
Abstract
The use of recombinant adeno-associated viruses (rAAVs) ushered in a new millennium of gene transfer for therapeutic treatment of a number of conditions, including congenital blindness, hemophilia, and spinal muscular atrophy. rAAV vectors have remarkable staying power from a therapeutic standpoint, withstanding several ebbs and flows. As new technologies such as clustered regularly interspaced short palindromic repeat genome editing emerge, it is now the delivery tool—the AAV vector—that is the stalwart. The long-standing safety of this vector in a multitude of clinical settings makes rAAV a selling point in the advancement of approaches for gene replacement, gene knockdown, gene editing, and genome modification/engineering. The research community is building on these advances to develop more tailored delivery approaches and to tweak the genome in new and unique ways. Intertwining these approaches with newly engineered rAAV vectors is greatly expanding the available tools to manipulate gene expression with a therapeutic intent.
Gene Replacement is the Standard Bearer of Gene Therapeutics
T

Broad categories of recombinant adeno-associated virus therapeutic strategies. (
RNA Interference Remains a Powerful Approach for Gene Knockdown
The capability of eliciting specific reduction of a mutant gene or virus has considerable appeal for gene therapy. RNA interference (RNAi) is an evolutionarily conserved mechanism to suppress endogenous and exogenous genes through complementarity between a small RNA and the target sequence. 9 Typically, primary microRNA transcripts are processed in two steps by Drosha 10 and Dicer, associate with Argonaute (Ago) proteins, and enter the RNA-induced silencing complex (RISC) where the resulting 21–24nt single stranded RNA is recruited to suppress target genes. 11 –13 Regions with complete complementary between the microRNA and mRNA target lead to most efficient mRNA knockdown through Ago2-mediated cleavage and degradation. 14 RNAi can be co-opted from a gene therapy perspective to suppress target genes of interest through use of synthetic double-stranded oligonucleotides or as genetic DNA templates from which hairpin RNAs are transcribed in the target cells. 15 Indeed, the discovery that genes could be targeted using the RNAi pathway was rapidly validated in mammals by the use of oligonucleotides 16 or small hairpin RNAs, 17 underscoring the potential therapeutic application of this technology. The ability to utilize rAAV vectors as delivery tools for RNAi-based gene knockdown has profound consequences for the treatment of disease.
Several gene knockdown strategies have been particularly effective in using rAAVs to deliver microRNAs (miRNAs) or short hairpin RNAs (shRNAs), especially in the preclinical evaluation of disease in rodents. 18 RNAi-based approaches are best suited for targeting mutations that confer gain-of-function properties, including expanded repeat disorders or to eliminate transcripts from pathogenic organisms. In the liver, delivery of shRNAs that target the hepatitis C virus (HCV) have been validated in nonhuman primate models of HCV infection 19 and attempted in clinical trials. However, because of the recent success of small molecule HCV inhibitors, interest in pursuing shRNA knockdown approaches has instead shifted to treating chronic hepatitis B. 17,20 The success of this approach, though, likely requires AAVs that can transduce the majority of hepatocytes, something that has yet to be achieved in humans.
Targeting the mutant SERPINA1 gene is a viable option for the treatment of the most common form of α-1 antitrypsin deficiency. 21,22 These patients can suffer from liver disease and/or early onset pulmonary emphysema. The former is the result of accumulated mutant protein in hepatocytes, while the latter is the consequence of a deficiency in the neutrophil elastase inhibitor. Liver disease can present at any time but commonly occurs in childhood and is the leading genetic cause for liver transplantation in the Western world. A combinatorial approach has thus been attempted whereby the administered vector can supply both the wild-type SERPINA1 coding sequence and an shRNA that specifically targets the mutant allele, reducing the mutant protein.
Delivery of shRNAs for gene knockdown has had success in other tissues, including the muscle for facioscapulohumeral muscular dystrophy 23 –25 and the eye for age-related macular degeneration and retinitis pigmentosa. 26,27 The brain represents a complex organ where shRNA-mediated removal of mutant RNAs and protein aggregates would have a profound impact on several neuronal disorders. For example, shRNAs have been administered to knock down Ataxin1 (Atxn1) harboring expanded CAG trinucleotide repeats that recapitulate Spinocerebellar ataxia in mice, ultimately reducing intranuclear inclusions and restoring cerebellar function. 28 Likewise, delivery of rAAVs bearing shRNAs that target CAG expansions in Huntingtin (Htt) have improved phenotypic outcomes in mouse models of Huntington's disease, 29,30 and rAAV-shRNAs have been used successfully to target superoxide dismutase 1 (Sod1) in rodent 31 –33 and monkey 34 models of amyotrophic lateral sclerosis and α-synuclein (Scna) in Parkinson's disease. 35 Together, the functionality of these approaches has been validated and can be translated to other related mutant genes and disorders.
Despite the success of rAAV-based RNAi, fewer clinical trials are in the pipeline than, for instance, antisense oligonucleotides (ASOs) or other nucleotide delivery mechanisms. The advantage of an AAV system is that a single administration can provide sustained expression of shRNAs, with levels of therapeutic oligonucleotide potentially increasing rather than decreasing longitudinally. Some of these discrepancies have been minimized through the development of optimized formulations of oligonucleotides and delivery conjugates that extend their potency and bioavailability and reduce cytotoxic responses. 36 Mammals are more amenable to repeated infusions of oligonucleotides at present, enabling a dosing regimen that can be titered and halted if necessary.
Several barriers still need to be overcome before the use of rAAV-shRNAs is broadly accepted. Complications including toxicity arising from high doses of shRNA delivery need to be recognized and avoided in order to ensure safe and sustained gene knockdown. This toxicity is associated with too high a dose of delivered shRNAs, irrespective of their sequence or the presence of an endogenous target. 20 In the context of the liver, the effect of endogenous miR-122 levels are an important indicator of sustained gene knockdown and unintended toxicity. 37 Several approaches have been designed to circumvent the severe effects of this toxicity. The most effective appears to be to design a sequence that first requires Drosha processing 38 in contrast to shRNAs that form a stem-loop structure that enters the RNA interference pathway at the Dicer cleavage step, bypassing Drosha processing but facilitating gene silencing. 16,39 –42 While the efficiency of Drosha processed miRNA-like structures may not approach shRNA expression, the level of target knockdown is sufficient to ameliorate many conditions. 38,43 Alternatively, a shorter pre-miRNA scaffold can be generated that bypasses Dicer processing and instead is generated via Ago2 cleavage. 44 Ago2 can also be co-delivered along with an shRNA to permit long-term gene suppression, 45 or RNA decoys can be co-administered to sequester sense strand by-products of shRNA processing. 46 shRNAs are continually being optimized with regards to expression in order to design a more robust processing site with fewer unintended small RNAs that are generated, reducing off-target effects. 47 Together, these refinements will permit the generation of safer and more effective vectors.
In the future, host genomes could be modified to express shRNAs that target a mutant gene or viral sequence continually. For instance, hepatocytes could be proliferated in the presence of shRNAs, targeting various viral sequences and preventing their replication. rAAVs could in theory be used to deliver the appropriate gene set to reprogram progenitor cells such as induced pluripotent stem cells (iPSCs), as well as expressing antiviral shRNAs prior to their transplantation to combat a badly damaged liver resulting from a chronic viral infection.
Gene Editing: Transcription Activator-Like Effector Nucleases and Zinc Finger Nucleases Lay the Groundwork for Targeted Genome Modifications
Classical rAAV provides an episomal genome in transduced cells. For quiescent tissues, this can provide long-term transgene expression, although the true length of persistence for each individual tissue is not yet known. In regenerative tissues or in infants, long-term expression is unlikely. 48 However, using any of the genome modification approaches would in most tissues provide lifelong expression. Hence, another advantage of the approaches to follow includes the possibility of treating neonates with a single vector dose administration. This is important because repeat administration of rAAVs may prove to be difficult due to the induction of humoral immune responses.
The zinc finger nucleases (ZFNs), 49 transcription activator-like effector nucleases (TALENs), 50 and engineered meganucleases 51 have provided an exciting starting point for the modification of host genomes. Broadly, they introduce a double-stranded break in the genomic DNA to be modified, followed by replacement of the desired sequence with a DNA donor template. Although TALENs and ZFNs may be considerably more time-consuming and laborious to construct than clustered regularly interspaced short palindromic repeat (CRISPR) gene editing (see below), this pales in comparison to the length and cost of a clinical trial, and should not necessarily be considered an impediment. In addition, there is greater difficulty in identifying a sequence that can be effectively targeted, but when found, it is possible that this affords a greater on versus off targeting rate. These approaches have translated into clinical trials, for instance by Sangamo Biosciences. Electroporation of ZFN mRNA targeting a safe harbor site such as the AAVS1 site on chromosome 19 and administration of a rAAV expressing a transgene of interest. One example is the ex vivo introduction of gp91phox in hematopoietic stem and progenitor cells for intervention in X-linked chronic granulomatous disease. 52 Likewise, the factor VIII or factor IX transgene can be inserted at the highly expressed albumin locus for amelioration of hemophilia A and B. 53,54 The open clinical trial on this indication is certainly of great interest, though concerns remain about the continued expression of a nuclease and the long-term consequences of potential off-targeting and/or immunogenicity associated with expression of a foreign protein. Provided the safety profile is acceptable and preclinical studies demonstrate few off-targeting events (and most importantly none that are harmful), this technology may be what ends up with the greatest translational potential.
CRISPR/Cas9 Opens a Pandora's Box of Gene Editing Prospects
The adaption of the CRISPR adaptive immune defense system in bacteria and archaea to mammalian systems has revolutionized molecular biology. 55 –57 CRISPR and the CRISPR-associated endonuclease Cas9 sample segments of phage or plasmid DNA to incorporate into CRISPR loci, 58 –60 which are then transcribed and processed into CRISPR RNAs (crRNAs). 61 These crRNAs guide Cas9 to homologous regions in invading sequences, facilitating their recognition and elimination. 62 The CRISPR system can be adopted and engineered to mediate double-stranded genomic breaks in any genomic sequence adjacent to a 5′-NGG protospacer-adjacent motif (PAM) through the association of a chimeric single-guide RNAs (sgRNA) with the Cas9 nuclease. 57 These breaks are typically rejoined through non-homologous end joining (NHEJ), and as this process is imperfect, short insertions and deletions (indels) at the targeted region are often generated. 63 In the event of an indel, the target region is no longer a perfect complement to the sgRNA, and the region becomes largely impervious to further editing by the sgRNA. Ideally, when employed correctly, this approach introduces frameshift mutations that lead to loss of expression of the target gene. Co-delivery of an oligonucleotide or rAAV bearing sequence homology to the target region can act as a template for homologous recombination (HR) mediated repair of the break to alter the resulting genetic sequence, often for the purpose of correcting a disease-causing mutation. 55,56 Together, while much of the groundwork for understanding the CRISPR/Cas9 complex in a natural and therapeutic setting has only recently been established, the work clearly reveals the remarkable potential of this system.
Just like with RNAi, the discovery that CRISPR/Cas9 approaches can be used to modify mammalian genomes has piqued interest into how this new technology can be extended from a powerful molecular biology approach to a therapeutic context. This is a nascent field, meaning that it will be intriguing to determine which low-hanging fruits (such as disrupting the HIV CCR5 co-receptor 64 ) are available for study. Researchers have already reported success in utilizing the CRISPR/Cas9 system and rAAV vectors for a multitude of expanding approaches. This includes correcting ornithine transcarbamylase (OTC) deficiency in newborn mice, 65 eliminating fumarylacetoacetate hydrolase (Fah)-positive hepatocytes by eliminating Fah splicing mutations, 66 reducing Pcsk9 levels for reduction of serum cholesterol, 67 introducing and correcting PRKAG2 mutations in mice for therapy of the cardiac Wolff–Parkinson–White syndrome, 68 ex vivo correction of the β-globin gene in sickle cell disease, 69 and skipping mutant exons in a mouse model of Duchenne muscular dystrophy. 70 –72 Proof-of-principle approaches have also shown the ability to disrupt multiple genes in the adult mouse brain. 73 Without question, CRISPR/Cas9 technology has already facilitated the development of animal models for diseases that previously were impractical or inconceivable to generate, for instance generation of several combined mutations causative for lung cancer. 74 Other technological developments include targeting non-genic regions to modulate gene expression. Potential allele-specific knockdown can be achieved when one allele is heavily methylated at CpG islands, for instance targeting the mutant, unmethylated, and expressed allele of p16INK4a in the HCT116 colorectal cancer cell line. 75 Allele-specific removal of an expanded CAG repeat has also recently been demonstrated in the HTT gene using adjacent common single nucleotide polymorphisms (SNPs) present within a patient that alter a PAM motif. 76 The use of common SNPs for allele-specific silencing is not new; this has been attempted previously for si/shRNA knockdown of several mutant genes, including HTT itself. 77 However, often the wild-type allele was simultaneously reduced. The definitive nucleotide sequence requirement of the PAM motif affords the allele specificity that was previously challenging. Several biotech companies have raced to develop therapeutic strategies employing CRISPR-mediated gene editing. While the operational success of these companies may hinge on the outcome of ongoing patent disputes, several strategies are already in the works. The results of these discoveries are eagerly anticipated and add an exciting new dimension to the American Society of Gene and Cell Therapy annual meeting.
Countless optimizations are still in the works to improve this system from a therapeutic standpoint (Fig. 2). Drawing parallels with oligonucleotide therapeutics, chemical modifications such as 2′-O-methyl and related analogs of the sgRNA can improve stability and bioavailability. 78 From a development standpoint, humanizing Cas9 to dampen immune recognition, as has been employed for antibody development, 79 and biasing to HR instead of NHEJ could present fewer complications. Mutating one of the HNH and RuvC nuclease domains to generate a Cas9 nickase enzyme 57,80 that cuts one DNA strand can facilitate specificity. 81 Delivering two guide RNAs that recognize adjacent genomic regions can vastly reduce the number of off-target cuts. Furthermore, the generation of a completely catalytically inert (dead) dCas9 with Asp10Ala and His840Ala mutations 57 can be used to tether proteins that modulate the genome. This takes advantage of the target specificity of the Cas9-gRNA complex. Theoretically, the consequence of targeting other genomic regions is also vastly minimized. For instance, combining dCas9 with a cytodine deaminase enzyme can introduce C-to-T or G-to-A edits without introducing DNA breaks, albeit with a more narrow range of targetable regions in the context of a PAM motif. 82 Activation or inhibition (CRISPRa or CRISPRi) of genomic loci can be accomplished by fusing dCas9 to transcriptional activators or repressors as a fascinating approach to fine-tune gene regulation. 83,84 Expanding the repertoire of approaches, the CRISPR/Cas9 complex has also been engineered to target single-stranded RNA. 85 Many of these combinatorial delivery approaches likely exceed the rAAV vector capacity and may require co-delivery or other clever tricks. Not all of these approaches will translate to the clinic either but serve as examples of modifications to the CRISPR/Cas9 system that can drive more exquisite modification of a target gene or genome and curb concerns of off-target genome editing.
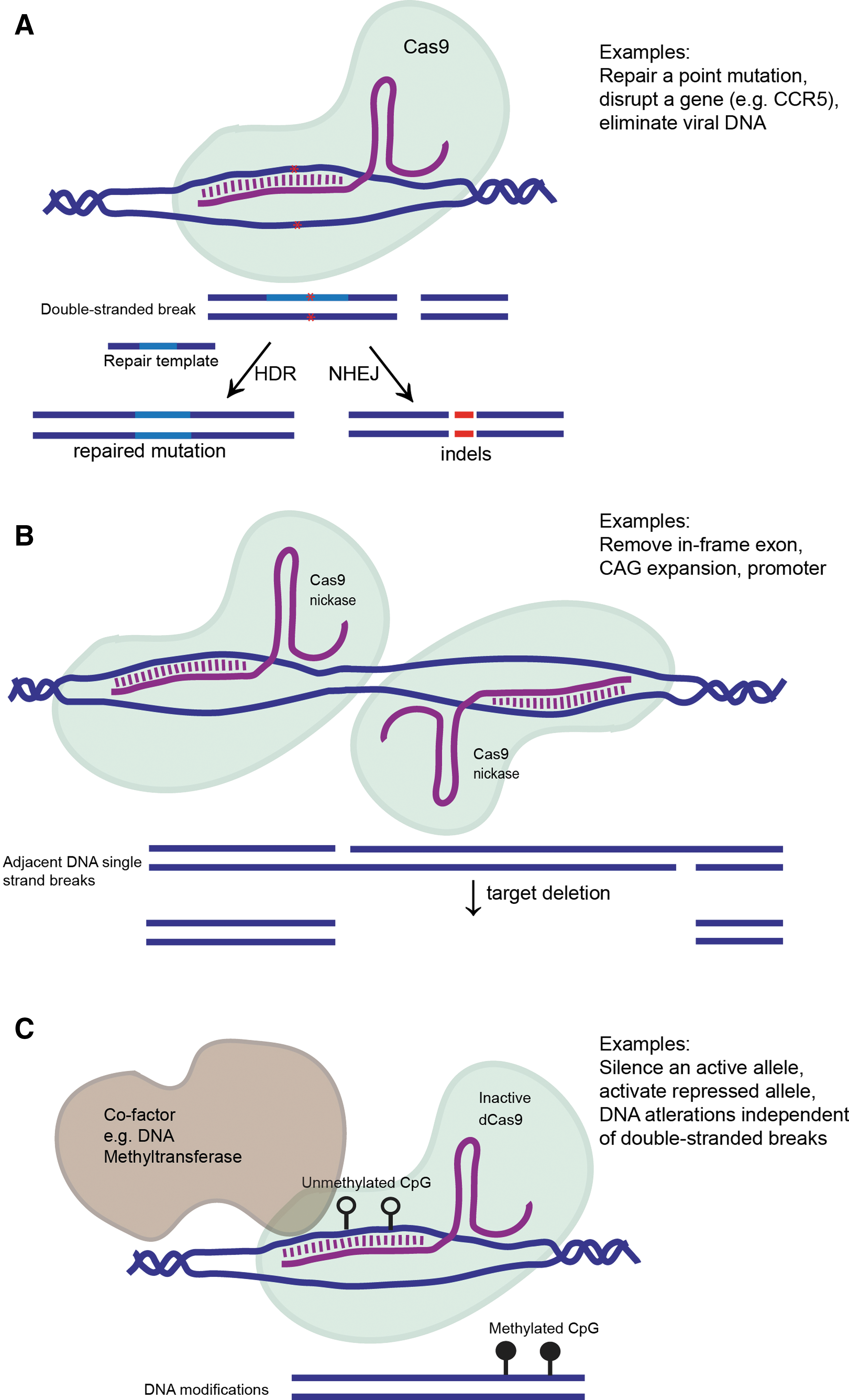
Broad categories of CRISPR/Cas9 genome editing strategies. (
The AAV research community should be heartened that the development of CRISPR/Cas9 approaches has embraced AAV vectors for delivery. The size limitation of rAAV vectors, however, has necessitated some optimization (as is usually the case) to ensure appropriate expression and gene editing. One approach to leave sufficient space for therapeutic transgenes is to use the Staphylococcus aureus (saCas9) system, reducing the length of Cas9 by about 1 kb from the 4.2 kb version derived from the widely used Staphylococcus pyogenes. This more manageably fits the 4.7 kb size constraints of AAV without compromising editing, initially requiring a modestly less abundant 5′-NNGRRT PAM motif, 67 which has been further optimized to confer additional targets using a NNNRRT PAM. 86 Importantly, using saCas9 permits delivery of Cas9 and one or more promoter-driven sgRNAs in the same vector. Another approach involves dual-rAAV delivery, including an intein for trans-splicing, again leaving sufficient space for sgRNAs. 87 The optimal approach likely will be to devise a mechanism to express Cas9 transiently, enabling hit-and-run edits at the intended target with next to no edits at unintended regions. Importantly, the stability (and clearly the functionality) of the sgRNA appears to be dependent on the presence Cas9. 78
For CRISPR/Cas9 gene editing technology to really take hold in a clinical context, several critical considerations need to be taken into account. This includes the consequences of off-target genome edits and immune responses to components of the CRISPR/Cas9 machinery, along with an acceptable delivery regimen, promoter choice, and sgRNA expression levels. It is impossible to overstate how dangerous it might be to have an endonuclease such as Cas9 continually present within the cell potentially editing more and more off-targets over time, leading to potentially drastic and dire consequences on a cell and therefore also in humans. Presently, this represents a formidable obstacle. Furthermore, it is exceedingly difficult to ensure that only the target genome region is edited, although optimized Cas9 nucleases have drastically reduced or undetectable off-targets based on current technology. 88,89 Even by high-resolution sequencing approaches, the consequences of potential off-target editing on each individual cell is difficult to ascertain. This is further complicated by cell-specific chromatin architecture and Cas9 accessibility precluding the direct translation of findings in cell culture to safety and success in animals and ultimately in humans. It should also be noted that most drugs have some level of off-targeting from their intended target, and ultimately how rare nuclease mediated off-targeting translate into truly detrimental events is not known.
Other concerns of the CRISPR/Cas9 system include the widespread use of the U6 Pol-III promoter to drive sgRNAs; this promoter can lead to toxicity when used to express shRNAs at high levels. 20,37 Situations that preclude the efficacy of this approach should be reported to ensure that the field is aware of scenarios that are prohibitive. A case in point is when attempting to edit and then correct using oligonucleotide-templated repair a hypomorphic sparse fur ash (spfash ) allele causing OTC in adult mice. The efficiency of repair was incomplete, resulting in complete loss of the transcript and a more adverse consequence than the initial relatively subtle mutation. 65 This underscores some of the difficulties in attempting multifaceted therapies such as combining editing and repair. Even if safe concentrations of CRISPR/Cas9 in vivo are achievable in humans, reaching effective levels to treat various disorders will need to be demonstrated. Finally, from an ethical standpoint, the field has monitored and attempted to discourage the potential for human embryo editing and other ramifications of the CRISPR/Cas9 system. 90 Over time, society will have to make the decisions on how far to move in this direction.
These challenges notwithstanding, the potential of CRISPR/Cas9 technology is remarkable, and contemplating the various uses is extremely exciting. For instance, one could envision a scenario whereby methods could be employed to modify somatic variants in a tumor back to the original host genomic sequence or correct a disease-causing mutation in carriers of a particular condition prior to symptom onset. These and other possibilities make for a promising future.
RNAi Knockdown and Nuclease-Mediated Knockout Comparisons
RNAi and nucleases such as CRISPR/Cas9 approaches differ in their consequence for gene targets, efficiency, and off-target effects. Genome editing has the potential to effectively knock out the resulting mRNA transcript, in contrast to gene knockdown with shRNAs that reduces and in some cases essentially eliminates existing transcripts. With shRNAs, the dose can be titrated to reduce but not necessarily eliminate a mutant transcript (or mutant and wild type, where it is problematic to affect only one allele exclusively). The convergence of hits or pathways from both complementary approaches can reveal scientifically meaningful insights. For instance, whole-transcriptome libraries can be generated using shRNAs or CRISPR-mediated edits of each gene. 91,92 These studies establish that results from one approach rarely if ever will perfectly match that of the other. Together they can cover more ground and reveal which genes prevent or accelerate phenotype when suppressed or completely eliminated. Both approaches could conceivably be combined to target a gene and knock down any residual or mutant transcripts. In the long term, it is likely that both RNAi and nuclease-mediated genome editing will find their specific therapeutic niches.
Advances in Modulating the Genome
Rather than gene knockdown or editing, a more traditional strategy involves the delivery of sequences complementary to a mammalian genome and the use of homologous recombination to integrate portions of the delivered viral sequence in vivo. 93 This can be used to model disease such as the generation of a porcine model of hereditary tyrosinemia type 1. 94 This also has several therapeutic applications, including the targeting of a disease gene to correct a mutant allele back to wild-type state, for instance the in vitro correction of a LAMA3 mutation in epidermolysis bullosa with the idea that this can then be reintroduced into patients. 95 Alternatively, rAAV-bearing homology arms flanking a coding and/or non-coding therapeutic sequences can be precisely integrated into a locus, allowing the expression of a completely new transgene of interest such as factor IX for the treatment of hemophilia. 96 In this example, the factor IX coding sequence was inserted near the 3′ end of the albumin locus but just upstream of the stop codon. Using a ribosomal skipping sequence, just 5′ of the factor IX coding results in a chimeric mRNA transcript that produces both albumin and factor IX proteins. Because the albumin promoter is so robust, up to 20% of the normal factor IX level was stably expressed from mouse liver, even though <1% of the albumin alleles were targeted. This “generide” approach is advantageous over classical AAV and nuclease-mediated gene therapy in that off-target integrations should have reduced capacity for expression given the avoidance of a promoter sequence. However, the low level of homologous recombination-mediated integration events 97,98 means the few successful integrants have to drive considerable transgene expression and mostly in conditions where incomplete gene replacement is sufficient to alleviate phenotypic consequences. However, improvements in gene targeting efficiency or utilizing a selection scheme can enhance the potential of this approach. 99 Recently, including an additional intron that encodes an shRNA into the factor IX sequence enabled a metabolic selection scheme when a short course of drug therapy was provided. This resulted in a stable 50-fold expansion of the gene-modified cells. 100 If the efficiency of this technology can be expanded considerably, the idea of directing rAAV to modulate genomic sequence can have a lasting effect on the cell, permitting selection of hepatocytes that have been corrected of a particular genetic mutation. In general, these approaches enable stable genomic modification and sustained intracellular effects.
The rare but non-negligible level of rAAV integration events, especially at unintended genomic intervals, needs to be monitored and addressed when contemplating the safety profile of rAAV therapy. Much has been discussed as to whether there is cause for concern over rAAV integration, especially at the mouse chr12qF1 Dlk1-Dio3 locus leading to hepatocellular carcinoma, including in this issue of Human Gene Therapy. The similarity in sequence conservation of all protein coding genes, noncoding RNAs, and most microRNAs along with parent-of-origin methylation status between the mouse chr12qF1 locus and the syntenic region on human chr14q32.2 (as is the case in all eutherian mammals) 101 means that findings in the mouse likely could have analogous complications in humans. The absence of a promoter in the delivered transgene can at least minimize complications and aberrant activation of genes neighboring these integrants.
Targeting Non-Coding RNAs
The expanding world of non-coding RNAs affords additional means of therapeutic intervention through the fine-tuning of their gene expression, and thus represents a new frontier of sequences to target or express. Several classes of RNAs from long non-coding RNAs (lncRNAs) to microRNAs are characterized. Other categories include a large complement of small nucleolar RNAs (snoRNAs) and small RNAs derived from tRNAs. The cellular roles of these RNAs are still being elucidated, and represent new facets of human biology and disease. rAAVs can be utilized to supplement microRNAs that are depleted in certain conditions such as miR-26a in liver cancer 102 or enhance decay of a target such as miR-196a in spinal and bulbar muscular atrophy. 103 rAAVs can also be exploited to administer a genomic sequence capable of sequestering microRNAs or RNA binding proteins, perhaps through the generation of a stable circular RNA such as ones that have been identified to be a sponge for miR-7. 104,105 Intricate interactions similarly exist between pseudogenes and their coding counterparts such as Pten. 106,107 These approaches can be contemplated for the removal of microRNAs or other non-coding RNAs that are elevated in various conditions, particularly in various forms of cancer. 108 Ultimately, as the non-coding RNA field matures and mechanisms of disease are established, the relationships between coding and non-coding elements of the genome can be exploited in a therapeutic context.
AAV Optimization for Enhanced Tropism and Delivery
While highly effective as a starting point, just like any drug, optimization of rAAV broadens the applicability and specificity of this technology. The differences in mice and humans in both transduction efficiencies of various rAAV serotypes 109 and immune responses mean a considerable amount of research is still warranted to ensure optimal safety and potency. At present, certain tissues are much more prone to rAAV transduction, meaning the study of associated diseases such as those affecting the liver can be studied with confidence. In other tissues, the ability for efficient targeting and transduction still presents a challenge. Consequently, the field of rAAV biology has graduated from a discovery-based approach for the identification of AAV serotypes 110 to the generation of shuffled capsid and peptide display libraries that can confer novel targeting properties 111 –114 to the rational design and engineering of novel serotypes. This is most notable in nervous system transduction where the ability of AAV to cross the blood–brain barrier and transduce neurons reveals tremendous potential for neuronal disease. Building on this work, groups have identified serotypes with peptide insertions that have been rigorously evaluated for enhanced neuronal transduction properties 115 or for retrograde transport to projection neurons. 116 Other approaches involve enhanced neuronal tropism via grafting the AAV9 galactose binding motif onto AAV2 117 through addition of a poly-alanine peptide at the N-terminus of the VP2 capsid gene 118 or through an AAV-capsid shuffling strategy. 119 rAAV serotypes have been generated that have optimized transduction in tissue culture 111 or that selectively infect human hepatocytes. 109 Additional strategies have been employed to enhance in vivo or ex vivo tropism through, for instance, incorporation of designed ankyrin repeat proteins in the AAV capsid protein VP2. 120 Work evaluating the crystal structure of AAV is imperative for comprehending how these modifications are tolerated and alter physiochemical properties of AAV. An understanding of some of the cellular receptors and machinery that permit cellular AAV entry can be modulated to enhance tropism of a vector or to de-target a particular organ (especially the liver), permitting more circulating AAV that can access an organ of interest. Notable examples include the identification of the KIAA0319L type I transmembrane (AAVR) receptor for AAV entry, 121 the heparan sulfate proteoglycan identified for AAV2 attachment, 122 and the N-linked terminal galactosyl residue binding specificity for AAV9. 123 Together, the combination of approaches will undoubtedly lead to a more diverse toolkit of rAAV vectors for selective transduction of one or more organs previously refractory or suboptimal to rAAV entry and transgene expression.
Conclusions
Just like the CRISPR/Cas9 field that has adopted rAAV vectors for delivery, future technologies will benefit from a gene delivery tool that has been studied for decades with established safety profiles. The field of gene therapy has benefited from a strong long-standing community. It is imperative that this collaboration continues to raise and adequately address issues associated with gene therapy approaches and AAV vectors. With these responsible approaches, the AAV community can enjoy continued successes and can be married to new technologies as they are developed and implemented.
Footnotes
Acknowledgments
We are grateful to members of the Kay lab for their critiques and suggestions. This work was supported by grants NIH 1R01 DK 078424, 1R01 AI11698, R01 HL064274 (M.A.K.).
Author Disclosure
No competing financial interests exist for Paul Valdmanis. Mark Kay is a scientific founder of Voyager Therapeutics, and founder of LogicBio Therapeutics.