enhancement of transgene expression (Fig. 2a). All of these results indicate that, as expected, the presence of intronic AUG sequences has no effect on gene expression.
It has recently been demonstrated that antibiotic resistance markers adjacent to AAV ITRs in plasmid backbone sequences are packaged into a significant percentage of AAV virus particles during production. To prevent contamination of clinical AAV vector particles with antibiotic resistance markers that may represent a theoretical safety concern to regulatory agencies, minicircle AAV vectors have been developed. 29 Alternatively, AAV vectors encoding the transgene expression enhancing 1.3 kb OIPR intron (pUC origin-RNA-OUT), 1 kb OIPR intron (minimalized pUC origin-RNA-OUT), or 545 bp R6K intron (R6K origin-RNA-OUT) reported herein can be modified to remove plasmid backbone sequences, since plasmid replication and antibiotic-free selection functions are encoded within these engineered introns. These MIP AAV vectors would have the same advantages as MC vectors in terms of AAV production and elimination of antibiotic resistance marker transfer during packaging, but with much simpler, high-yield manufacture and dramatically improved AAV vector encoded transgene expression. Inclusion of the R6K-RNA-OUT or ColE2-RNA-OUT introns within the AAV transcription unit should present no safety issues for future clinical use. Unlike the pUC origin, the R6K or ColE2 bacterial replication origins are not replication competent, so there is no risk of vector replication after inadvertent transfer to a patient's endogenous gram negative microbial flora. Additionally, the RNA-OUT antibiotic-free selection marker is a noncoding RNA so there is no risk of expression of a cryptic bacterial peptide that might create T-cell responses in treated patients.
Recombinant AAV vectors are showing promise in clinical applications, albeit the ability to enhance gene expression would potentially reduce the dose required for many indications. The fact that our engineered intron can enhance rAAV-mediated transgene expression by up to 40- to 100-fold is remarkable. The ability to reach the same level of transgene expression, with a 40- to 100-fold lower dose would reduce the cost of manufacturing and perhaps reduce the immune response observed after systemic administration of the vector in humans. 14,15 Moreover, these small intronic sequences seem to work in both DNA plasmids and viral vectors providing a universal addition that may enhance transgene expression from any vector.
Footnotes
Author Disclosure
James Williams and Jeremy Luke have an equity interest in Nature Technology Corporation. Jiamiao Lu, Feijie Zhang, Kirk Chu, and Mark Kay declare that no other competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.