
Abstract
While therapeutic expression of coagulation factors from adeno-associated virus (AAV) vectors has been successfully achieved in patients with hemophilia, neutralizing antibodies to the vector and inhibitory antibodies to the transgene severely limit efficacy. Indeed, approximately 40% of mice transduced with human factor VIII using the AAV8 serotype developed inhibitory antibodies to factor VIII (FVIII inhibitor), as well as extremely high titers (≥1:500) of neutralizing antibodies to AAV8. To correct hemophilia in these mice, AAV9, a serotype with low in vitro cross-reactivity (≤1:5) to anti-AAV8, was used to deliver mouse-activated factor VII (mFVIIa). It was found that within 6 weeks of systemic administration of 2 × 1013 particles/kg of AAV9/mFVIIa, hemophiliac mice with FVIII inhibitors and neutralizing antibodies (NAb) to AAV8 achieved hemostasis comparable to that in wild-type mice, as measured by rotational thromboelastometry. A level of 737 ng/mL mFVIIa was achieved after AAV9/mFVIIa adminstration compared to around 150 ng/mL without vector treatment, and concomitantly prothrombin time was shortened. Tissues collected after intra-articular hemorrhage from FVIII-deficient mice and mice with FVIII inhibitors were scored 4.7 and 5.5, respectively, on a scale of 0–10, indicating significant pathological damage. However, transduction with AAV9/mFVIIa decreased pathology scores to 3.6 and eliminated hemosiderin iron deposition in the synovium in most mice. Collectively, these results suggest that application of alternative serotypes of AAV vector to deliver bypassing reagents has the potential to correct hemophilia and prevent hemoarthrosis, even in the presence of FVIII inhibitor and neutralizing antibodies to AAV.
Introduction
H
Unfortunately, broad application of AAV vectors to deliver FVIII or FIX is limited by the development of inhibitory autoantibodies to the missing protein and of neutralizing antibodies to AAV. The development of inhibitory autoantibodies, which render subsequent infusions with FVIII and FIX ineffective, 6,7 is a critical issue that affects up to 30% of patients. In recent prospective, randomized, and controlled trials to investigate the risk of forming inhibitory autoantibodies, the cumulative incidence ranged from 26.8% for plasma-derived FVIII/von Willebrand Factor, to 44.5% for recombinant FVIII. 6 On the other hand, >95% of humans are naturally exposed to AAV, of whom about half produce neutralizing antibodies that prevent gene delivery via AAV.
To overcome inhibitory autoantibodies, large doses of clotting factors are typically administered to elicit immune tolerance. However, this approach is generally ineffective in patients with high antibody titers. In such patients, treatments with alternative products, including activated prothrombin complex and recombinant factor VIIa (FVIIa), have been attempted. Hemostasis is generally achieved in this manner but at lower efficiencies than with replacement of deficient clotting factors. Other less promising approaches include antibody depletion using a combination of immunoabsorption, cyclophosphamide, intravenous immunoglobins, large doses of FVIII, and antibodies against CD20.
To escape AAV neutralizing antibody-mediated clearance of AAV vector, numerous strategies have been investigated. AAV capsids have been engineered in attempts to evade recognition by neutralizing antibodies. For example, AAV vectors have been coated with polymers such as polyethylene glycol, 8 –10 directionally evolved in vitro and in vivo in the presence of neutralizing antibodies to obtain novel AAV capsid variants, 11 –13 or rationally mutated to eliminate antibody recognition sites. 14 Other AAV serotypes that exhibit little to no cross-reactivity with pre-existing neutralizing antibodies 15 –19 have also been explored. Alternatively, several clinical approaches have been used to deplete neutralizing antibodies, including plasmapheresis, infusion with anti-CD20 (Rituximab), 20 and simultaneous delivery of an excess of empty AAV capsids as decoy. 21
This study found that about 40% of hemophiliac mice transduced with AAV8 to express human FVIII eventually developed FVIII inhibitor and neutralizing antibodies to AAV8. An attempt was then made to correct hemophilia by delivering mouse FVIIa via AAV9, which was found in vitro to cross-react poorly with neutralizing antibodies to AAV8. Remarkably, transduction with FVIIa improved hemostasis and mitigated joint damage.
Material and Methods
Animal care
FVIII knockout mice (FVIIIKO), from which exon 16 of FVIII had been deleted, were provided by H. H. Kazazian Jr. (University of Pennsylvania, Philadelphia, PA) 22 and were then bred in-house. Plasma samples were collected from the retro-orbital plexus into 3.2% sodium citrate, and stored at −80°C until analysis. Animal protocols were approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
Construction of AAV vectors
A cassette to express human FVIII 23 was kindly provided by St. Jude Children's Research Hospital and packaged into an AAV8 capsid. Mouse FVIIa was synthesized at Genscript, with the furin cleavage site as linker between the heavy and light chains. Both constructs are driven by the liver-specific transthyretin (TTR) promoter, as described previously. 24 Mouse FVIIa was packaged into an AAV9 capsid. All vectors were produced using a triple transfection protocol and titered at the Virus Vector Core Facility at University of North Carolina at Chapel Hill as described previously. 25
Quantification of FVIII activity and anti-FVIII
Human FVIII activity in mouse plasma was quantified with one-stage FVIII activity assay (FVIII-specific aPTT) using recombinant human factor VIII (Advate®; Baxter, Westlake Village, CA) to prepare the standard curve. Titer of anti-human FVIII inhibitory antibody is measured by the Bethesda assay, as previously described, 26 using a STart® 4 coagulation analyzer (Diagnostica Stago, Asnières, France).
Quantification of mFVIIa expression and prothrombin time
The prothrombin time (PT) assay was performed by adding 50 μL of citrated mouse plasma diluted in Owren-Koller buffer (Diagnostica Stago) at a 1:50 dillution to 50 μL of hFVII-deficient plasma (George King Bio-Medical, Inc., Overland Park, KS), and the time to clot formation was recorded on the STart 4 coagulation analyzer after adding 100 μL of Neoplastine CL Plus reagent (Diagnostica Stago).
For the quantification of mFVIIa expression in mouse plasma, a second enzyme-linked immunosorbent assay was used with minimal cross-reactivity to mFVII zymogen, as described, 27 with a slight modification that included a rat anti-mFVII antibody (Abcam, Cambridge, MA) used to coat the plate. The recombiant mouse FVIIa for the preparation of standard curve was kindly provided as a gift by Dr. Dougald M. Monroe at the Department of Medicine, University of North Carolina at Chapel Hill.
Induction of FVIII inhibitor and AAV8 capsid neutralizing antibodies
To elicit both FVIII inhibitor and neutralizing antibodies against AAV8, a human FVIII expression cassette packaged into AAV8 was administered intravenously at a dose of 4 × 1012 particles/kg body weight into FVIII-deficient mice. This dose has previously been shown to induce peak FVIII activity at 200–300% of normal activity and FVIII inhibitor development in 40% of treated mice (unpublished).
In vitro analysis of neutralizing antibodies
Inhibition of AAV transduction by neutralizing antibodies was assessed in vitro according to published methods. 28 Briefly, Huh7 cells were seeded in a 48-well plate at 1 × 105 cells/well, and cultured with 1 × 108 particles of AAV8 or AAV9/luciferase that had been preincubated for 2 h at 4°C with a serial dilution of sera from mice with neutralizing antibodies to AAV8. Cells were lysed 48 h later, and luciferase activity was measured with a Wallac1420 Victor 2 automated plate reader. Neutralizing antibody titers were defined to be the highest dilution of mouse serum that reduced luciferase activity by 50% in comparison to that in cells transduced with AAV9/luciferase vectors that had been preincubated with phosphate-buffered saline (PBS).
In vivo analysis of neutralizing antibodies
AAV8 or AAV9/luciferase particles (3 × 109) were incubated for 2 h at 4°C in PBS or in 6 μL of undiluted pooled serum from mice with neutralizing antibodies to AAV8. The vectors were then injected directly into the hind-leg muscle of 6- to 8-week-old C57BL/6 mice. Transgene expression was assessed by imaging 2 weeks thereafter.
Rotational thromboelastometry
Clotting was assessed by rotational thromboelastometry (ROTEM), as described previously. 29 Briefly, whole blood was collected from the inferior vena cava at sacrifice, mixed at a ratio of 9:1 with 3.2% sodium citrate, and then 300 μL of the resulting mixture was coagulated with 20 μL of 0.2 M CaCl2 in a pre-warmed rotational thromboelastometer.
Tail transection
Bleeding was assessed in vivo, as described, 30 with a slight modification by transecting 3 mm instead of 4 mm of the distal tail and placing the proximal tail into a pre-warmed pre-weighed tube. Blood loss per gram of body weight was determined 40 min after the tail was clipped, or at the time of death.
Joint pathology
Intra-articular bleeding was assessed, as described, 31 by creating a ∼0.5 mm incision in the skin overlying the patella and then inserting a Hamilton syringe with a 30.5 gauge needle into the knee joint. Knee joints were collected 2 weeks thereafter by sectioning the femur and tibia/fibula 1 cm from the joint. Specimens were fixed and decalcified using routine histology. Sagittal sections were prepared and stained with hematoxylin and eosin to score hemophilic synovitis on a scale of 0–10, as previously described. 32 Additional sections were stained with Prussian Blue to assess iron deposits, as described previously. 33,34
Statistical analysis
Data are presented as mean ± standard error of the mean and were analyzed by one-way analysis of variance in GraphPad Prism for Windows v7 (La Jolla, CA). An adjusted p-value of <0.05 was considered statistically significant.
Results
Characterization of mouse FVIIa in vitro and in vivo
In this study, hemostasis corection was first measured by ROTEM to see if this assay could serve as an alternative option to measure the hemostasis correction in the absence of a reliable commercial mFVIIa detection kit. Different doses of recombinant human FVIII protein were infused into FVIII knockout mice intravenously via the tail vein. As shown in Fig. 1, even the lowest tested dose (2 IU/kg) significantly improved coagulation parameters measured on ROTEM. At doses ≥5 IU/kg, coagulation was comparable to that in wild-type mice. Collectively, the data indicate that ROTEM is a sensitive assay suitable for detecting <5 IU/kg of circulating recombinant FVIII or its equivalent.
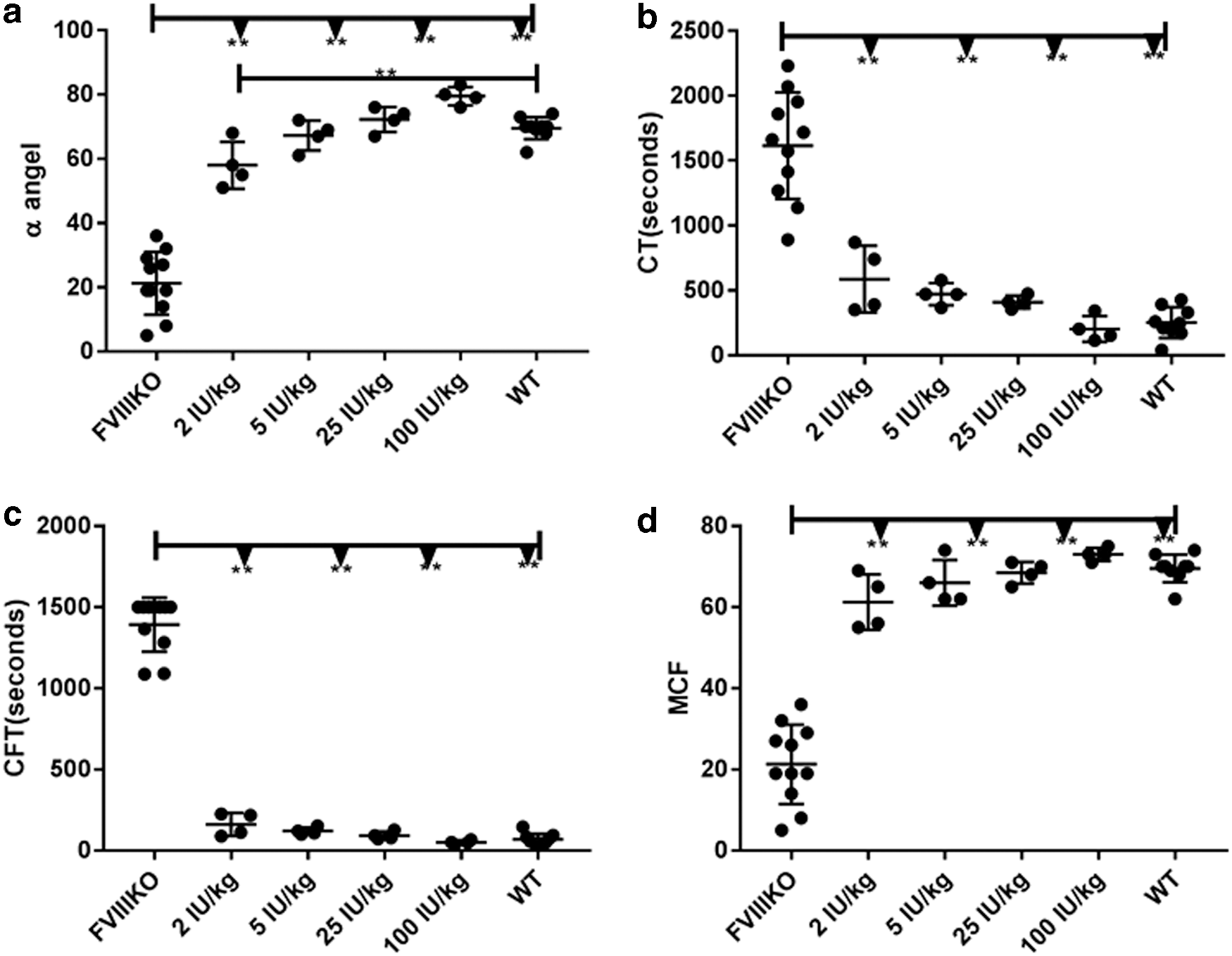
Rotational thromboelastometry (ROTEM) after infusion of recombinant human factor VIII (FVIII). Escalating doses of recombinant human FVIII from 2 to 100 IU/kg were administrated intravenously into FVIII knockout mice, with untreated FVIII knockout mice (FVIIIKO) and wild type (WT) mice as the controls. Citrated whole blood was collected 15 min later from the inferior vena cava, and analyzed by ROTEM to measure
To express mouse FVIIa transgenically, an AAV9 cassette was constructed with the furin cleavage site, RKRRKR, as a linker between the heavy and light chains (Fig. 2A). The well-defined liver-specific promoter TTR was used to drive expression, 24 noting that AAV9, like AAV8, exhibits liver tropism in mice. 35 Doses of AAV9/mFVIIa from 8 × 1011 to 2 × 1013 particles/kg were then intravenously administered to FVIII knockout mice via the tail vein. Dose-dependent improvements in hemostasis were observed 4 weeks thereafter (Fig. 2B). In particular, α-angle and maximum clot firmness increased, while clotting time and clot formation time decreased. These parameters were similar at doses between 4 × 1012 and 2 × 1013 particles (Fig. 2B) but were strikingly comparable to those of wild-type mice at 2 × 1013 particles/kg body weight. Due to the dynamic range limitation of ROTEM analysis (detection of hemostasis correction is saturated at equivalent to >5 IU/kg recombinant hFVIII) and to evaluate further the hemostastic correction, blood-loss analysis was also carried out by transecting the tail in these mice. At 8 × 1011 particles/kg, blood loss due to tail clipping was similar to that in untreated FVIII knockout mice. Blood loss gradually decreased with the dose of AAV9/mFVIIa but remained above that in wild-type mice, even at 2 × 1013 particles/kg (Fig. 3a). Interestingly, all mice transduced with AAV9/mFVIIa survived tail clipping, regardless of dose (Fig 3b), while 2/10 untreated FVIII knockout mice died. Collectively, these results indicate that mouse FVIIa is functional in vivo, although a high dose is required to improve hemostasis.

ROTEM after escalating doses of AAV9/mouse FVIIa.
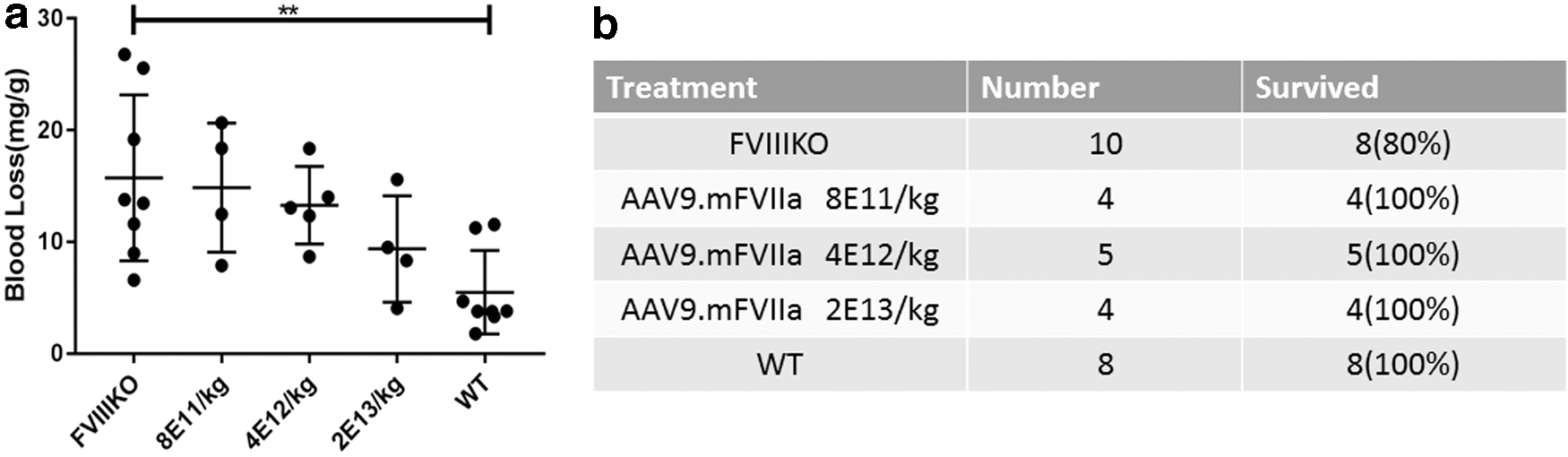
Blood loss due to tail transection after AAV9/mFVIIa administration. Doses of AAV9/mFVIIa from 8 × 1011 to 2 × 1013 particles/kg were administered into FVIII knockout mice (FVIIIKO) via the tail vein. FVIII knockout (FVIIIKO) and wild-type mice (WT) injected with normal saline were used as control. Tails were transected at week 4, and
FVIII activity and inhibitors in mice treated with AAV8/hFVIII
To elicit both FVIII inhibitor and AAV8 capsid neutralizing antibodies, AAV8/hFVIII was administered intravenously once at a dose of 4 × 1012 particles/kg body weight into FVIII-deficient mice (Fig. 4). Administration of this dose induced peak FVIII activity at 200–300% of normal activity, and FVIII inhibitor developed in a subset of injected mice. Citrated plasma was then harvested at 2, 4, and 8 weeks post transduction, and assayed for FVIII activity, titer of FVIII inhibitor, and anti-AAV8 neutralizing antibodies. Notably, 20/34 mice presented stable FVIII activity at 2–8 weeks without developing inhibitor antibodies (Fig. 5a). However, FVIII inhibitors were detected at titers 0.75–42 BU/mL in the other 14 mice (∼40%; Fig. 5b), in which FVIII activity peaked at week 2 before gradually decreasing and dropping below detectable levels at week 8 (Fig. 5a). Subsequently, mice with inhibitory antibodies were randomly assigned to receive further intravenous normal saline or intravenous AAV9/mFVIIa. It was noted that titers of FVIII inhibitory antibodies were comparable between these groups (n = 7 vs. n = 6; Fig. 5b).

Design of in vivo studies. A human FVIII expression cassette packaged into AAV8 was administered intravenously at 4 × 1012 particles/kg body weight into FVIII knockout mice. Citrated blood was collected 2, 4, and 8 weeks thereafter. Samples at 8 weeks were assayed for inhibitory antibodies to FVIII, and mice with such antibodies were randomly assigned to receive AAV9/mFVIIa or normal saline (NS) subsequently. Four weeks after the second treatment with AAV9/mFVIIa or normal saline, a needle was inserted into the left knee joint capsular to induce intra-articular hemorrhage. After another 2 weeks, whole blood was harvested from the inferior vena cava and analyzed for hemostasis correction by ROTEM, quantification of mFVIIa expression, and prothrombin time (PT). Knee joint tissues were evaluated for pathology changes by histology.
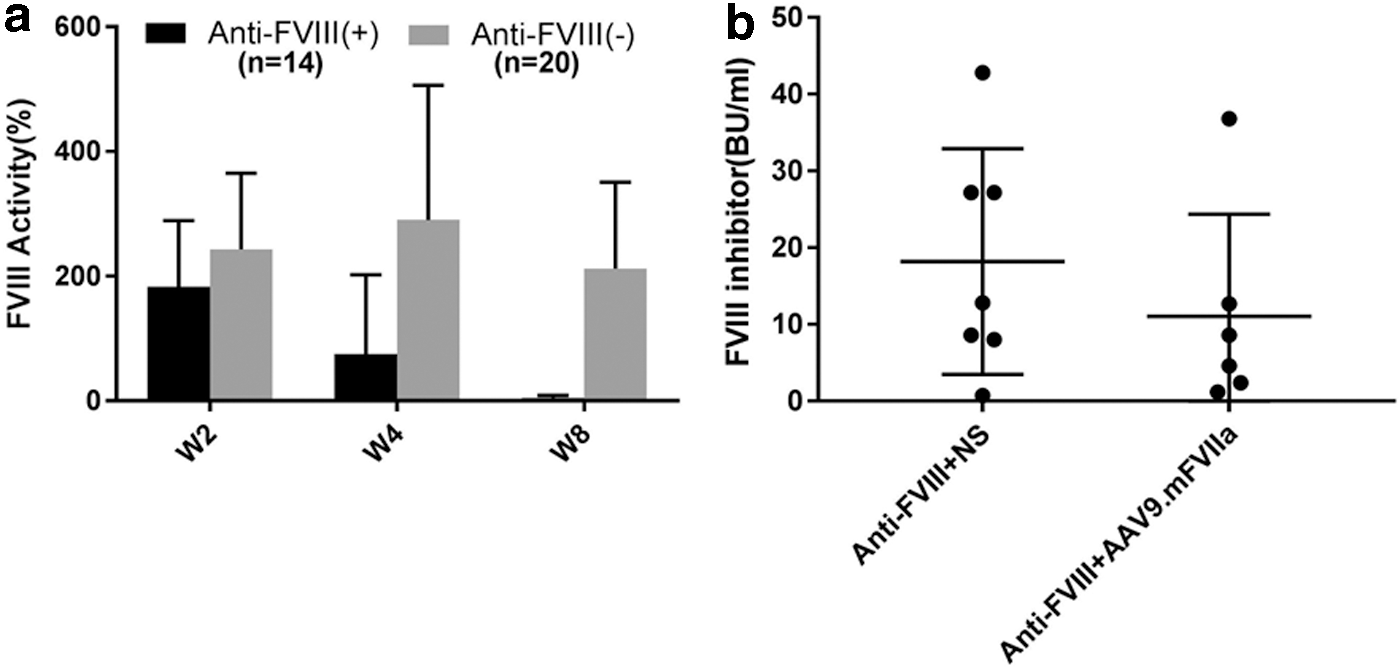
In vivo remodeling of inhibitory antibodies to FVIII. AAV8/hFVIII was administered intravenously at 4 × 1012/kg body weight in mice with hemophilia A to generate inhibitory antibodies to FVIII. Plasma was collected 2, 4, and 8 weeks later, and assayed for
Cross-reactivity of AAV8 neutralizing antibodies to AAV9
The activity of neutralizing antibodies to AAV8 was characterized in vitro to investigate the potential of AAV9 as a vector to deliver mFVIIa in attempts to correct hemophilia A in mice with FVIII inhibitor. Approximately 13/14 mice that developed FVIII inhibitor also developed neutralizing antibodies to AAV8 (Figs. 5 and 6). AAV-neutralizing antibodies in animals and humans have been shown to have little or no cross-reactivity among AAV serotypes. 14,36 As shown in Fig. 6a, very high titers of neutralizing antibodies (from 500- to 5,000-fold dilution) against AAV8 were observed in mice transduced with AAV8/hFVIII but with low cross-activity to AAV9 (up to fivefold dilution). Since FVIIa is not as potent as FVIII in correcting hemophilia A, it was anticipated that high doses would be needed to achieve therapeutic effects. At a test dose of 2 × 1013 particles/kg, the concentration of AAV9 in the plasma would be approximately 2 × 1010 particles/μL, given that the average AAV8-treated mouse weighed about 25 g, of which 4% (∼1 mL) would be plasma. To keep the same ratio of vector to plasma, 3 × 109 particles of AAV8 OR AAV9/luciferase were incubated in 6.0 μL of PBS or in 6.0 μL of pooled sera from mice transduced with AAV8/hFVIII for 2 h at 4°C. The samples were then injected in each leg in the same mouse, and mice were imaged after 2 weeks to measure the inhibitory activity of pooled sera against AAV9 transduction (Fig. 6b and c). It was found that the sera almost completely blocked transduction of AAV8 but not AAV9, suggesting that therapeutic transgene expression may be achieved, even in the presence of low titers of cross-reactive neutralizing antibodies by systemic administration of at least 2 × 1013 particles/kg.
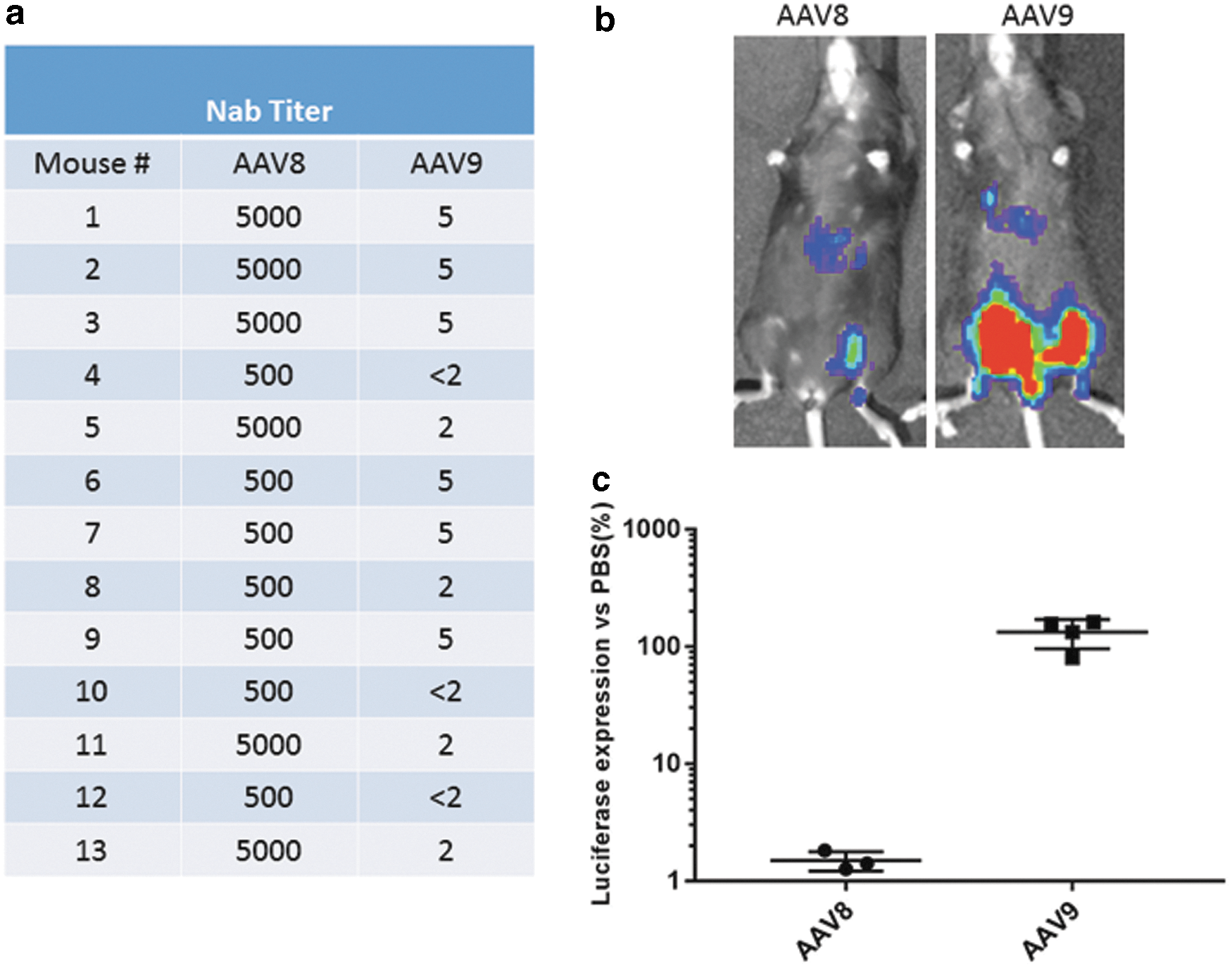
In vitro and in vivo analysis of AAV neutralizing antibodies. Animals treated with AAV8/hFVIII, as described in Fig. 4, were examined for neutralizing antibodies against AAV8 or AAV9 by
Hemostasis correction in the presence of FVIII inhibitor and anti-AAV8
Based on the previous result, 2 × 1013 particles/kg of AAV9/mFVIIa were administered into mice with both FVIII inhibitor and anti-AAV8. As shown in Fig. 4, whole blood was harvested from the inferior vena cava at sacrifice and analyzed by ROTEM. mFVIIa protein expression and prothrombin time (PT) were also detected from plasma at the sacrifice. As shown in Fig. 7, in the presence of both anti-FVIII inhibitor and anti-AAV8 NAbs, AAV9-mediated gene transfer of mFVIIa corrected all coagulation parameters, most of which improved relative to those of FVIII knockout mice (with or without inhibitory antibodies to FVIII). Indeed, coagulation was comparable in the three groups of mice with FVIII inhibitor treated with AAV9/mFVIIa (AAV8.hFVIII + AAV9.mFVIIa), and in mice treated only with AAV9/mFVIIa (AAV9.mFVIIa) and wild-type control mice. At the end time point of 6 weeks since AAV9/mFVIIa administration, the mFVIIa level achieved a range of 500–900 ng/mL (737 ± 167 ng/mL), while the mFVIIa level in the rest of the animals without AAV9/mFVIIa treatment was around 150 ng/mL (Fig. 8a). Concomitantly, the PT was shortened to around 15 s after AAV9/mFVIIa treatment compared to around 22 s with AAV9/mFVIIa treatment gene therapy (Fig. 8b).
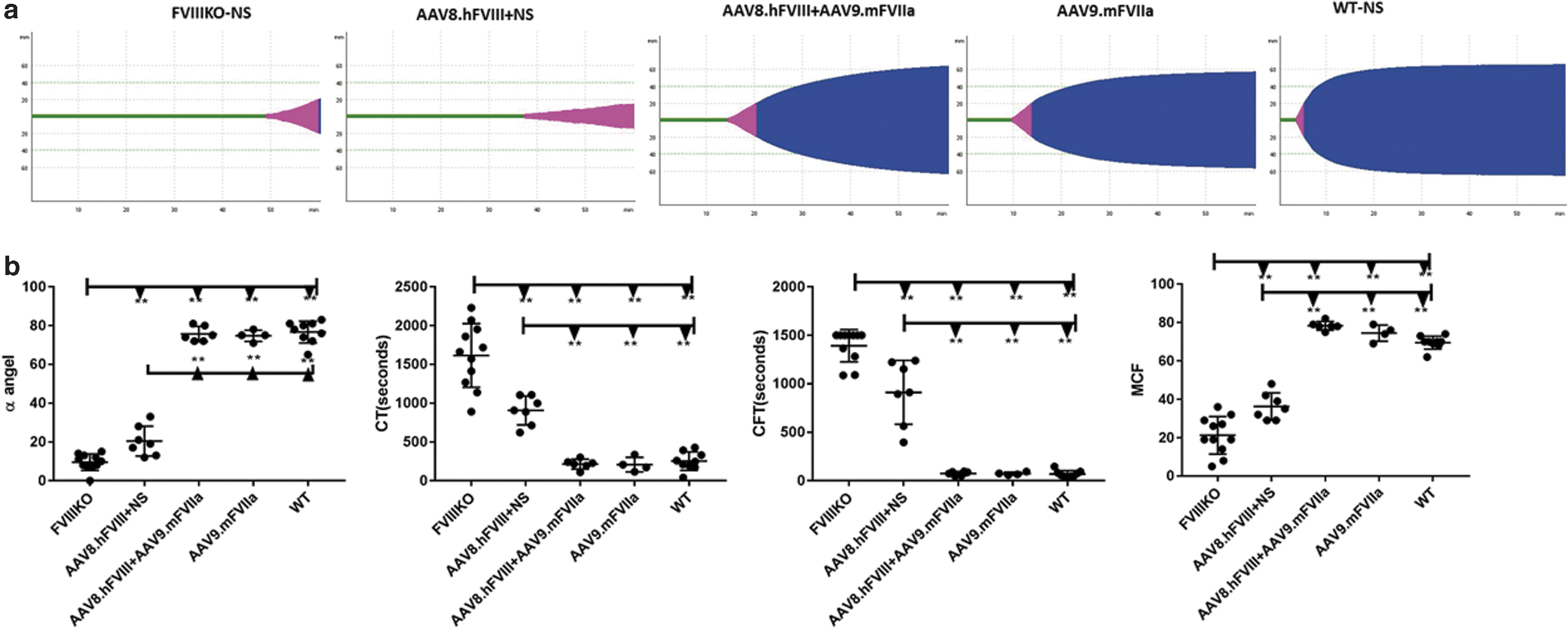
Improved coagulation in the presence of both antibodies to FVIII and AAV8 after AAV9-mediated mFVIIa gene therapy. Mice with pre-existing antibodies to FVIII and AAV8 (n = 13), as described in Fig. 4, were intravenously injected with normal saline or AAV9/mFVIIa. Intra-articular hemorrhage was induced 4 weeks later by inserting a needle into the left knee joint capsule. After another 2 weeks, whole blood was harvested from the inferior vena cava and tested by ROTEM.
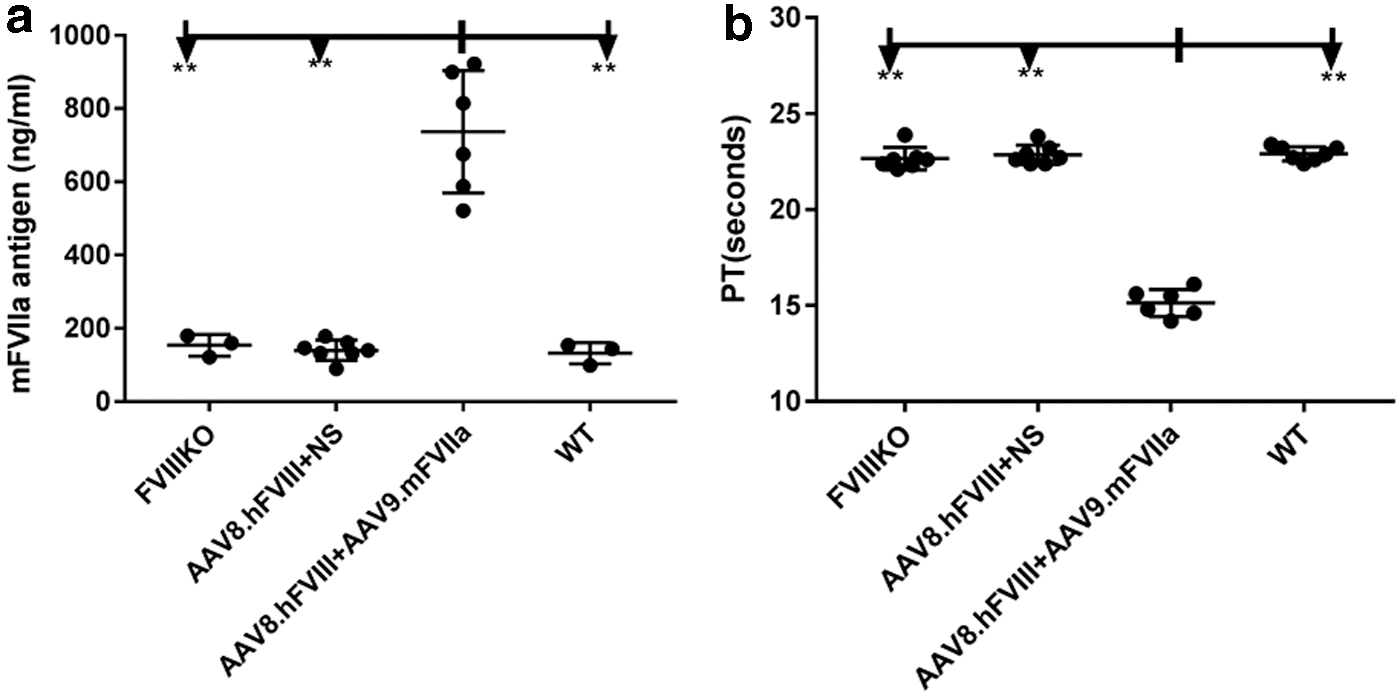
Efficacy of AAV9/mouse FVIIa gene therapy in hemophiliac mice with antibodies to FVIII and AAV8. Plasma from AAV9/mFVIIa gene therapy treated animals, as described in Fig. 7, were measured for
Improved hemoarthrosis after transduction of AAV9/mFVIIa into mice with FVIII antibodies and neutralizing antibodies to AAV8
To investigate whether systemic administration of AAV9/mFVIIa could prevent joint damage from hemoarthrosis, a major complication in severe hemophilia patients, intra-articular hemorrhage was induced by inserting a needle into the left knee joint capsule 4 weeks after AAV9.mFVIIa treatment. Mice were sacrificed 2 weeks thereafter to assess joint damage by histology (representative images shown in Fig. 9a). As expected, joint bleeding resulted in pathological damage in FVIII-deficient mice with anti-FVIII inhibitor (Fig. 9b; score 5.5/10) or without anti-FVIII inhibitor (score 4.7/10). AAV9-mediated gene therapy with FVIIa mitigated but did not completely reverse the damage (score 3.6/10) compared to wild-type mice (score 0.51/10). In addition, hemosiderin ferritin in the synovium, a hallmark of erythrocyte lysis due to hemoarthrosis and a trigger of joint inflammation, was observed in only one mouse transduced with AAV9/mFVIIa, but was present in most FVIII knockout mice with or without FVIII antibodies (Fig. 9c). These results indicate that AAV9 delivery of FVIIa not only improves hemostasis in mice with antibodies to FVIII and AAV8, but also mitigates hemoarthrosis after joint injury. However, optimization of the FVIIa expression cassette could further improve efficacy and should be investigated.
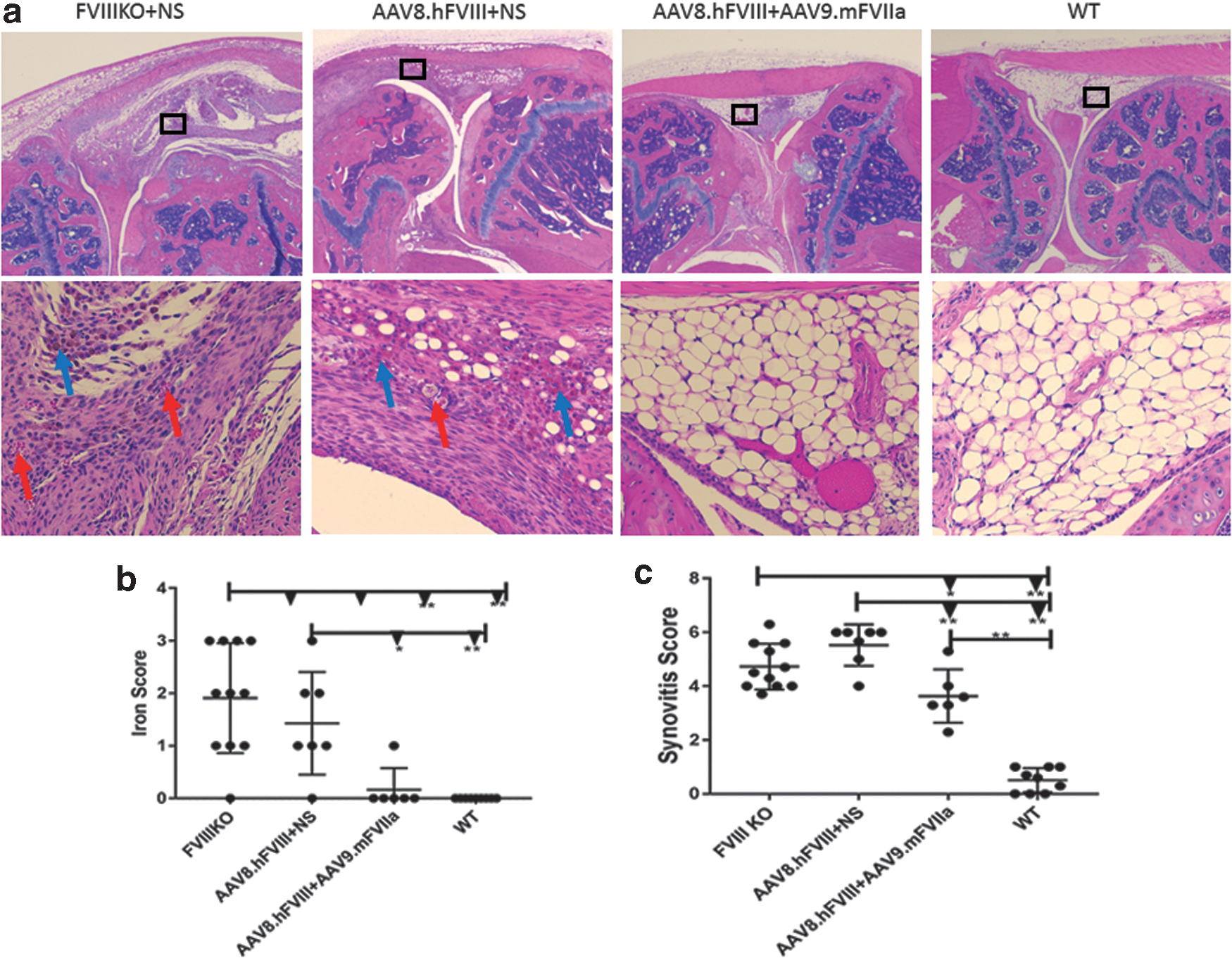
AAV9/mouse FVIIa gene therapy mitigates joint damage from hemoarthrosis in hemophiliac mice with antibodies to FVIII and AAV8. Knee joints were collected from mice described in Fig. 7, and assessed based on a murine synovitis scoring system.
Discussion
Hemophiliac mice transduced with AAV8/hFVIII eventually developed high titers of neutralizing antibodies against the virus and inhibitory antibodies to FVIII. As inhibitory antibodies would essentially reverse the therapeutic effects of transgenic FVIII, the potential benefits of delivering FVIIa using an alternative AAV serotype-AAV9, a serotype that evades neutralizing antibodies to AAV8, was investigated. 37 Although neutralizing antibodies to AAV8 showed low cross-reactivity to AAV9 in vitro, these antibodies did not block AAV9 transduction in vivo when a high dose of AAV9 was used. Importantly, systemic administration of 2 × 1013 particles/kg of AAV9/mFVIIa improved hemostasis, even in mice with FVIII inhibitor and neutralizing antibodies to AAV8, and prevented hemoarthrosis after joint injury.
After extensive studies in preclinical animal models, gene therapy based on AAV vectors has shown promise in clinical trials as treatment for hemophilia A and B, as has been reviewed. 38,39 After administraion of AAV vectors encoding human FIX or FVIII to the human liver via systemic administration, therapeutic level of coagulation factors has been achieved. 40 Unfortunately, patients with neutralizing antibodies to the AAV serotype used are routinely excluded from clinical trials. Generally, approximately 30–50% of humans and 20–50% of hemophilia patients have pre-existing neutralizing antibodies to AAV. 39 In any case, neutralizing antibodies are less common in younger patients, 36 who should thus be eligible to receive AAV gene therapy.
To achieve the goal in patients with AAV neutralizing antibodies, either AAV capsids have to be engineered to eliminate antibody binding sites, 41 or antibody titers must be depleted from the circulation. 20 Among these strategies, alternative AAV serotypes that neutralizing antibodies do not recognize can be used. For example, in utero gene transfer of scAAV5-LP1-hFVII-coop elicited production of anti-AAV5 in monkeys, but AAV8-mediated gene therapy 1 year later also induced expression of therapeutic levels of human FVII. 42 In the same way, this study attempted to apply AAV9 for delivering therapeutic transgene into mice with neutralizing antibodies to AAV8. Even though some cross-reactivity was observed against AAV9, a high dose of AAV9 was sufficient to correct a hemophilic phenotype and prevent pathological sequelae in an injured knee joint. Based on the results from neutralizing antibody analysis and achievement of therapeutic effect after administration of high dose of AAV9 vectors in mice with neutralizing antibody to AAV8, it is emphasized that in vivo neutralizing antibody assays are necessary to determine whether a patient with neutralizing antibodies can potentially benefit from this approach, especially when neutralizing antibody is positive based on an in vitro assay.
Although hemophilia management has significantly evolved in the past several years, with long-lived coagulation factors for infusion coming to market and with clinical trials of gene therapy ongoing, 5,43 FVIII inhibitor development after coagulation factor infusion is still a big challenge for clinical practice, especially in patients with hemophilia A. Development of inhibitory antibodies elevates the risk of serious bleeding, accelerates joint degradation, decreases quality of life, increases mortality, and severely complicates disease management. 44,45 Prophylactic or on-demand agents that bypass inhibitory antibodies, including recombinant activated FVII and plasma-derived activated prothrombin complex concentrate (FVIII inhibitor bypass activity), are the few options available. 46 Recombinant FVIIa has been used safely for two decades to treat bleeding episodes and prevent bleeding during surgery and other invasive procedures in patients with congenital hemophilia A or B (with or without inhibitory antibodies). The safety profile of recombinant FVIIa is generally attributed to the confinement of its activities within local injured sites, to which it is specifically recruited by exposed tissue factor. 47 Nonetheless, it has been reported that persistent over-expression of mFVIIa (>2,000 ng/mL) was associated with premature mortality in transgenic mice, 27 and further investigation into the complications of long-term FVIIa expression after AAV vector mediated delivery is necessary. In addition, FVIIa can directly activate FX to FXa without binding to tissue factor 48 and cycles between the circulation and the extravascular space. 49 According to clinical results from application of rFVIIa, gene therapy using an AAV vector to deliver FVIIa has been tested in animal models with hemophilia, and phenotypic correction has been achieved. 27,42,50 Consistent with previous studies, improved hemostasis was observed when AAV9 encoding mouse FVIIa was administered into hemophiliac mice with FVIII inhibitor. Collectively, these results suggest that delivery of FVIIa utilizing alternative AAV serotypes without cross-reactivity is a novel approach to treat hemophilia in patients with pre-existing neutralizing antibodies to AAV and inhibitory antibodies to the deficient coagulation factor.
In comparison to replacing deficient factors, a major concern of bypass coagulation agents is the low efficiency to ameliorate hemostasis. Hence, a high dose is generally required to achieve therapeutic effects, as demonstrated in this study. However, administration of AAV vectors may elicit a dose-dependent capsid-specific cytotoxic T-cell response that may, in turn, eliminate transduced cells. 4,5 To help address this potential issue, a lower dose of AAV could be used if the half-life of exogenous FVIIa could be extended, for example by expressing FVIIa as a fusion with albumin and Ig-Fc fragment, which would compensate for reduced expression. Other approaches should also be exploited in the future, including use of strong promoters and optimization of the transgene cassette.
In summary, administration of AAV8/hFVIII induced antibodies to the transgene in some mice and to AAV8 in all mice. However, systemic administration of AAV9/mFVIIa corrected the hemophilia phenotype in mice with such antibodies, although a high dose was required. Importantly, this approach also mitigated hemoarthropathy after joint injury. These results highlight the potential of alternative AAV serotypes that evade pre-existing neutralizing antibodies to deliver coagulation factors and treat hemophilia in patients with inhibitory antibodies to the infused coagulation proteins.
Footnotes
Acknowledgments
We thank Nikita E. Hall and Charles Askew for their help in editing the manuscript and providing critical assessment. The authors acknowledge the UNC Biomedical Research Imaging Center (BRIC) Small Animal Imaging (SAI) facility for assistance of mouse imaging. This work was partly supported by National Institutes of Health Grants R01AI117408 (to C.L. and R.J.S.), R01HL125749 (to C.L.), P01HL112761 and R01AI072176 (to R.J.S.), P30-CA016086-35-37 and U54-CA151652-01-04 (to the BRIC SAI facility), and a research grant from Asklepios BioPharmaceutical (to J.S.). H.B.L. is the recipient of Beijing Municipal Natural Science Foundation (No. 7162151). We would also like to thank Editage (
Author Disclosure
R. Jude Samulski is the founder and a shareholder at Asklepios BioPharmaceutical. He receives research support through the University of North Carolina from Asklepios BioPharmaceutical. He holds patents that have been licensed by UNC to Asklepios Biopharmaceutical, for which he receives royalties. He has consulted for Baxter Healthcare and has received payment for speaking. Other authors have no conflict of interest to declare.