
Abstract
Recent studies have demonstrated that angiotensin-converting enzyme 2 (ACE2) plays an important role in the pathogenesis of abdominal aortic aneurysms (AAAs). However, few studies have reported the direct effect of ACE2 overexpression on the aneurysm. This study hypothesized that the overexpression of ACE2 may prevent the pathogenesis of aneurysms by decreasing RAS activation. Thirty-nine mice were randomly assigned to three groups (n = 13 in each group): the Ad.ACE2 group, the Ad.EGFP group, and a control group. After 8 weeks of treatment, abdominal aortas with AAAs were obtained for hematoxylin and eosin staining, Verhoeff Van Gieson staining, immunohistochemistry, and Western blotting. The incidence and severity of AAAs, macrophage infiltration, and MMP protein expression were all recorded. The results showed that ACE2 gene transfer significantly decreased the occurrence of AAAs and inhibited AAA formation in ApoE−/– mice by inhibiting the inflammatory response and MMP activation, and the mechanisms may involve decreased ERK and Ang II–nuclear factor kappa B signaling pathways.
Introduction
A
Recent studies have demonstrated that the renin-angiotensin system (RAS), especially Ang II, plays an important role in the pathogenesis of AAAs. Daugherty et al. 2 reported that subcutaneous infusion of Ang II for 28 days gives rise to AAA formation in ApoE−/– mice.
This result suggested that short-term exposure to elevated Ang II could induce and potentiate the vascular pathology. 2 Daugherty et al. also demonstrated that the AT1 receptor antagonist losartan abolished the formation of AAAs induced by Ang II. In contrast, the AT2 receptor antagonist PD123319 increased the incidence and severity of AAAs. 3 These results suggest that the imbalance of RAS is involved in the pathogenesis of AAAs.
Angiotensin-converting enzyme 2 (ACE2) is a homologue of angiotensin-converting enzyme (ACE) and plays an important role as the negative regulator of RAS. ACE2 may degrade ANG II to generate Ang-(1–7), and existing studies indicate that ACE2 acts against ACE under physiological and pathological states. A recent study demonstrated that ACE2 plays an important role in the pathogenesis of atherosclerosis, coronary artery disease, hypertension, left ventricular hypertrophy, and diabetic cardiomyopathy. 4 A previous study showed that ACE2 overexpression inhibited atherosclerosis plaque evolution and increased the stability of atherosclerotic plaques in a rabbit model. 5,6 Recently, Daugherty and Cassis found that the administration of diminazene aceturate, an activator of ACE2, increased ACE2 activity and significantly decreased aortic lumen diameters, therefore reducing the incidence of AAAs. The results indicated that increased ACE2 activity may play an important role in the pathogenesis of AAAs. 7 However, few studies have reported the direct effect of ACE2 overexpression on the aneurysm.
This study hypothesized that the overexpression of ACE2 may prevent the pathogenesis of aneurysms by decreasing RAS activation. Based on this hypothesis, the effects of ACE2 overexpression on the pathogenesis of aneurysm were determined in ApoE−/– mice.
Methods
Recombinant adenovirus construction
Adenovirus containing murine ACE2 cDNA was produced, as previously described. 5,6 The recombinant adenovirus carrying murine ACE2 (Ad.ACE2) or a control transgene (Ad-EGFP) was prepared by use of the AdMax system.
Animal model
Male ApoE−/– mice on a C57BL/6J background strain were purchased from the animal center of Shandong Medical University. Mice were housed individually and then fed a high-fat diet (0.25% cholesterol and 15% cocoa butter). All animal care and experimental protocols complied with the animal management rule of the Ministry of Public Health, P.R. China, and the protocol was examined and approved by the Animal Ethics Committee of Shandong University. All surgeries were performed under intraperitoneal injections of ketamine and xylazine anesthesia and local anesthesia of the inguinal region by lidocaine.
Thirty-nine mice (24 weeks old, male) were assigned randomly to three groups (n = 13 in each group): the ACE2 group, the
Histological analysis and diagnosis of aneurysms
All animals were euthanized by an intraperitoneal injection of ketamine and xylazine. The aortas of the mice were dissected from the surrounding connective tissue, and the aortas were perfused with phosphate-buffered saline and fixed with 10% buffered formalin solution. The maximal aortic diameter was measured with a biological dissection microscope. Aneurysms were diagnosed with according to the literature. A commonly used clinical standard to diagnose aneurysms, where an aneurysm is defined as when the maximum diameter of the aorta exceeds 50% of the normal diameter, was used.
Histology and immunohistochemistry
Abdominal aortas containing AAAs were embedded in paraffin. Paraffin-embedded arteries were cut into 4 μm cross-sections and stained with Verhoeff Van Gieson and hematoxylin and eosin according to previously described methods.
To identify macrophage infiltration and MCP-1 and MMP expression in the aortic wall, rabbit anti-mouse antibodies for CD68, MCP-1, MMP-2, and MMP-9 (Abcam, Cambridge, United Kingdom) were used, and the biotinylated secondary antibody followed by avidin–biotin amplification was used for detection. The slides were incubated with 3,3′-diaminobenzidine and counterstained with hematoxylin. Quantification of the positive staining area was executed by two blinded investigators. Each cross-section was analyzed in eight randomly selected areas, and the average result was calculated using an automated image analysis system (Image-Pro Plus v5.0, Media Cybernetics, Rockville, MD).
Western blot
ERK1/2 and nuclear factor kappa B (NF-κB) protein expression were detected by Western blot analysis. Mice abdominal aortas containing AAAs were homogenized in RIPA lysis buffer (Sigma–Aldrich Chemie, Steinheim, Germany), and the total protein was extracted. The protein sample was electrophoresed on 14% sodium dodecyl sulfate polyacrylamide gel and transferred to a polyvinylidene difluoride membrane for 120 min at 250 mA. Following incubation in blocking solution (4% nonfat milk; Sigma-Aldrich Chemie), membranes were hybridized with 1:250 dilution of the primary antibodies anti-ACE2 (Santa Cruz Biotechnology, Dallas, TX), anti-ERK (Cell Signaling Technology, Danvers, MA), and anti-NF-κB (Abcam). The membranes were then incubated with 1:1,000 diluted secondary antibodies for 60 min at room temperature. Then, the membranes were incubated with enhanced chemiluminescence reagent, and relative intensities of protein bands were detected by use of an MSF-300G Scanner (Microtek Lab, Nikon, Japan).
Measurement of blood pressure and serum cholesterol, low-density lipoprotein, and triglyceride
The blood pressure of the mice was measured with a noninvasive tail-cuff system (BP2010AUL; Softron, Mississauga, Canada) in the morning. To avoid anxiety and tension, mice were acclimated to the blood pressure monitor for 6 days before the actual measurement. Starting on the seventh day, blood pressure was measured once every 2 days. The remaining 12 values were averaged and used for analysis. The serum cholesterol, low-density lipoprotein, and triglyceride levels were measured using an enzymatic assay kit.
Ang-(1–7) level by enzyme-linked immunosorbent assay
The concentration of Ang-(1–7) in the mouse abdominal aortas containing AAAs was measured using a commercial enzyme-linked immunosorbent assay (ELISA) kit (Bachem, Torrance, CA) at the end of the experiment. The abdominal aortas were mechanically homogenized with 0.045 N HCl in ethanol containing 0.90 μmol/L p-hydroxymercuribenzoate, 50 μmol/L 1,10-phenanthroline, 0.032% EDTA, 0.90 μmol/L phenylmethylsulfonyl fluoride, 1.75 μmol/L pepstatin A, and 0.0043% protease-free bovine serum albumin on ice. The samples were centrifuged at 3000 g for 10 min at 4°C. The level of Ang-(1–7) was determined by ELISA.
Statistical analysis
Data analysis was completed using SPSS Statistics for Windows v11.5 (SPSS, Inc., Chicago, IL). Pearson's chi-square test was performed to compare the aneurysm subtype variables and the incidence of aneurysms. One-way analysis of variance was used to test the difference of means among three groups for continuous variables. A p-value of <0.05 was considered statistically significant.
Results
Delivery of Ad.ACE2 induced by ACE2 overexpression and the activity in aortic tissue in ApoE−/– mice
The efficacy of Ad.ACE2 gene transfer and the expression of ACE2 protein in aortic tissue in ApoE−/– mice were determined. Two weeks after Ad.ACE2 injection, ACE2 expression in the abdominal aortic tissue was abundant, as determined using Western blotting. ACE2 protein was highly expressed in the aortic tissue of the Ad.ACE2 group but was weakly expressed in the aortic tissue of the Ad.EGFP and control groups (Fig. 1A and B). These results demonstrated that Ad.ACE2 transfection in vivo was successful.

ACE2 protein expression and ACE2 overexpression suppressed Ang II–induced AAA formation in ApoE−/–
mice.
ACE2 overexpression suppressed AAA formation in ApoE−/– mice
The effect of ACE2 overexpression on Ang II–induced AAA progression was evaluated, and the results showed that Ang II infusion significantly increased the incidence of AAA formation in the Ad.EGFP and control groups (Fig. 1C and Fig. 2), with 12/13 (92%) mice in the Ad.EGFP group and 11/13 (85%) mice in the control group exhibiting aneurysms (Fig. 3A). However, 6/13 (46%) mice in the Ad.ACE2 group exhibited aneurysms, indicating that ACE2 gene transfer significantly decreased the occurrence of AAAs.

ACE2 overexpression prevented remodeling of the aortic wall.

ACE2 overexpression reduces the incidence and severity of Ang II–induced AAA formation in ApoE−/–
mice.
Furthermore, the maximal aortic diameters were measured in three groups. The diameter of abdominal aortas in the Ad.EGFP group (1.94 ± 0.37 mm) and the control group (1.95 ± 0.35 mm) were significantly higher than that in the Ad.ACE2 group (1.26 ± 0.40 mm, n = 13; Fig. 3B). The results demonstrated that ACE2 treatment obviously attenuated the incidence and severity of Ang II–induced AAA formation in ApoE−/– mice.
To determine elastin degradation, the grades of elastin degradation were used, as described previously. The grades were judged and defined as follows: grade 1, no degradation; grade 2, mild elastin degradation; grade 3, severe elastin degradation; and grade 4, aortic rupture. 10
The ratio of aneurysm rupture was also evaluated in each group. The ratios of aneurysm rupture were 75% in the Ad.EGFP group and 73% in the control group. However, the ratio of aneurysm rupture (40%) by ACE2 gene treatment was significantly lower in the Ad.ACE2 group than in the Ad.EGFP and control groups.
ACE2 overexpression decreased the inflammatory response
The results showed that Ang II infusion not only increased MCP-1 expression but also induced macrophage infiltration, as evaluated by immunohistochemical staining (Fig. 4). Conversely, ACE2 overexpression significantly decreased MCP-1 expression and macrophage infiltration. These results suggested that ACE2 overexpression inhibited the inflammatory response in aortic aneurysms.

ACE2 overexpression inhibited the expression of macrophage infiltration and MCP-1 protein expression.
ACE2 overexpression inhibited MMP-9 and MMP-2 expression
The results showed that the protein expression of MMP-9 and MMP-2 detected by immunohistochemistry was extensive in the Ad.EGFP and control groups compared to that of the Ad.ACE2 group (Fig. 5). The protein expression of MMP-9 and MMP-2 was significantly lower in the Ad.ACE2 group than in the Ad.EGFP and control groups.

ACE2 overexpression inhibited MMP-9 and MMP-2 expression.
ACE2 regulates ERK and NF-κB signaling pathways
To explain the signaling pathways in aortic aneurysms, ERK1/2 signaling pathways were evaluated. ERK1/2 phosphorylation in aneurysm tissue was significantly lower in the Ad.ACE2 group than in the Ad.EGFP and control groups (Fig. 6A and B), and the results suggest that ERK1/2 plays an important role in the pathogenesis of AAAs. Additionally, NF-κB protein expression was also significantly lower in the Ad.ACE2 group than in the Ad.EGFP and control groups (Fig. 6C and D), and the results indicated that ACE2 overexpression inhibited NF-κB protein expression.
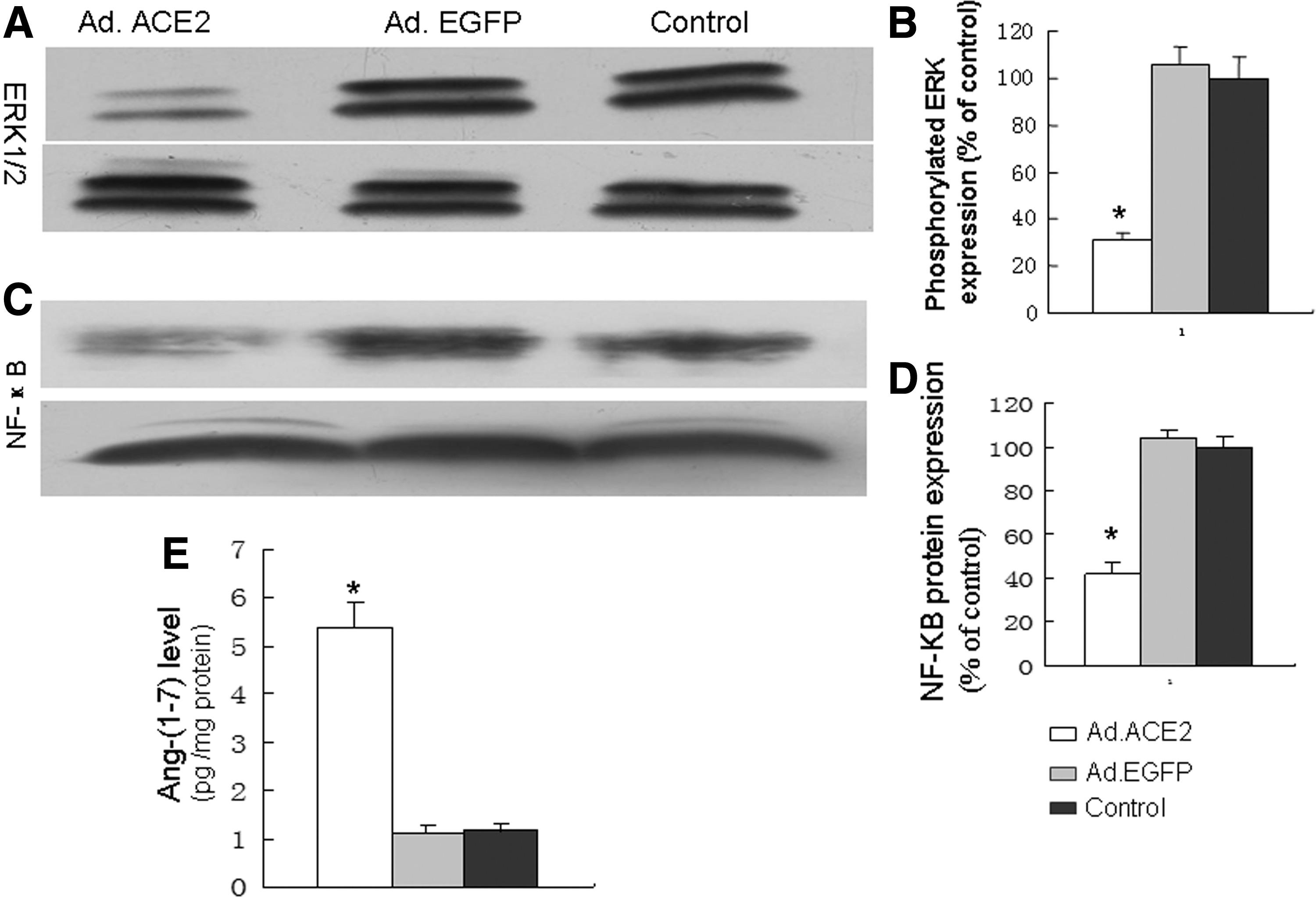
ACE2 overexpression inhibited MAPK kinase and NF-κB expression.
ACE2 overexpression modulates the Ang-(1–7) level
The concentration of Ang-(1–7) was measured by a commercial ELISA kit. The results showed that the Ang-(1–7) level was significantly higher in the Ad.ACE2 group than in the Ad.EGFP and control groups (Fig. 6E).
Biological measurements and blood pressure
There were no significant differences in lipid profiles and body weight among the Ad.ACE2, Ad.EGFP and control groups, but ACE2 gene transfer lowered the blood pressure in the Ad.ACE2 group relative to the Ad.EGFP and control groups (Table 1).
Blood pressure, serum total cholesterol, low-density lipoprotein (LDL), and triglyceride levels in the three groups studied
p < 0.01 versus Ad.EGFP and control group.
Discussion
The major finding of this study was that ACE2 overexpression significantly decreased the incidence and severity of aneurysms in ApoE−/– mice. The underlying mechanisms may include the reduction of macrophage infiltration, decreased MMP-2 and MMP-9 expression, and prevention of ERK phosphorylation and NF-κB expression in AAA.
The pathology of AAAs is rather complicated. Studies have shown that many pathological factors, including smoking, hypertension, high lipid level, ageing, male sex, and chronic inflammation are involved in the pathogenesis of AAAs. 10 –12 A recent study 5,6,12 showed that ACE2 activity and expression were aberrant in atherosclerosis, diabetic cardiomyopathy, acute myocardial infarction, and so on. 5,6,12 Strategies to increase ACE2 activity have been demonstrated to be protective against cardiovascular diseases, including anti-atherosclerosis and anti-left hypertrophy, increasing the stability of plaques and the inhibition of myocardial fibrosis. 5,6,12
A recent study by Daugherty and Cassis showed that the whole-body deficiency of ACE2 increased the severity of AAAs, and the administration of the activator of ACE2 (diminazene aceturate) reduced the incidence of AAAs, but this study did not observe the direct effect of ACE2 overexpression on the aneurysm. 7 Here, ACE2 overexpression was used to increase ACE2 activity, and the protective effects of ACE2 on Ang II–induced AAA evolution were demonstrated. The results showed that Ang II infusion significantly increased the severity and incidence of AAAs. Conversely, ACE2 gene transfer significantly decreased the occurrence and severity of AAAs, and the results demonstrated that ACE2 overexpression inhibited AAA formation.
The effects of ACE2 overexpression on the inhibition of AAA evolution may be involved in various and complicated mechanisms. A chronic inflammatory response in the aortic wall is a main characteristic of AAAs. 13 –16 A study showed that macrophage infiltration is highly expressed in the AAAs. 15–16 Activated macrophages can produce many inflammatory cytokines and chemokines. Macrophages can also produce MMP, which can degrade the extracellular matrix and produce pro-apoptotic mediators, which induce SMCS apoptosis. These could give rise to an imbalance between matrix degradation and synthesis and result in AAA formation.
A large body of research has demonstrated that RAS plays an important role in the pathogenesis of atherosclerosis and human AAAs. Ang II not only plays a role in macrophage activation but also stimulates the secretion of the inflammatory response in atherosclerosis and aneurysms. This study found that the infusion of Ang II induced marked inflammatory responses in AAA, which manifests as increased macrophage infiltration and MCP-1 expression. However, the administration of ACE2 overexpression led to a significant decrease in macrophage infiltration and MCP-1 secretion. Another author found that AAAs in ACE2-deficient mice exhibited pronounced macrophage infiltration at sites of medial degeneration. 7 The results suggested that ACE2 overexpression inhibited the inflammatory response in AAAs.
MMPs play an important role in the degradation of extracellular matrix and the impairment of the structural integrity of the vascular wall, which contribute immeasurably to AAA development. 17 MMPs, especially MMP-2 and MMP-9, have been demonstrated to play important roles in human disease. The positive relationship between increased MMP activity and aneurysm evolution has been reported. Yali et al. 8 found that MMP-2 and MMP-9 activity was significantly increased in AAAs in ApoE−/– mice, and the administration of simvastatin decreased MMP-2 and MMP-9 activity. Another study demonstrated that treatment with the MMP inhibitor doxycycline prohibited the development of the AAA model induced by calcium chloride. 8 Our study showed that MMP-2 and MMP-9 protein levels were significantly higher in AAAs, and ACE2 overexpression decreased the expression of MMP-2 and MMP-9. These results indicate that decreased MMP-2 and MMP-9 protein expression may be an important mechanism behind ACE2 overexpression.
Previous studies demonstrated that ERK signaling pathways are activated by AngII, and Zhang et al. found that simvastatin inhibited ERK phosphorylation and aneurysm formation. Furthermore, the administration of the ERK inhibitor CI1040 mimicked the effect of simvastatin on the aneurysm induced by AngII, and this result suggested that ERK is involved in the pathogenesis of aneurysm. 8 A previous study found that MAPK activation has been associated with macrophage infiltration and the activation of MMPs in the aneurysm. 18 Ghosh et al. found that the MAPK pathway is an important modulator of MMPs during the formation of AAAs. Accumulating evidence indicates that the MAPK pathway takes part in the pathogenesis of AAAs by increasing MMP production and the inflammatory response. 18 This study found that ERK phosphorylation was significantly higher in the Ad.EGFP and control groups than in the Ad.ACE2 group. This suggested that ACE2 inhibited the ERK signaling pathways.
A recent study showed that NF-κB expression was significantly higher in the intracranial aneurysm wall tissue than in the normal intracranial arterial tissues. 14 Ang II has properties consistent with the activation of the NF-κB, and activation of this pathway increases the transcription of both MCP-1 and adhesion molecules. In close concert, these effectors could promote the entry of white blood cells into the arterial wall and take part in the pathogenesis of AAAs. 19 Saito et al. 15 found that endothelial NF-κB promotes adhesion molecule expression, triggers macrophage infiltration, and increases the inflammatory response in aneurysm formation. These studies suggest that NF-κB plays an important role in vascular remodeling and aortic aneurysm formation. The current results showed that Ang II induced NF-κB activity in aneurysms, and ACE2 overexpression inhibited NF-κB activity. A previous study also found that the overexpression of ACE2 reversed the imbalance of ACE2/ACE, decreased the Ang II level, and increased Ang-(1–7) levels. 5,6 Another study demonstrated that Ang II can activate NF-κB activity. 15 Thus, it is speculated that the mechanism of ACE2 overexpression against NF-κB activities may be due to its inhibition of Ang II–induced NF-κB activity.
Combining the present results with those of other studies, it is speculated that complex signaling pathways, including MAPK and NF-κB, MMP activation, and the inflammatory response may be involved in the pathogenesis of aneurysms. The mechanism of ACE2 overexpression to reduce the onset of AAAs may have two aspects: the direct effect of ACE2 overexpression itself and the indirect effect of ACE2 overexpression by signaling circuits. The former refer to that ACE2 overexpression decreased Ang II and increased Ang-(1–7) levels, which prohibited the pathogenesis of aneurysms. The latter suggests that ACE2 overexpression inhibited the Ang II–induced MAPK and Ang II-ROS-NF-κB signaling pathways, which produced protection against the formation of AAAs.
In summary, this study demonstrated that ACE2 overexpression significantly prevented the pathogenesis of aneurysms by inhibiting the inflammatory response and MMP expression. The mechanisms may involve decreased ERK and Ang II-ROS-NF-κB signaling pathways, as well as decreased Ang II levels and increased Ang-(1–7) levels. However, the exact downstream signaling pathways mediating the therapeutic effects are still unclear and require further investigation. These results indicate that ACE2 may provide a new and unique therapeutic target in the prevention and treatment of aneurysms.
Footnotes
Acknowledgments
This study was supported by the National 973 Basic Research Program of China (no. 2013CB530700), the National Natural Science Foundation of China (no. 81570729, 81170207), the Shandong Provincial Science Foundation of China (no. 81170207, 03BS37), and the Program of State Chinese Medicine Administration Bureau (no. JDZX2012113).
Author Disclosure
No competing financial interests exist.