
Abstract
Chronic granulomatous disease (CGD) is characterized by defects in the production of microbicidal reactive oxygen species (ROS) by phagocytes. Testing of gene and cell therapies for the treatment of CGD in human hematopoietic cells requires preclinical transplant models. The use of the lymphocyte-deficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/ SzJ (NSG) mouse strain for human hematopoietic cell xenografts to test CGD therapies is complicated by the presence of functional mouse granulocytes capable of producing ROS for subsequent bacterial and fungal killing. To establish a phagocyte-defective mouse model of X-linked CGD (X-CGD) in NSG mice, clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 was utilized for targeted knockout of mouse Cybb on the X-chromosome by microinjection of NSG mouse zygotes with Cas9 mRNA and CRISPR single-guide RNA targeting Cybb exon 1 or exon 3. This resulted in a high incidence of indel formation at the CRISPR target site, with all mice exhibiting deletions in at least one Cybb allele based on sequence analysis of tail snip DNA. A female mouse heterozygous for a 235-bp deletion in Cybb exon 1 was bred to an NSG male to establish the X-CGD NSG mouse strain, NSG.Cybb[KO]. Resulting male offspring with the 235 bp deletion were found to be defective for production of ROS by neutrophils and other phagocytes, and demonstrated increased susceptibility to spontaneous bacterial and fungal infections with granulomatous inflammation. The establishment of the phagocyte-defective NSG.Cybb[KO] mouse model enables the in vivo assessment of gene and cell therapy strategies for treating CGD in human hematopoietic cell transplants without obfuscation by functional mouse phagocytes, and may also be useful for modeling other phagocyte disorders in humanized NSG mouse xenografts.
Introduction
C
Allogeneic transplant of hematopoietic stem/progenitor cells (HSPCs) can cure CGD, but many patients lack a suitable donor, and graft-versus-host-disease remains a significant risk. Gene-therapy approaches for the correction and transplant of autologous CGD patient HSPCs lack these issues. Such approaches include retrovirus 2 –4 and lentivirus vectors 5,6 and, more recently, nuclease-mediated methods of targeted gene insertion or correction. 7 –9 For X-CGD, autologous HSPC gene therapy using retrovirus vector derived from murine spleen focus forming virus has demonstrated clinical benefit for life-threatening infections, but long-term gene marking was low, and myelodysplasia due to retroviral insertional mutagenesis was observed. 3,4 This demonstrates the need for additional gene-correction strategies, which require preclinical models of X-CGD gene therapy in patient cells to test their efficacy in a transplant setting.
An in vivo model of X-CGD was previously established in the C57BL/6 mouse background by Dinauer's group through homologous recombination-mediated disruption of exon 3 of the mouse Cybb gene, 10 which resulted in defective ROS production and increased susceptibility to experimental infection with Staphylococcus aureus and Aspergillus fumigatus in affected Cybb knockout male mice. Although this mouse strain allows for modeling of the X-CGD phenotype and gene and cell therapy approaches for treatment using mouse hematopoietic cells, 11 –14 alternate models are necessary for preclinical testing of human gene and human cell therapies for CGD, such as targeting human HSPCs as tested in vivo in a xenograft animal model.
The use of lymphocyte-deficient mouse strains, such as NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJ (NSG) mice, 15 enables the transplant of human HSPCs to reconstitute mice with human hematopoietic cells, including mature functional neutrophils and other phagocytic cells. However, NSG mice also produce functional mouse neutrophils and other phagocytes, which complicate the functional analysis of engrafted human phagocytes. Crossbreeding of the already established X-CGD C57BL/6 mouse strain into the genetically complex NSG background would be a lengthy and complicated breeding program. Alternatively, here, the generation of an X-CGD mouse model is described in the NSG mouse strain by using clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease 16 –18 to knock out the mouse Cybb gene in zygotes from NSG mice through induction of indels (insertions or deletions) at the CRISPR target site. Cybb knockout NSG mice exhibited the defect in ROS production and susceptibility to microbial infections with granulomatous inflammation that are characteristic of CGD, and maintained the capacity for human HSPC engraftment, enabling human CGD therapeutic strategies to be tested without the background of functionally oxidase-positive mouse phagocytes.
Materials and Methods
Approvals for human blood and animal use
CD34+ HSPC-enriched granulocyte colony-stimulating factor (G-CSF) mobilized peripheral blood was obtained from healthy volunteers and X-CGD patients after written informed consent following the Declaration of Helsinki under the National Institute of Allergy and Infectious Diseases (NIAID) Institutional Review Board–approved protocol 94-I-0073.
The use of lymphocyte-deficient NSG mice (JAX Stock No: 005557; obtained from The Jackson Laboratory, Bar Harbor, ME) and Cybb knockout NSG (NSG.Cybb[KO]) mice was approved by the NIAID Animal Care and Use Committee (ACUC) under animal use protocol LHD 3E and by the National Heart, Lung, and Blood Institute ACUC under animal use protocol H-0125R2.
Generation of Cybb knockout NSG mice
Cybb knockout mice were generated directly on the NSG genetic background using CRISPR/Cas9 methodology. 19 Briefly, two CRISPR guide RNA sequences, targeting Cybb exon 1 (GGTACTTACAATGACAAAGA) or exon 3 (GATTTCGACACACTGGCAGC), were separately cloned into the pDR274 vector (Addgene plasmid #42250). 20 Corresponding sgRNAs (single-guide RNAs) were syntheized by in vitro transcription using MEGAshortscript T7 transcription kit (Invitrogen; Thermo Fisher Scientific, Waltham, MA). Cas9 mRNA was synthesized by in vitro transcription from the plasmid pMLM3613 (Addgene plasmid #42251) 20 using the mMESSAGE mMACHINE T7 kit (Invitrogen). Either exon 1 or exon 3 sgRNA was individually mixed with Cas9 mRNA at a concentration of 50 ng/μL of Cas9 mRNA and 20 ng/μL of sgRNA in nuclease-free microinjection buffer (10 mM of Tris, pH 7.5, 0.1 mM of EDTA) and microinjected into the cytoplasm of fertilized eggs collected from NSG mouse mating pairs. The injected zygotes were cultured overnight in M16 medium (EMD Millipore, Billerica, MA) at 37°C in 5% CO2. The next morning, embryos that had reached the two-cell stage of development were implanted into the oviducts of pseudopregnant foster mothers (Swiss Webster mice; Taconic Biosciences, Hudson, NY).
The mice born to the foster mothers were genotyped by polymerase chain reaction (PCR) of Cybb exon 1 or 3 using tail snip DNA using Q5 high-fidelity DNA polymerase (New England Biolabs, Ipswich, MA), and PCR products were sequenced commercially (Macrogen, Rockville, MD). Primers used for exon 1 PCR and sequencing included mCybb1-565-FWD: 5′-GTTGGAAGAGCCTGTGAGAAGA-3′, mCybb1-851-FWD: 5′-GGGAACAGCCTTTCAGTTGG-3′, mCybb1-908-FWD: 5′-CCTATTGCCCCAAAGCTGCT-3′, mCybb1-1192-BWD: 5′-ACCGAATCTACCTGCAAGCA-3′, mCybb1-1232-BWD: 5′-AAGGCGTGCTGGGATTAAGA-3′, mCybb1-1467-BWD: 5′-TGCACTCTGGTAAATGCTGG-3′. Primers used for exon 3 PCR and sequencing included mCybb3-3905-FWD: 5′-AGGATAGGAGTTCTTGCCGC-3′, mCybb3-4213-FWD: 5′-AACTGTGGTCTGGGAGGTGA-3′, mCybb3-4431-FWD: 5′-TGGGGAAGAGGAATACGGGT-3′, mCybb3-4694-BWD: 5′-ACAGAGCATTGCTTGGCTCT-3′, mCybb3-4858-BWD: 5′-TCCAGGAACTTTTGCCCTCA-3′, mCybb3-5156-BWD: 5′-TGCTTGTCTCAGGCTCCTTA-3′.
A female mouse heterozygous for a 235 bp deletion in Cybb exon 1 was bred to an NSG male to establish the NSG.Cybb[KO] mouse strain. Genotyping of NSG.Cybb[KO] mice was performed using the mCybb1-565-FWD and mCybb1-1192-BWD primer set, resulting in a 628 bp product for the wild-type allele and a 393 bp product for the 235 bp deletion. NSG.Cybb[KO] mice were maintained by crossing of heterozygous females with NSG males both to avoid genetic drift from the NSG background and to protect breeders from spontaneous bacterial or fungal infections to which CGD mice are susceptible. In order to reduce the incidence of infections, breeding mice and offspring were provided with water containing 0.67 mg/mL of sulfamethoxazole and 0.13 mg/mL of trimethoprim (Sulfatrim; STI Pharma, Langhorne, PA), and regular cage changes were performed for these mice before those of other mice housed in the facility.
Predictions of likely deletions resulting from microhomology-mediated end joining
21
(MMEJ) at the exon 1 and exon 3 CRISPR cut sites were performed using the Microhomology-Predictor tool of the Center for Genome Engineering, Institute for Basic Science, Korea (
Dihydrorhodamine assessment of ROS activity in NSG.Cybb[KO] mice
For dihydrorhodamine (DHR) assay of ROS activity on mouse peripheral blood, 100–200 μL of mouse blood was collected by tail venisection, lysed with ACK lysis buffer (Quality Biological, Gaithersburg, MD) for 5–7 min at 37°C, and washed with Hank's balanced salt solution without calcium or magnesium (HBSS; Thermo Fisher Scientific). Cells were resuspended in 400 μL of HBSS, 130 μM of DHR (Molecular Probes; Thermo Fisher Scientific), and 500 IU of catalase (Sigma–Aldrich, St. Louis, MO), and samples were incubated at 37°C for 5 min. Samples were then stimulated for ROS production by addition of 100 μL of 400 ng/mL phorbol myristate acetate (Sigma–Aldrich) in HBSS containing calcium and magnesium (Thermo Fisher Scientific), followed by incubation at 37°C for 14 min. Cells were analyzed by flow cytometry for DHR fluorescence using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA), as previously described. 22
NSG.Cybb[KO] mouse pathology analysis
For histopathology, mouse tissues were fixed in 10% buffered formalin and embedded in paraffin, cut at a thickness of 5 μm on a microtome, placed on slides, and stained with hematoxylin and eosin. Each section was examined by a board-certified veterinary pathologist. Images were obtained using a Zeiss Observer 1 microscope (Zeiss, Thornwood, NY) and ZEN blue software (Zeiss).
For bacterial and fungal identification from mouse infections, isolates were cultured on blood agar, phenylethyl alcohol (PEA) agar, MacConkey agar, chocolate agar, anaerobic reducible blood agar, anaerobic reducible PEA agar, anaerobic reducible LKV agar, Sabouraud dextrose agar, inhibitory mold agar slants, thioglycollate broth, or brain heart infusion broth (Remel; Thermo Fisher Scientific), and were further identified based on analysis using spot indole reagent (Remel), Biolog GEN III MicroPlates (Biolog, Hayward, CA), or Vitek 2 identification cards for Gram positive bacteria, Gram negative bacteria, or yeast (bioMérieux, Durham, NC), according to the manufacturers' protocols.
Transduction of human CD34+ HSPCs
CL20i4r-EF1a-gp91OPT lentiviral vector 23 for constitutive expression of gp91phox was produced through transient transfection of 293T cells (ATCC, Manassas, VA) with pCL20i4r-EF1a-gp91OPT plasmid and gag-pol, rev-tat, and VSV-G packaging plasmids. 24 Virus was harvested daily for 3 days, filtered (0.22 μm; EMD Millipore), concentrated by centrifugation at 18,600 g for 3 h at 4°C, and reconstituted in X-VIVO 10 medium (Lonza, Walkersville, MD). Titer of CL20i4r-EF1a-gp91OPT lentivirus was measured in transduced K562 cells (ATCC) based on flow cytometry analysis of antibody staining for gp91phox expression, as described below.
Cryopreserved mobilized human CD34+ HSPCs were thawed 24 h prior to first transduction, and cultured in X-VIVO 10 medium containing 1% human serum albumin (Talecris Biotherapeutics, Research Triangle Park, NC) and 50 ng/mL each of human stem cell factor, Flt3-ligand, and thrombopoietin (PeproTech, Rocky Hill, NJ). Up to 5 million mobilized human CD34+ HSPCs from a patient with X-CGD were transduced twice on consecutive days with CL20i4r-EF1a-gp91OPT lentivirus at a multiplicity of infection of 10 viruses per cell, by spinoculation on RetroNectin (Lonza)-coated plates at 1360 g for 30 min at 32°C, followed by overnight culture at 37
For in vitro analysis of transduction efficiency at 3 weeks after the final transduction, HSPCs (naïve X-CGD, transduced X-CGD, or normal donor) were cultured in medium containing 50 ng/mL of G-CSF (Neupogen; Amgen Mfg., Thousand Oaks, CA), then fixed with 2% paraformaldehyde (Sigma–Aldrich) in phosphate-buffered saline (PBS) for 5–10 min, permeabilized with 0.1% saponin (Sigma–Aldrich) in PBS, and stained with an unconjugated antibody to human gp91phox (7D5 antibody) 2 for 15–20 min, followed by staining with a fluorescein isothiocyanate (FITC)-conjugated secondary antibody for 5–10 min. Analysis of gp91phox expression was then performed with a FACSCalibur flow cytometer.
Transplant of human CD34+ HSPCs in NSG.Cybb[KO] mice
NSG.Cybb[KO] mice at 6–10 weeks of age were treated with 20 mg/kg of busulfan by intraperitoneal injection 24 h before transplant. Thereafter, mice were provided with water containing 2.5 mg/mL of neomycin (VetOne, Boise, ID) instead of Sulfatrim. After the final round of overnight transduction (i.e., no more than 3 days total culture after thaw), one million human HSPCs (naïve X-CGD, transduced X-CGD, or normal donor) were transplanted per mouse by tail-vein injection. At 6 weeks post transplant, blood was collected, and mice were euthanized for analysis of bone marrow.
Analysis of human CD34+ HSPCs engraftment and phagocyte activity
Blood and bone-marrow cells were collected from transplanted mice for analysis of human cell engraftment and human X-CGD donor cell correction. Blood samples were lysed with ACK lysis buffer prior to analysis, as described above. Blood cells were stained for ROS production by DHR assay, as described above, to detect functional human phagocytes. In parallel, marrow cells were co-stained with allophycocyanin (APC)-conjugated antibody to human CD45 pan-leukocyte marker (BD Biosciences) to detect engrafted human hematopoietic cells, or phycoerythrin (PE)-conjugated antibody to human CD45 together with APC-conjugated antibody to human CD13 myeloid marker (BD Biosciences) and unconjugated human gp91phox antibody with FITC-conjugated secondary antibody, as described above. For some transplanted mice, additional marrow cells were stained with FITC or APC-conjugated antibodies to CD13, CD14, CD19, or CD56 (BD Biosciences) for analysis of hematopoietic reconstitution of mice with human myeloid cells, monocytes/macrophages, B cells, or NK cells, respectively. Peripheral blood and spleen cells were collected from additional mice at 10 weeks post transplant and stained with PE-Cy5, APC, or FITC-conjugated antibodies to human CD3, CD4, and CD8 (BD Biosciences), for analysis of reconstitution with human T cells. Samples were analyzed with a FACSCalibur flow cytometer.
Results
Establishment and characterization of NSG.Cybb[KO] mouse strain
Injection of NSG mouse zygotes with CRISPR/Cas9 targeting either exon 1 or exon 3 of the mouse Cybb gene on the X-chromosome resulted in highly efficient induction of deletions in Cybb at the CRISPR target site in the resulting mice (Supplementary Table S1; Supplementary Data are available online at

Cybb mutations identified in mice arising from clustered regularly interspaced short palindromic repeats (CRISPR) targeting of Cybb exon 1 or exon 3. (
One female mouse was heterozygous for a 235 bp deletion encompassing all of exon 1 and 190 bp of the upstream Cybb promoter, with the second allele containing a 240 bp deletion (Fig. 1A and Supplementary Table S1). Breeding of the female Cybb knockout founder mouse to an NSG male resulted in germline transmission of the deletions, as confirmed by PCR analysis. Male offspring with the 235 bp deletion in Cybb exon 1 were confirmed to completely lack ROS activity in peripheral blood phagocytes by DHR assay (Fig. 2A) in contrast to wild-type NSG control mice (Fig. 2B). The NSG.Cybb[KO] mouse strain was established and maintained through breeding of females heterozygous for one 235 bp deleted allele and one wild-type allele with wild-type NSG males, generating heterozygous or wild-type females and wild-type or knockout male offspring.
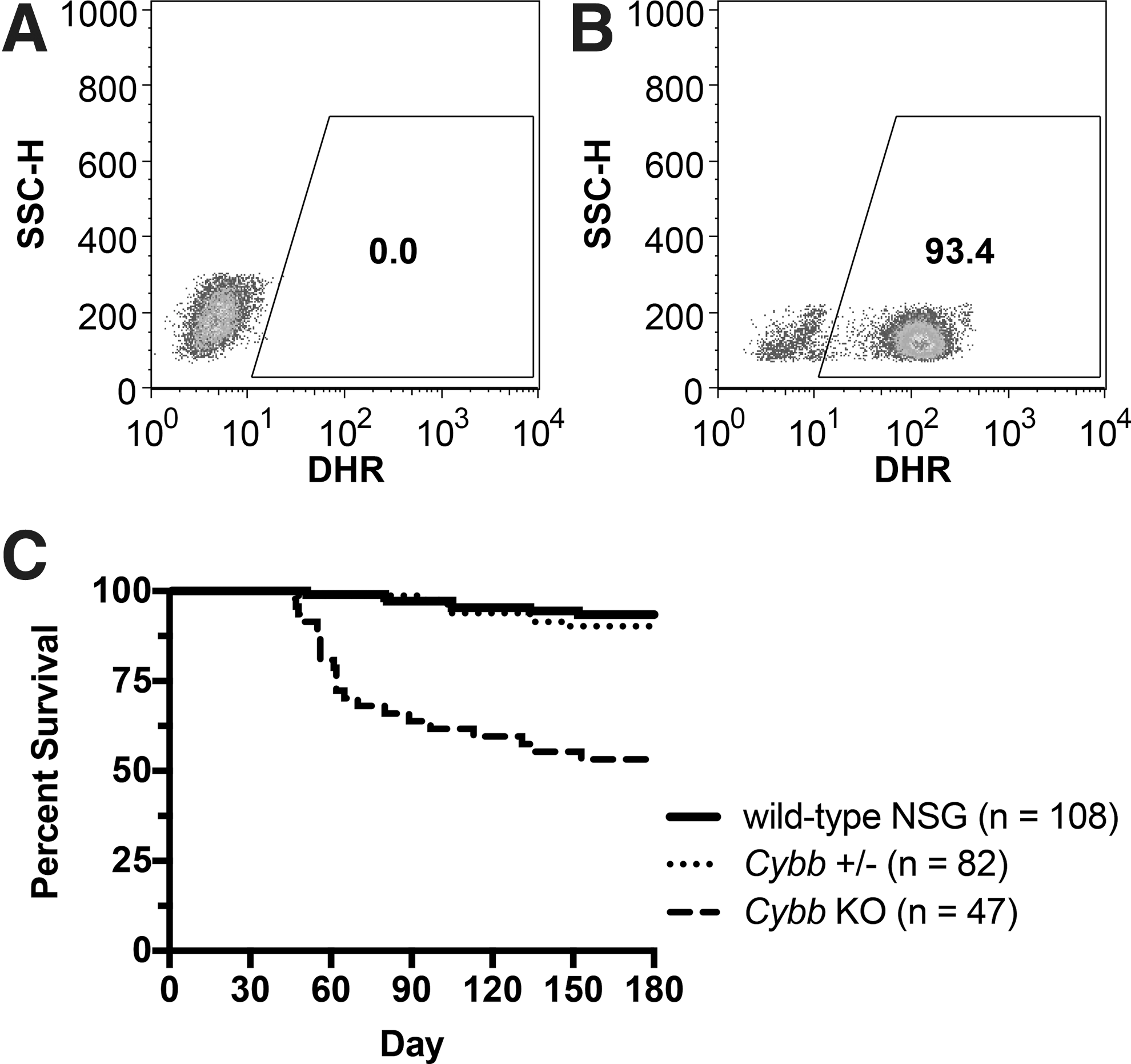
NSG.Cybb[KO] mouse phenotype. (
Despite maintenance of mice with Sulfatrim antibiotic, increased morbidity due to spontaneous bacterial and fungal infections was observed in Cybb knockout mice, along with a significant decrease in mouse survival compared with heterozygous or wild-type NSG mice (Fig. 2C; p < 0.000001 based on log-rank statistical analysis 25 using the Kaplan–Meier estimator), 26 demonstrating that NSG.Cybb[KO] mice exhibit the characteristic susceptibility to microbial infections of human CGD. Microorganisms identified in mouse infections included Staphylococcus xylosus, Staphylococcus hominis, Klebsiella oxytoca, Escherichia coli, Aerococcus viridans, Enterobacter cloacae, and Candida albicans, with multiple infectious species detected in some mice. Further analysis of bacterial cultures established from a K. oxytoca infection confirmed its antibiotic resistance to Sulfatrim. Infections in NSG.Cybb[KO] mice were typically systemic, occurring at multiple tissues in the same mouse. Infection sites included the mouth, esophagus, stomach, lymph node, lung, and liver. Granulomatous inflammation was also observed in knockout mice at sites of microbial infections (Fig. 3), consistent with the phenotype of human CGD. No other causes of morbidity were identified in NSG.Cybb[KO] mice.

Histology of NSG.Cybb[KO] mouse infections. (
Human HSPC transplants in NSG.Cybb[KO] mice
In order to test the utility of NSG.Cybb[KO] mice as a human xenograft model, CD34+ HSPCs from normal, healthy donors or X-CGD patients were transplanted into knockout mice preconditioned with busulfan. As expected for the lymphocyte-deficient NSG mouse background, NSG.Cybb[KO] mice could be engrafted with human HSPCs to give rise to human CD45+ hematopoietic cells of multiple lineages, including CD13+ myeloid cells, CD14+ monocytes/macrophages, CD19+ B cells, CD56+ NK cells, and CD3+ T cells (Fig. 4). Normal donor HSPC transplants resulted in engraftment with phagocytes capable of ROS production in both bone marrow and peripheral blood (Fig. 5A and Supplementary Fig. S1A), while engrafted X-CGD patient cells lacked ROS activity in bone marrow and peripheral blood (Fig. 5B and Supplementary Fig. S1B). The lack of DHR activity by mouse phagocytes in the NSG.Cybb[KO] background allowed for unambiguous detection of even small numbers of human phagocytes that are functional for ROS production.

Multi-lineage human hematopoietic cell engraftment in NSG.Cybb[KO] mice. Shown are bone marrow cells stained for antibodies to human CD45 (pan-leukocyte), CD13 (myeloid cells), CD14 (monocyte/macrophages), CD19 (B cells), and CD56 (NK cells), for (
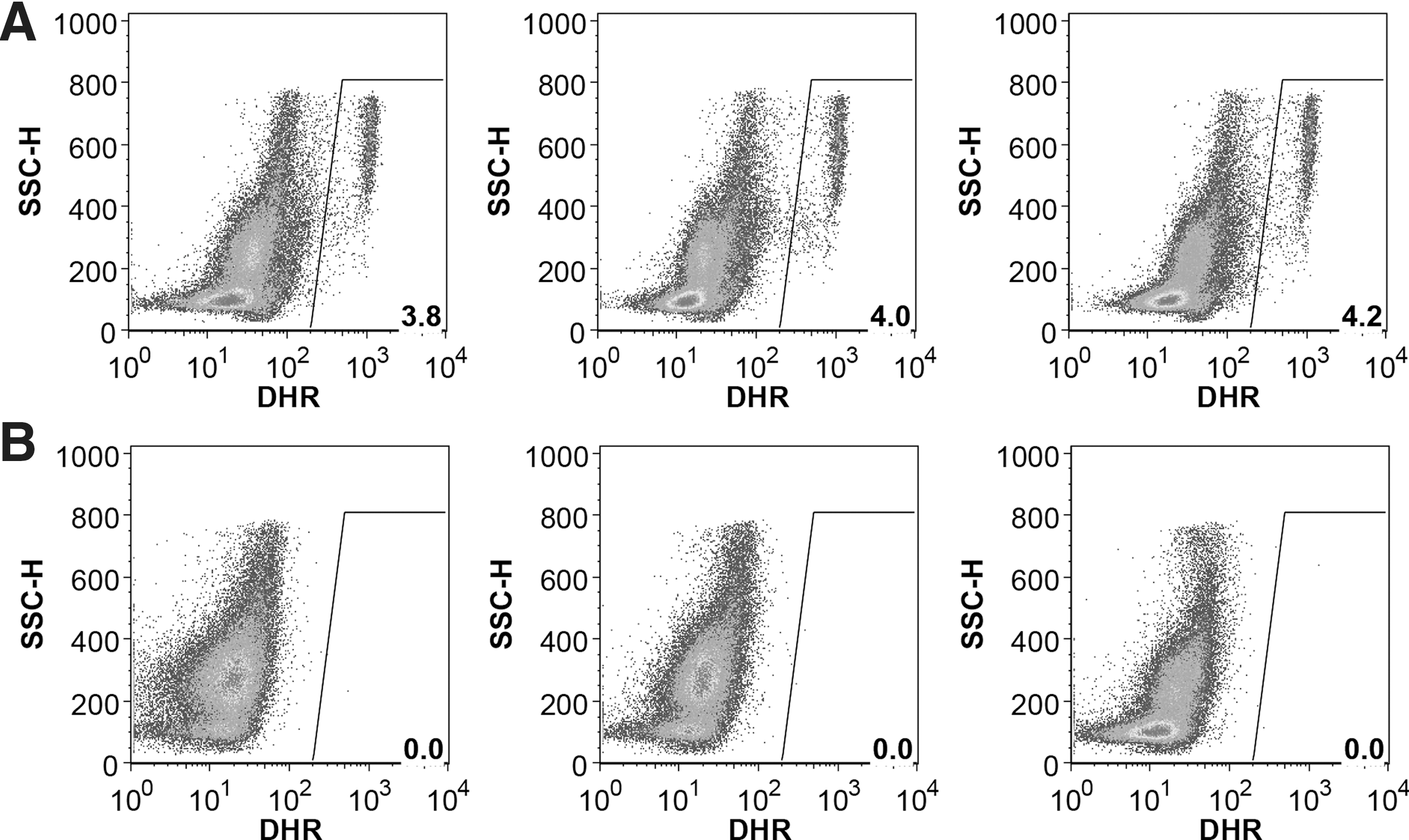
ROS production from engrafted human cells in NSG.Cybb[KO] mice. Shown are DHR assays of ROS production in bone-marrow samples from NSG.Cybb[KO] mice at 6 weeks post transplant with (
To test further the utility of the NSG.Cybb[KO] mouse strain for preclinical modeling of gene therapy approaches to CGD in human cell transplants, X-CGD patient HSPCs were transduced with a self-inactivating CL20i4r-EF1a-gp91OPT lentiviral vector expressing a codon-optimized human CYBB cDNA from the elongation factor-1a (EF1a) short promoter (Fig. 6A). Lentiviral transduction of X-CGD patient HSPCs was confirmed by flow cytometry analysis of gp91phox antibody staining after 3 weeks of in vitro culture in medium containing G-CSF for granulocyte differentiation, which demonstrated the expected defect in gp91phox expression in untransduced patient cells (Supplementary Fig. S2A), while lentivirus-transduced patient cells (Supplementary Fig. S2B) and untransduced normal donor cells (Supplementary Fig. S2C) expressed gp91phox. Transplant of transduced X-CGD patient HSPCs or normal donor HSPCs into NSG.Cybb[KO] mice immediately following the final day of transduction resulted in bone-marrow engraftment and myeloid reconstitution with human hematopoietic cells (Fig. 6B), including transduced patient cells that constitutively expressed gp91phox at levels approximately 70–80% of endogenous gp91phox expression in engrafted normal donor HSPC-derived phagocytes (Fig. 6C) based on mean fluorescence intensities of the positive populations. Corrected human phagocytes in bone marrow produced ROS at mean fluorescence intensities comparable to normal human phagocytes by DHR assay (Fig. 6D), with similar levels observed in peripheral blood phagocytes (Supplementary Fig. S2D).

Transduction and transplant of X-CGD patient HSPCs with lentivirus expressing gp91phox. (
Discussion
This study describes the CRISPR/Cas9-mediated knockout of mouse Cybb to generate a phagocyte-defective model of X-CGD in the lymphocyte-deficient NSG mouse strain, enabling engraftment of human cells to test gene and cell therapies for all forms of human CGD without the background of mouse phagocytes capable of ROS production. Efficient gene targeting using CRISPR/Cas9 in lymphocyte-deficient mice was previously shown in NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl /SzJ (NRG) mice by Li et al., 27 who reported a 100% mutation rate at the Fah gene following CRISPR/Cas9 microinjection into mouse zygotes for the purposes of establishing a model for human hepatocyte and hematopoietic cell xenografts. The similar highly efficient induction of Cybb mutations by CRISPR/Cas9 to generate the NSG.Cybb[KO] model of X-CGD in the current study further demonstrates the utility of CRISPR technology for establishing humanized disease models involving human cell transplants in immunodeficient mice.
The Cybb CRISPRs used in this study were designed without prior microhomology analysis of the target sites to predict deletions that could result from the MMEJ DNA repair pathway. Based on the microhomology deletion pattern scoring metric described by Bae et al., 21 approximately half of the mice in the current study exhibited in-frame mutations that matched three of the top five predicted deletion patterns for the Cybb exon 1 site or the top predicted deletion pattern for the exon 3 site. This high incidence of predictable in-frame deletions lends support for the use of microhomology analysis in selecting nuclease target sites that reduce the likelihood of in-frame deletions in favor of frameshift mutations for more effective knock out of a target gene.
The functional defect in ROS production of the NSG.Cybb[KO] mice resulted in increased sensitivity to spontaneous microbial infections, including bacterial Staphylococcus and Klebsiella species and the fungus C. albicans, which are also common infections in CGD patients. 1 The infections that were identified in moribund animals were all from catalase-positive microorganisms, as expected for CGD infections, except for A. viridans, which is classified as catalase-negative or weakly catalase-positive, and which has previously been reported as a spontaneous pathogen in NOD.Cg-Prkdcscid (NOD/SCID) mice. 28 Morbidity due to infection was also observed in a smaller number of Cybb heterozygous and wild-type NSG mice, including littermates and cage mates of Cybb knockout mice, which is consistent with a previous report of spontaneous bacterial infections, including K. oxytoca, that have been observed in NSG mice at the Jackson Laboratory and other independent breeding colonies. 29
To the authors' knowledge, the NSG.Cybb[KO] strain is the most immunodeficient mouse strain to be engineered to date, since it completely lacks lymphocyte immune functions and is defective in myeloid oxidative killing function. Consequently, this strain requires particular care in order to maintain mouse health and survival. Morbidity of NSG.Cybb[KO] mice due to spontaneous infections was reduced when routine changing of mouse cages was performed prior to handling of other mice in the facility, and use of microisolator cages or other barrier facilities is recommended.
The demonstrated susceptibility of NSG.Cybb[KO] mice to spontaneous microbial infections would also allow for experimental infection studies in these mice, as were performed in the prior C57BL/6 mouse X-CGD model. 10 –14 This aspect of the NSG.Cybb[KO] model will enable the CGD research community to begin assessing the effectiveness of corrected CGD patient HSPCs or other therapeutic human cells for in vivo protection from new infections or clearing of existing infections, expanding the current assays available for preclinical testing of human CGD therapies in a transplant setting. Due to its functional defect in killing of select microorganisms by phagocytic cells, this mouse strain may also be useful for in vivo modeling of other human phagocyte deficiencies or for infection studies involving human cell transplants.
Footnotes
Acknowledgments
This research was supported by the Intramural Research Program of NIAID under intramural project numbers Z01-Al-00644 and Z01-Al-00988. The authors thank the Diagnostic and Research Services Branch of the Division of Veterinary Resources of NIH for pathology and bacteriology analysis, the Department of Laboratory Medicine of the NIH Clinical Center for additional bacteriology analysis, and Crystal Thomas of the Laboratory of Animal Medicine Section of NIAID for advice and assistance in maintaining the health of NSG.Cybb[KO] mice.
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.