
Abstract
Recombinant adeno-associated viruses (AAVs) are quickly becoming the preferred viral vector for viral gene delivery for the treatment of a wide variety of genetic disorders. However, since their use in a clinical trial targeting hemophilia B patients 10 years ago, immune responses to the AAV capsid appear to have hampered some of the early clinical gene transfer efficacy. Indeed, AAV-based gene transfer has been shown to reactivate capsid-specific memory T cells, which have correlated with a decline in AAV-transduced tissue in some patients. Importantly, clinical trials have also shown that this reactivation can be quelled by administering time-course taper of glucocorticoid steroids before or after dosing. More recently, two clinical studies have shown that AAV gene transfer is not only able to induce a deleterious immune response, but also can result in the initiation of a tolerance to the AAV capsid mediated by regulatory T cells and exhausted T cells. This article reviews clinical trials describing immune responses to AAV, as well as the mechanisms responsible for immune tolerance in chronic infections and how it could apply to AAV-based gene transfer. A better understanding of both cytotoxic and tolerogenic immune responses to recombinant AAV will lead to safer gene transfer protocols in patients.
Introduction
A
Indeed, AAV vectors had been considered as non-immunogenic viral vectors until a clinical trial on hemophilia B patients described a cytotoxic immune response to the AAV capsid mediated by CD8+ T cells. 2,3 Despite a proof of concept of persistent expression in studies with factor IX (FIX)-deficient mice 4 and dogs, 5 it was only in humans that Manno et al. 2 first appreciated the transient expression of FIX. This loss of FIX expression was related to an asymptomatic elevation of transaminases and detection of AAV2 capsid-specific T cells secreting interferon gamma (IFN-γ) between 4 and 6 weeks after dosing. A second clinical trial in hemophilia B patients by Nathwani et al. 6 using an AAV8 vector also showed an immune response marked by a transient elevation in transaminases and positive IFN-γ ELISpot responses to the capsid. However, unlike the AAV2 trial described by Manno et al., in this case transgene expression was diminished but sustained. They reported an elevation of transaminases between 7 and 10 weeks post dosing in addition to a transient cellular immune response to the AAV capsid. Importantly, this trial had as part of its clinical protocol the use of a brief immune suppressive regimen, which is thought to have quelled the immune response to the capsid, evidenced by a return to basal transaminase levels and persistence of transgene expression. More recently, two clinical trials were initiated by Spark Therapeutics and uniQure with rAAV encoding a codon-optimized FIX. In the Spark Therapeutics trial, nine patients were injected intravenously (i.v.) with a proprietary rAAV capsid designated as SPK-9001 encoding a high-activity FIX (Padua mutant) at a dose of 5 × 1011 viral genome (vg)/kg. They have shown persistence of transgene expression above the FIX therapeutic threshold up to 12–31 weeks (31.8% of normal FIX level) in the absence of any immunosuppressive regimen except for 2/9 patients who showed asymptomatic liver enzyme elevation and developed immune response to the AAV capsid (Table 1). 7 In the uniQure trial, two cohorts of five patients were injected with a rAAV5 encoding for a codon-optimized FIX at doses of 5 × 1012 vg/kg and 2 × 1013 vg/kg. In comparison to the Spark Therapeutics trial, they have reported persistence of transgene expression above the FIX therapeutic threshold up to 6 months but at lower levels (5.1–13% of normal FIX level depending on the dose), despite much higher vector doses and in the absence of any immunosuppressive regimen, except for 3/10 patients (Table 1). 8 The results of these two studies suggest that at least in the context of hemophilia B optimization of the expression cassette, the dose and/or the choice of AAV serotype can influence the initiation of an expression limiting immune response.
Clinical trials using rAAV and implications of a capsid-specific immune response on gene transfer efficacy
ALT, alanine transaminase; AST, aspartate transaminase; CK, creatine kinase; co, codon optimized; IFN-γ, interferon gamma; rAAV, recombinant adeno-associated virus; ss, single-stranded; sc, self-complementary.
Finally, and contrary to the results obtained by Manno et al. and Nathwani et al. in hemophilia B patients, two clinical trials have shown that rAAV gene transfer may not only be reactivating cytotoxic T cell responses but also may be able to initiate a regulatory T cell response. This was first described by Mueller et al. 9 in a Phase 2b trial for alpha-1 antitrypsin deficiency (AATD) and later confirmed in a clinical trial with lipoprotein lipase (LPL)-deficient patients. 10 In both cases, patients received intramuscular (i.m.) injections of a rAAV1, which led to the finding of infiltrated regulatory T cells (Tregs) in situ. These studies have highlighted a new research area on AAV immunology, including the understanding of Treg activation and T cell exhaustion. Below we provide a brief review of Treg and exhausted T cell responses and how these fit in the clinical experience without AAV.
Regulatory T Cells
The importance of CD4+ CD25+ T cells in tolerance was described in 1995 when mice depleted in CD25+ T cells showed T cell self-reactive responses and elicited autoimmune diseases. 11 Since then, the exploration of these commonly called regulatory T cells has expanded, and they are now considered a central mechanism of immune regulation.
Two major subsets of Tregs have been described: the natural Treg (nTreg) and induced Treg (iTreg). Natural Tregs are differentiated in the thymus and educated on self-antigens, whereas iTregs develop in the periphery and are generated from naive T cell populations. 12 Subtypes of Tregs are characterized but are not limited to expression of CD4, CD25, CTLA-4, HLA-DR, or CD39 and downregulation of CD127 IL-7 receptor on mouse and/or human Tregs. 13 –17 In addition to extracellular markers, intracellular markers such as the FoxP3 (forkhead box P3) transcription factor has been described as the most specific Treg marker in the thymus and in periphery. Foxp3 has shown its importance in the thymic development and suppressive activity of regulatory T cells. Indeed, the last point was supported by data suggesting that its expression in naïve T cells confers regulatory phenotype and function to those cells. 18 –20 Another marker that has been recently identified is Helios, a member of the Ikaros transcription factor family that is expressed in natural and activated Tregs and is also involved in Treg suppressive activities. 21,22
During the past years, in vitro and in vivo studies have demonstrated that Tregs can mediate tolerance by interacting with cells in an APC-dependent or -independent manner, as well as through the secretion of regulatory cytokines (Fig. 1). Tregs are able to interact with the CD8+ effector T cells by preventing proliferation and IFN-γ secretion by CD8+ T cells without any interaction with APC. 23 They can also induce effector T-cell death through the granzyme and perforin-dependent pathways. 24 –26 In some cases, the immune regulation can be APC dependent; Tregs have shown the ability to prevent dendritic cell (DC) maturation in vitro through downregulation of CD80/CD86 costimulatory receptor expression by affecting the activation of effector T cells. 27 –29 Moreover, regulatory T cells are also able to decrease the time of interactions between the CD4+ T cells and DC in vivo, as shown by two-photon laser-scanning microscopy. 30,31 Finally, the cytokine environment plays an important role in immune regulation. In fact, the regulatory cytokines interleukin-10 (IL-10), transforming growth factor beta (TGF-β), or IL-35 have shown the ability to convert naïve T cells to regulatory T cells by activating, for example, the Foxp3 pathway in these cells. 32 –34
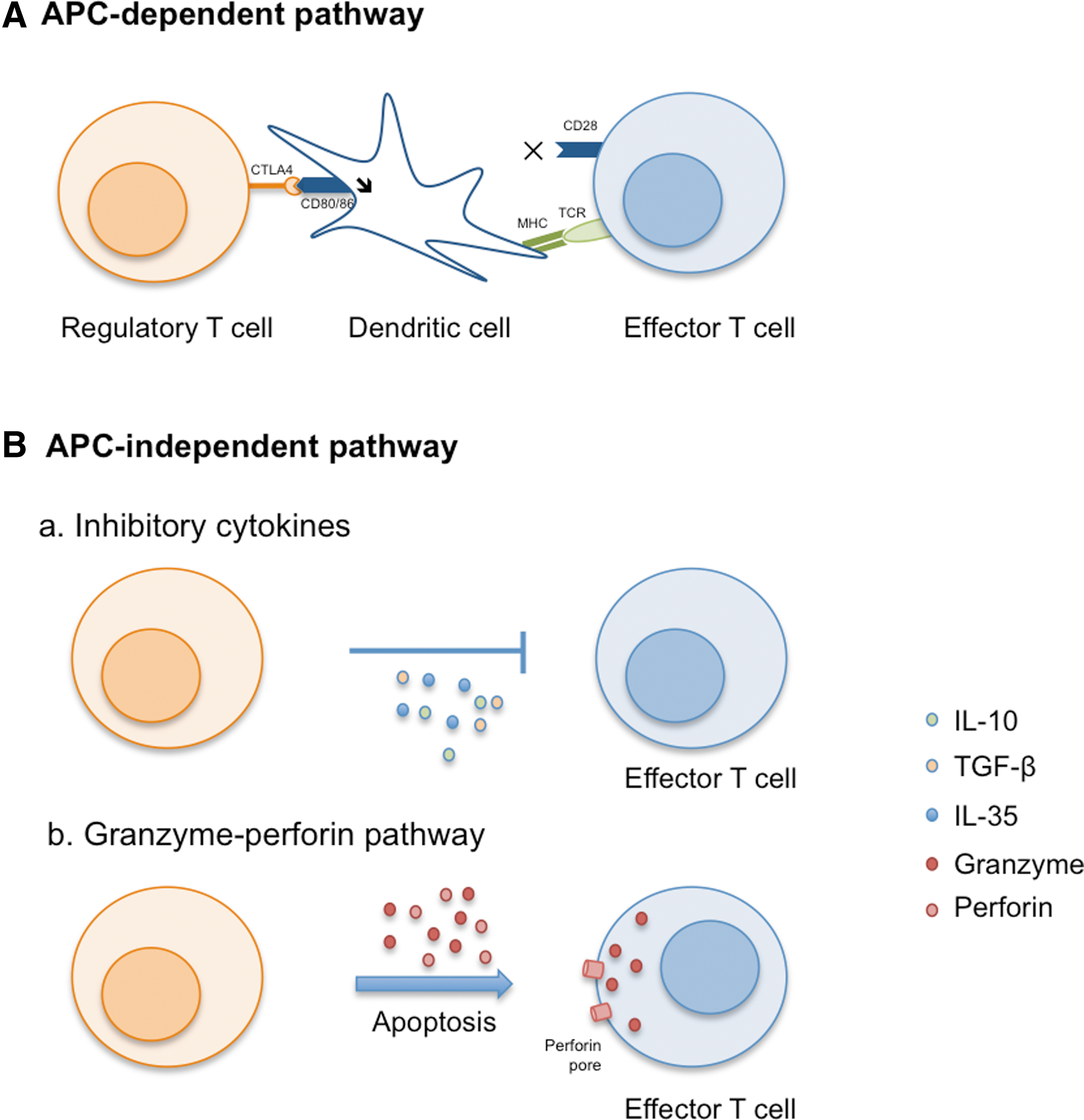
Mechanisms used by regulatory T cells to mediate tolerance. Tolerance can be mediated by antigen-presenting cell–dependent (
Despite the fact that regulation mechanisms are not completely understood, regulatory T cells play a major role in peripheral tolerance maintenance and autoimmune disease prevention. However, several studies have demonstrated that the induction or presence of preexisting Treg cells can also prevent effective virus clearance, especially during chronic infections.
Regulation Mechanisms in Chronic Infections
Regulatory T cell responses were initially described as regulators in a context of self-reactive antigen modulation in the thymus and autoimmune diseases. However, as mentioned above, they have also been shown to be deleterious in animal models of chronic viral infection. In fact, in some cases, chronic viral infections are associated with tolerance to viral antigens mediated by Tregs, which in turn leads to uncontrolled viral replication. It is thought that in these cases, Tregs suppress the activity of antigen-specific CD4+ T cells through secretion of IL-10 and TGF-β; this mechanism has been widely observed in patients infected by human immunodeficiency virus (HIV) receiving antiretroviral therapy, as well as patients with chronic hepatitis B virus or papillomavirus infections. 35 –37 Moreover, suppressive assays have shown that Tregs suppress effector T cells by inhibiting their proliferation and IFN-γ secretion. Their depletion leads to an increase in IFN-γ secretion after peripheral blood mononuclear cell re-stimulation in vitro. Interestingly, Treg induction is not the only mechanism responsible for immunoregulation in chronic infections. In fact, hepatitis C virus (HCV) chronic infected patients have presented induction of Tregs secreting IL-10 and TGF-β, 38 as well as T cell exhaustion. This exhaustion was revealed by a lower frequency of specific cytotoxic T cells in long-term HCV-seropositive patients. 39 Exhausted T cells were first described in a lymphocytic choriomeningitis virus mouse model where activated virus-specific CD8 T cells without effector function were detected. 40 These cells were actually expressing activation markers associated with TCR signaling, such as CD69 after antigen restimulation in vitro, but were unable to elicit cytotoxic cytokine secretion such as IFN-γ leading to an unresolved viral infection.
To date, three different pathways involving exhaustion have been described: exhaustion due to interactions with the regulatory cells, exhaustion due to influence of the cytokine milieu, or exhaustion due to activation of inhibitory receptors (Fig. 2). Whereas it is still unclear whether Tregs directly affect T cell exhaustion, it is clearly established that involvement of inhibitory receptors such as PD-1 and LAG3 can drive T cell exhaustion. Indeed, several studies have shown the reversal of the exhaustion by in vitro blockade of these inhibitory pathways. 41,42

Mechanisms leading to T-cell exhaustion. During infection, T cells are primed by antigen, co-stimulation signals, and inflammatory cytokines, and they differentiate into effector T cells. These cells show a cytotoxic (IFN-γ, interleukin-2, and tumor necrosis factor alpha secretion) and cytolytic (perforin and granzyme release) phenotype and a high capacity of proliferation (top panel). During chronic infection and/or recombinant adeno-associated virus–mediated gene transfer, antigens persist in situ. T cells progressively lose their effector functions leading to T cell exhaustion. This exhaustion is driven by different pathways: interactions between regulatory receptors (PD-1 and LAG-3) and their ligands expressed by dendritic cells and/or target cells and interactions with immunoregulatory cell types such as Tregs (bottom panel).
Altogether, these results show the importance of Treg and exhausted T cells in modulating immune response in chronic infections. Results described in mouse studies using rAAV as a viral vector or vaccine have led us to question whether AAV-based gene transfer is not mirroring a chronic viral infection and whether it could induce tolerance mechanism. CD8+ T cell exhaustion was actually described after i.m. AAV administration in a mouse model where CD8+ T cells failed to secrete IFN-γ and bound annexinV at the site of injection, suggesting they received a signal for activation-induced death. 43 Other studies using rAAV as a vaccine to HIV-1 epitopes have also suggested T cell exhaustion after administration. 44,45 Despite that, a transgene-specific cytotoxic T cell response was induced after the first exposure to rAAV, and the cells failed to proliferate after a booster immunization with an adenoviral vector encoding for the same antigen due to their inability to differentiate into memory T cells. It turns out these cells were also expressing inhibitory markers, including PD-1 and CTLA-4, suggesting exhaustion. More recently, exhausted CD8 cells in addition to Tregs have been described in clinical trials using AAV vectors. 9,10
First Evidence of Immune Tolerance to Raav in Clinical Trials
In 2011, Flotte et al. reported that in AATD patients injected i.m. with rAAV1 (6 × 1012 vg/kg) expressing AAT presented with an IFN-γ+ ELISpot immune response to the AAV capsid without loss of transgene expression 3 months after dosing. 46 In 2013, Mueller et al. published a follow-up to this clinical study showing the first evidence of Treg activation in response to the AAV capsid after gene transfer in humans. 9 AATD patients have been injected i.m. with an AAV1 vector encoding for the AAT protein. Patients injected with the highest dose of vector (6 × 1012 vg/kg) presented with a transient elevation of creatine kinase at 30 days post injection and a partial decrease in transgene expression. 46 However, the AAT expression persisted for >1 year after dosing, despite the detection of cellular infiltrates in the injected muscle. The characterization of muscle biopsy sample by immunofluorescence staining and analysis bisulfite-specific polymerase chain reaction of the methylation of Treg-specific demethylated region showed that ∼10% of the T cells in the biopsy material was actually composed of Tregs (CD4+CD25+ FoxP3+). Moreover, an in vitro re-stimulation assay revealed that the patients had peripheral Tregs that were reactivated when stimulated with AAV1-capsid peptides, demonstrating they were AAV1 capsid specific. The presence of the Helios marker on peripheral AAV-specific Tregs suggests that this population is natural Treg. Another important finding in that study was that the i.m. injection of AAV1 may be mimicking a chronic infection. While unexpected, it was reported that intact AAV1 capsids were still present in the biopsy material at both 3 months and 1 year post administration. In fact, much as would be expected during a chronic infection, the muscle biopsies showed high levels of staining for PD-1 and PD-L1 markers in situ, suggesting initiation of T cell exhaustion. More recently, this was confirmed in an update on this trial, where the authors described sustained expression of AAT protein 5 years after dosing with continued evidence of infiltrated Treg and, more importantly, the presence of exhausted CD8 T cells in situ, all concomitant with a persistent IFN-γ positive ELISpot to the AAV capsid. 47
For the first time, the AAV1 trials by Flotte et al. have shown that a tolerogenic response could be initiated after i.m. AAV-based gene transfer in humans. However, the mechanism remains to be established as to whether it is due to the dose, the transgene, and/or the administration route. There is some evidence to suggest that the mode of delivery and transgene may not be the only factors inducing this tolerance phenomenon. A previous clinical trial targeting LPL-deficient patients injected i.m. with a rAAV1 showed a loss of gene transfer efficacy after 18–31 months post dosing, 48 probably related to an IFN-γ immune response to the AAV1 capsid (Table 1). 49 On the other hand, the same clinical trial performed more recently on patients transiently immunosuppressed described a continued transgene expression based on increased chylomicron turnover at 52 weeks and positive biopsies and showed evidence of infiltrated Treg in situ (Table 1). 10
Consequences of Immunosuppressive Treatment in Aav-Based Gene Transfer
Since a deleterious immune response to the AAV capsid was described in patients in 2006, efforts were made to develop new strategies preventing this phenomenon. One of the strategies established was the use of immunosuppressant drugs. These protocols were first tested in animal models. They were inspired from immunosuppressive protocols developed to prevent graft rejection in transplantation and have shown mixed results. More recently, glucocorticoid steroid immunosuppression has also been used in the context of human trials and has shown promising results after rAAV gene transfer.
Immunosuppression protocols in animal models after AAV-based gene transfer
Preclinical studies have shown the importance of immunosuppressant choice and combination. In one study, an anti-thymocyte globulin, cyclosporine, and mycophenolate mofetil (MMF) cocktail inducing T cell depletion, blocking transcription of cytokine genes, and targeting activated T cells and primary antibody response, respectively, was used in Duchenne muscular dystrophy (DMD) dogs injected i.m. with an AAV6 vector encoding for the canine microdystrophin. 50 This cocktail allowed transgene expression persistence, even after treatment interruption in areas without cellular infiltrates. These results were then confirmed in the same model after injection in several muscles; transgene expression was maintained until 1 year post dosing after immune suppressive treatment was stopped. 51
On the contrary, the immune suppressive treatment might sometimes have a negative impact on gene transfer efficacy, as described in a nonhuman primate (NHP) study. NHPs received an AAV2 vector encoding for FIX and were assigned to one of three groups receiving no immunosuppression, a combination of MMF, sirolimus (a potent inhibitor of antigen-induced proliferation of T cells and B cells) and daclizumab, and a combination of MMF and sirolimus 1 week prior to dosing and maintained 10 weeks after liver-directed gene transfer. Surprisingly, the animals on the three-drug immunosuppressive (IS) regimen developed high titer inhibitory antibodies to FIX, whereas monkeys receiving the vector alone or the two-drug IS regimen did not and had sustained FIX expression. The three-drug regimen also had a negative impact on antibodies to AAV2 capsid by showing higher antibody titer than monkeys receiving no IS or the two-drug IS regimen where the titers were the lowest. In this study, the three-drug IS regimen not only showed a boosting effect on the B cell response, but also had a deleterious effect on the cellular immune response through a dramatic drop of Treg percentage. This was not the case when they received the same regimen without the daclizumab. This phenomenon was explained by the fact that this drug is an antagonist of the CD25 receptor highly expressed on Tregs. 52
These studies highlight the importance of optimizing the immune suppressive regimens before protocol translation to humans. Immunosuppressive treatments have shown the ability to prevent immune responses to the AAV capsid in humans affected by muscular dystrophies or metabolic disorders.
Use of immune suppressive drugs and/or regimen in AAV-based gene transfer clinical studies
The first evidence of the efficiency of immune suppressive drugs on immune response to the AAV capsid in patients was described in 2011. Hemophilia B patients were injected with an AAV8 vector encoding for codon-optimized FIX transgene at different doses. Contrary to the preclinical studies described above or clinical trials cited below, the administration of the immunosuppressive drug was not initially planned and occurred because patients receiving the highest dose (2 × 1012 vg/kg) have shown an elevation of liver transaminases 7–10 weeks post vector administration, as already described. 2 These patients received a transient immune suppression based on injection of prednisolone during at least 4 weeks, and then until transaminases returned to baseline. This corticosteroid treatment was justified by its efficiency in severe autoimmune hepatitis where cytotoxic T lymphocytes (CTL) are directed to hepatocytes. This transient immune suppression led to the resolution of the elevated alanine transaminase (ALT) level (range 2–35 days) and the disappearance of capsid-specific T cell response, demonstrating the effect of prednisolone on cellular immune response to the AAV capsid occurring after gene transfer. 6 The last update was published in 2014, and the patients are still expressing FIX 3 years after gene transfer. 53 Given that this was not a controlled clinical trial with two arms to explore the effectiveness of the prednisolone on curbing the immune clearance of AAV transduced hepatocytes, it is yet to be determined if this intervention can be applied broadly.
The efficacy of this immunosuppressive regimen was also described in clinical trials targeting neuromuscular disorders. However, in this context, the trials were designed to prevent or suppress immune responses to the transgene protein product and not necessarily to the AAV capsid. Patients suffering from muscular dystrophies are generally treated with glucocorticoids known to inhibit inflammation-associated molecules such as cytokines and chemokines, and prednisolone treatment in these patients has shown improvements in motor function and increased ambulation up to 1–3 years. However, the mechanism remains unknown. 54 Prednisolone was also used in two different clinical trials in limb-girdle muscular dystrophy (LGMD) and DMD patients to prevent cellular immune responses. The LGMD patients were off glucocorticoid treatment during the trial and 3 months before gene therapy. They received a boost of methylprednisolone 4 hr prior to dosing (2 mg/kg i.v.) to prevent inflammation due to the needle. In patients injected with 3.25 × 1011 vg of an AAV1 vector, no IFN-γ response to the AAV capsid was detected, despite CD4+ and CD8+ cell detection in situ and transgene expression being observed for 3 months. 55 Regarding the immune response to the transgene, a clinical trial was performed on DMD patients using an i.m. injection of an AAV1 vector encoding for the minidystrophin while patients underwent a standard glucocorticoid therapy and received methylprednisolone prior to dosing, as described previously. Surprisingly, 2/6 patients showed an IFN-γ cellular immune response to the dystrophin protein. These cells were already detected before injection, suggesting that these patients had a pre-existing immunity to the dystrophin probably due to the expression of this protein in revertant muscle fibers. 56 Thus, this immunosuppressive treatment does not seem to prevent the reactivation of specific T cells to the transgene. The authors did not describe the cellular immune response to the capsid in this study.
More recently and in a different patient population, a transient prednisolone treatment resolved the episode of transaminase elevation in addition to a high IFN-γ positive response to AAV9 capsid in spinal muscular atrophy (SMA) patients, which appeared 2–3 weeks after i.v. administration 57 (Table 1), as already described in hemophilia B patients.
All these trials described a persistence of transgene expression with a transaminase elevation episode resolved and a loss of IFN-γ-secreted T cell detection after transient immunosuppressive treatment. However, the mechanism(s) responsible for induction of tolerance to the AAV capsid, and in some cases to the transgene, remain to be explored. Only one study in LPL-deficient patients has shown evidence of Treg induction after AAV gene transfer associated with an immune suppressive regimen.
As described earlier, the first clinical trial performed on LPL-deficient patients receiving an AAV1 vector encoding the LPL protein showed an elevation of creatine kinase correlated with a dose-dependent activation of a capsid-specific T cell response limiting the duration of transgene expression. 49 The results led to the development of a new clinical trial including an immune suppressive regimen pre- and post-vector administration. The vector used in this trial was the first approved by the European Medicines Agency. The regimen consisted of administration of cyclosporine and MMF starting before injection and continued for 12 weeks in addition to a bolus of methylprednisolone 30 min before dosing. This led to long-term transgene expression and clinical improvement of the metabolic disorder. Regarding the immune response to the AAV capsid, patients have shown a transient IFN-γ positive response to the AAV capsid without any clinical consequences. As already described, after i.m. injection, the patients presented infiltrated Treg (CD4+ FoxP3+) cells in situ in addition to CD8+ cells. In accordance with the AAT clinical trial results where T cells expressed PD-1 and PD-L1 regulatory markers, 9 , the infiltrated CD8+ T cells do not seem to present characteristics of cytotoxicity due to absence of expression of Granzyme B and Fas ligand. A further study regrouping analysis of exhausted T cell markers would be interesting to characterize the mechanism of tolerance involved.
Conclusion and Future Prospects
In the past decade, immunological research on AAV vectors has evolved considerably. Before translation to humans, rAAVs were considered as non-immunogenic vectors due to their low ability to transduce APC. 1 Now, it is known that they are able to trigger an innate immune response through Toll-like receptor (TLR) signaling pathways 58 as well as transduce DC in vitro 59 –61 and in vivo. 62 This parameter could play a major role in the initiation of the immune response or reactivation of the pre-existing humoral and/or cellular immunity 63,64 after gene transfer. Previous studies have shown that DC transduction efficiency is dependent on AAV serotype and genome; self-complementary (sc) vectors are more efficient than single-stranded (ss) vectors, and rAAV serotypes 1, 2, and 6 are described as transducing murine 59 and human monocyte- or CD34+-derived DC, 60,61 whereas serotype 8 presents a lower tropism to DC, at least in vitro. 65 This difference in tropism for DC between the different serotypes and the genome conformation (ss vs. sc vector) could explain why some serotypes are more immunogenic or conversely more tolerogenic than others. Further investigation of DC phenotype or functional maturation after rAAV transduction would be helpful to understand the mechanisms involved in immune response initiation or reactivation and to modulate immune responses, as described by Pandya et al., in an antitumor vaccine context. 66,67 Despite that, immune-activating properties of rAAV are much reduced relative to other viral vectors such as adenoviral or non-viral vectors.
A number of strategies have emerged to prevent adaptive immune responses to antigens such as the capsid and the transgene product by transient immune modulation. The duration of these treatments was based on studies suggesting that the rAAV capsid does not persist more than 6–8 weeks, depending on serotypes, after gene transfer. 68 However, past and recent studies have demonstrated that the AAV capsid can be detected up to 6 years after gene transfer, depending on the administration route. Studies in NHP have shown that 6 years after subretinal injections, rAAV2 and rAAV5 capsids were still detected in the retina. 69 More recently, Mueller et al. described the AAV1 capsid persistence in the muscle 1 year after i.m. injection. 9 These observations support that AAV vector gene transfer could mimic a chronic infection due to the epitope persistence in situ. This hypothesis is also supported by the detection of Tregs and exhausted T cells in situ, as described in two different clinical trials. 9,10 However, the mechanisms initiating the regulation are still unknown. To date, it cannot be concluded whether the persistent antigen exposure leads to T cell exhaustion and/or Treg initiation or whether the Tregs are initiating the exhaustion. Moreover, the administration route cannot be the only factor inducing the immune regulation either. Indeed, in LPL clinical trials, regulatory T cells were observed when an immunosuppressive regimen is administered, and in the AAT context the influence of the transgene itself cannot be excluded. AAT is actually described as immunomodulatory by promoting IL-10 production by DC and facilitating Treg expansion in mouse models. 70 Another way to promote tolerance to the AAV capsid would be the use of molecules targeting DC maturation and/or Treg initiation. Nowadays, the only immune suppressive treatment administered in patients injected with a rAAV is glucocorticoids prior to or post dosing. Further studies using molecules altering DC maturation and differentiation such as FLT3 71 and GSK-3 72 inhibitors and Wtn 73 in a β-catenin-dependent or -independent signaling pathways or drugs promoting Treg induction such as rapamycin 74 would merit investigation in a rAAV-based gene transfer context.
This review has shown that the administration route seems to play an important role in immune response modulation to the AAV capsid, but additional factors are described to modulate the immune response initiation after gene transfer, including the AAV serotype, the promoter (ubiquitous vs. tissue-specific), and/or the dose, as illustrated in Table 2. Some AAV serotypes are described as being more tolerogenic than others, 65 and the dose also appears important, as observed in several clinical trials experimenting with a dose escalation. Most of the time, low or intermediate doses do not lead to enzyme elevation or to an immune response to the AAV capsid. However, the main limitation—therapeutic level—is not reached at the lower doses. Finally, contrary to the clinical trial in hemophilia B patients reported by Manno et al., subsequent trials at a lower (5 × 1011 vg/kg; Spark Therapeutics) or equivalent dose (5 × 1012 vg/kg; uniQure) and in absence of any suppressive regimen have resulted in patients expressing 31.8–5.1% of normal FIX level, respectively, and most of them do not present any elevation of ALT/ AST transaminases and/or cellular immune response to the AAV capsid. Clearly, there is broad variability in immune responses mounted by patients, and to date there is not a clear predictor of which patents will mount an expression-limiting response. Given the complexity of the human immune system and immense diversity of the TCR and MHC repertoire of each individual, it may take some time before it can be properly predicted which patients are likely to present with a self-limiting AAV capsid immune response. The field has been fortunate that while the immune response to AAV are far more complex than initially appreciated (i.e., involving Tregs, CTL, and exhausted T cells) in the clinical setting, it remains one that is amenable thus far to immune modulation with prednisolone and rather mild when placed in the context of other gene therapy approaches.
Factors involved in immune responses to the AAV capsid in clinical trials
CNS, central nervous system; MPSIIIA, Mucopolysaccharidosis Type IIIA; LCA2, Leber's congenital amaurosis type 2; SMA, spinal muscular atrophy; AATD, α1-antitrypsin deficiency; LGMD2D, limb-girdle muscular dystrophy type 2D; LPLD, lipoprotein lipase deficiency; n/a: not analyzed.
In conclusion, during the past decade, this field has evolved considerably, and long-term transgene expression is now able to be observed in patients when immune responses to capsid and/or transgene are avoided or under control. However, cytotoxic as well as tolerogenic immune response initiation still need to be investigated and understood in order to develop safer gene transfer strategies.
Footnotes
Acknowledgments
C.M. is supported by P01-HL131471, R01-DK098252, R24-OD018259, and R01-NS088689.
Author Disclosure
C.M. is an inventor on various patents that may be entitled to royalty payments in the future. J.M.W. is an advisor to REGENXBIO, Dimension Therapeutics, and Solid Gene Therapy, and is a founder of, holds equity in, and has a sponsored research agreement with REGENXBIO and Dimension Therapeutics. In addition, he is a consultant to several biopharmaceutical companies and is an inventor on patents licensed to various biopharmaceutical companies. G.G. declares no competing financial interests.