
Abstract
Previous research has proven that disruption of either the CCR5 or the CXCR4 gene confers resistance to R5-tropic or X4-tropic human immunodeficiency virus type 1 (HIV-1) infection, respectively. However, the urgent need to ablate both of the co-receptors in individual post-thymic CD4+ T cells for dual protection remains. This study ablated the CCR5 and CXCR4 genes in human CD4+ cell lines and primary CD4+ T cells simultaneously using clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9, a well-developed, highly efficient genetic engineering tool. The efficiency of gene modification is as high as 55% for CCR5 and 36% for CXCR4 in CD4+ cell lines through infection of a single lentiviral vector (LV-X4R5), which were markedly protected from both HIV-1NL4-3 (X4-using strain) and HIV-1YU-2 (R5-using strain) infection. Importantly, approximately 9% of the modified GHOST (3) CXCR4+CCR5+ cells harbor four bi-allelic gene disruptions in both the CXCR4 and CCR5 loci. Moreover, co-delivery of two single-guide RNAs loaded with Cas9: ribonucleoprotein (sgX4&R5 Cas9RNP) disrupted >12% of CCR5 and 10% of CXCR4 in primary human CD4+ T cells, which were rendered resistant to HIV-1NL4-3 and HIV-1YU-2 in vitro. Further, the modified cells do not show discernible mutagenesis in top-ranked off-target genes by the Surveyor assay and Sanger sequencing analysis. The results demonstrate the safety and efficacy of CRISPR/Cas9 in multiplex gene modification on peripherally circulating CD4+ T cells, which may promote a functional cure for HIV-1 infection.
Introduction
T
Clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 is a class of RNA-guided endonucleases from bacteria and archaebacterial adaptive immune systems, which could be easily targeted theoretically to any genomic loci of choice by a short guide RNA (sgRNA). 20 –23 The CRISPR system consists of a short non-coding guide RNA (gRNA) composed of a target complementary CRISPR RNA (crRNA) and an auxiliary trans-activating crRNA (tracrRNA). The gRNA guides the Cas9 endonuclease to a specific genomic locus via base pairing between the crRNA sequence and the target sequence, and it cleaves the DNA to create a double-strand break. Recently, the CRISPR/Cas9 system has been modified to fit various research fields from genetic engineering to transcriptional modulation, 24 which significantly improves the development of translational medicine. By delivering a panel of sgRNAs targeting different genes simultaneously, a single round of transduction of CRISPR/Cas9 vectors can achieve multiplex gene modification in a certain cell, 20,25 which is much more efficient and more convenient than other custom nucleases such as ZFNs and TALENs. Therefore, CRISPR/Cas9 as a versatile tool is promising in defeating diseases caused by multiple genetic factors, such as complex, polygenic disorders and HIV-1 infection.
This study utilized two independent methods to deliver CRISPR/Cas9 into mammary CD4+ cells to modify CXCR4 and CCR5 simultaneously. First, GHOST (3) CXCR4+CCR5+ cells were transduced with lentivirus-vectored CRISPR/Cas9, and the gene knockout efficacy was assessed between single sgRNA (sgX4/sgR5) and dual sgRNAs (sgX4 and sgR5). It was found that delivery of a single vector expressing both sgX4 and sgR5 yielded high-efficient ablation of both CCR5 and CXCR4. Importantly, a fraction of the modified cells obtained homologous gene deletions in both genes. Then, the assay was replicated on two CD4+ T-cell lines and similar results were obtained, confirming the potency of CRISPR/Cas9. Further, primary CD4+ T cells were transduced with Cas9 protein and in vitro transcribed sgRNAs by electroporation. The results showed modified CD4+ T cells resistant to both tropic HIV-1 infections. An off-target assay in modified GHOST cells proved the safety of CXCR4 and CCR5 ablation for CD4+ T-cell-based HIV-1 therapy.
Materials and Methods
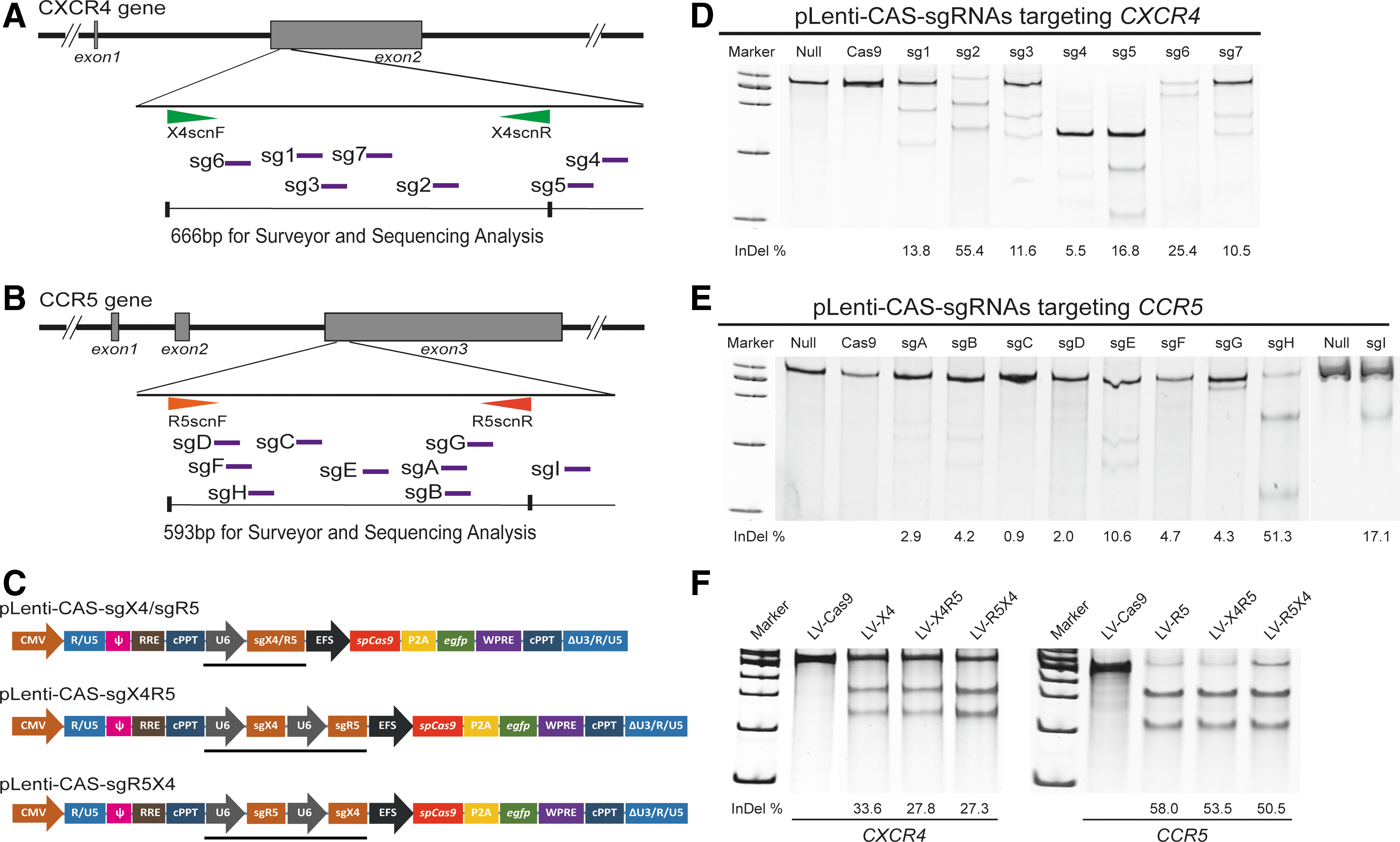
Construction of the pLenti-CAS-sgX4/pLenti-CAS-sgR5 and pLenti-CAS-sgX4R5/R5X4 vectors
For the single-sgRNA expression vectors, the cDNAs encoding sgRNAs for seven targets in the CXCR4 gene and eight targets in the CCR5 gene were designed and synthesized to construct the pLenti-CAS-sgX4 or pLenti-CAS-sgR5 transfer vectors, as previously described.
26,27
Briefly, the 24 bp forward and reverse primers, including 20 bp of the target sequence and BsmbI sticky end, were annealed and inserted into the lentiCRISPR-v2 plasmid (Addgene #52961) digested with BsmBI (NEB). For the dual-sgRNA expression vector, the U6 promoter-sgR5-H cassette was PCR amplified from pLenti-CAS-sgR5-H and then tandemly linked with the 3′ end of the sgX4-2 sequence of EcoRI-digested pLenti-CAS-sgX4-2 using a Clone Express II One Step Cloning Kit (Vazyme). This new plasmid was named pLenti-CAS-sgX4R5. The other dual-sgRNA expression plasmid, in which the sgR5-H cassette precedes sgX4-2, was also constructed following the same protocol except for PCR amplification of the U6 promoter-sgR5-H cassette and insertion into the pLenti-CAS-sgX4-2 backbone (Fig. 1C). All of the sgRNA target sites and primer sequences are shown in Supplementary Fig. S1 (Supplementary Data are available online at
Clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9-mediated editing of the human CXCR4 and CCR5 genes in HeLa-CD4 cells.
In vitro transcription and purification of sgX4-2 and sgR5-H
For in vitro transcription, a multiple-segment PCR, including a target primer pair corresponding to sgX4-2 or sgR5-H, Tracr fragment, and T7 Primer Mix, was used to produce sgX4-2 and sgR5-H coding templates. The in vitro transcription was conducted following the protocol of the GeneArt™ Precision gRNA Synthesis Kit (Thermo Fisher) and purified with a gRNA Clean Up Kit (Thermo Fisher). The concentration and the quality of the transcribed sgRNAs were measured with a Nanodrop 8000 (Thermo Fisher). For long-term storage, the transcribed sgRNAs were aliquoted and stored at −80°C.
Packaging and purification of CRISPR/Cas9 lentiviruses
CRISPR/Cas9 lentivirus production was carried out as previously described. 28,29 Briefly, twelve 145 mm petri dishes of 1 × 107 HEK293T cells (ATCC) were seeded the day before transfection in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum (FBS; DF-10; Gibco). Transfection was performed using linear PEImax (Polysciences). For each dish, 20 μg of pLenti-CAS-sgRNA plasmid, 15 μg of psPAX2, and 10 μg of pMD2.G (Addgene) was diluted in 1 mL of Opti-MEM (Gibco) and mixed gently. For each dish, the PEImax stock solution (7.5 mM) was diluted in 1 mL of Opti-MEM, and after 5 min, it was added to the mixture of DNA. The complete mixture was incubated for 20 min before being added to the cells. After 6 h, the medium was replaced with 20 mL of serum-free UltraCULTURE (Lonza) medium supplemented with 0.6 mg/mL of glucose (Sigma–Aldrich) and 1% GlutaMAX (Gibco). After an additional 60 h, a large amount of syncytia of fused cells was observed, and approximately half of the cells were still attached. All of the cell supernatant was collected and centrifuged at 500 g at 25°C for 10 min to pellet the cells and large debris. The supernatant was filtered through a 0.45 μm low protein binding membrane (Merck Millipore). To achieve a 300 × concentration of the CRISPR/Cas9 lentiviruses, the virus was ultracentrifuged (Becman) at 20,000 g for 2 h at 4°C and then re-suspended overnight at 4°C in phosphate-buffered saline (PBS) without Ca2+ and Mg2+. Aliquots were stored at −80°C. For primary CD4+ T gene modification, CRISPR/Cas9 lentiviruses underwent extra measures to purify the virus and increase the titer in order to improve the CRISPR/Cas9 efficacy on primary CD4+ T cells. First, the filtrated supernatant was subjected to Pellicon XL Cassette Bimax Membrane, an ultrafiltration (UF) tangential flow filtration (TFF) system (Merck Millipore), for replacement of media with PBS without Ca2+ and Mg2+. The filtrated supernatant was also passed through an anion exchange chromatography membrane to exclude impurities further. Other improvement measures included adjusting the pre-stimulation condition, adding as much virus as possible, strengthening the attachment of virions to the cell surface by pre-coating RetroNectin and spin infection for the purpose of promoting the genome editing of CRISPR/Cas9 lentiviruses in primary CD4+ T cells.
Titration of CRISPR/Cas9 lentiviruses based on the number of vector copies integrated into cellular genome
To determine the titer of concentrated lentiviruses, a real-time quantitative PCR method was utilized 29 with some modifications. Approximately 1 × 105 HeLa cells were cultured for 24 h and then infected with 0.5, 5, or 50 μL of prediluted stock lentiviruses (100 × ). Polybrene (final concentration of 8 μg/mL; Sigma) was added to increase infection. Twenty hours after initiating the transduction, the medium was replaced with 0.5 mL of DF-10 containing DNaseI (final concentration of 10 IU/mL; Takara). Cells were incubated for 15 min at 37°C. The medium was changed to fresh DF10, and the cells were incubated for another 48 h before being collected for DNA extraction with a DNeasy Blood and Tissue Kit (Qiagen). Real time quantitative PCR was conducted with transduced and normal cell DNA as templates. For viral WPRE detection, the primer pair forward: 5′-CCTTTCCGGGACTTTCGCTTT-3′, reverse: 5′-GCAGAATCCAGGTGGCAACA-3′, and the probe 5′-FAM-ACTCATCGCCGCCTGCCTTGCC-TAMRA-3′ were used. As an internal reference, RNaseP was used to normalize the WPRE copies per cell. The primer pair and probe for RNaseP were included in the TaqMan RNaseP control reagent kit (Applied Biosystems). The PCR cycling conditions were set as follows: 1 min at 95°C, 45 cycles of 95°C for 15 s, and 60°C for 15 s with plate read, then 10 s at 4°C. Real-time PCR was performed with a CFX96 real-time PCR system (Bio-Rad Laboratories). The vector copy numbers in HeLa cells were normalized to human RNase P gene copies and presented as proviral copies per genome equivalent. Titers were calculated (integration units per mL [IU/ml]) according to the following formula: IU/mL = (P × N × D × 1,000)/V, where P = proviral copies per genome, N = number of cells subjected to transduction, D = dilution of stock viruses, and V = volume of diluted viruses added in each well for transduction. To obtain a precise titer, average values were gained from three vector dilutions. A linear relationship exists between the number of integrated viral copies per genome and the amount of lentivirus added at fewer than five viral copies per genome.
Cell line cultures and transduction of CRISPR/Cas9 lentiviruses
HEK293T cells (Clontech) were cultured in DF-10 medium supplemented with 1% GlutaMAX and 1% penicillin–streptomycin (Gibco). HeLa-CD4-LTR-β-gal cells (NIH AIDS Reagent Program) were cultured in DF-10 medium supplemented with 0.2 mg/mL of G418 and 0.1 mg/mL of hygromycin B and passaged every 3 days. GHOST (3) CXCR4+CCR5+ cells (NIH AIDS Reagent Program) were cultured in DF-10 medium supplemented with 0.5 mg/mL of G418, 0.1 mg/mL of hygromycin B, and 1 μg/mL of puromycin.
For cell-line transduction, 5 × 104 cells were seeded onto 12-well plates on day 0. CRISPR/Cas9 lentiviruses or the GFP lentivirus control were added to the cell culture at a multiplicity of infection (MOI) of 2.5–5 on day 1. Twenty-four hours later, the medium was changed to fresh DF-10 with cell type-adapted antibiotics added. Cells were trypsinized for phenotypic determination or DNA extraction after an additional 3–5 days of culture.
Primary CD4+ T-cell culture and electroporation with Cas9RNPs
Whole-blood samples were obtained from healthy donors under the auspices of the Guangzhou Medical University affiliated first hospital (Guangzhou, China). The human peripheral blood mononuclear cells (PBMCs) were isolated from the whole blood by centrifugation with LymphoPrep™ (Stemcell). The human primary CD4+ T cells were enriched using the Dynabeads® CD4 Positive Isolation Kit (Thermo Fisher) according to the manufacturer's instructions and then maintained in Optimizer full medium (basal medium with added supplement, 10% heat inactivated FBS, 2 mM of L-Glutamine, and 100 IU/mL of rhIL-2). The purity of enriched CD4+ T cells was determined by flow cytometry with FITC-labeled anti-CD4 (BD Biosciences). Before transduction, CD4+ T cells were pre-activated for 24 h with anti-CD3/anti-CD28-bonded microbeads in the presence of recombinant human interleukin-2 (100 IU/mL; Peprotech). One hour before electroporation, the Cas9 nuclease stock solution (GeneArt™ Platinum™ Cas9 Nuclease; 1 μg/μL) and purified in vitro transcribed sgRNAs (sgX4-2 and sgR5-H) were diluted to a working concentration of 20 μmol/L each. Then, the two components were mixed at a ratio of 1:1 to form a preassembled Cas9RNP complex according to the manufacturer's instructions. A total of 1 × 105 T cells were washed three times with PBS before resuspension in 8 μL of buffer T (Neon kit; Invitrogen). Cas9RNP (4.5 μL of 10 μM Cas9 protein without sgRNA or 1.8–4.5 μL Cas9RNP complex; final concentration: 2–4 μM) was added to the cell suspension to a final volume of 11 μL (adjusted with Cas9 storage buffer) and mixed. Ten microliters of the suspension was electroporated with a Neon electroporation device (Invitrogen; 1,600 V, 10 ms, 3 pulses). Then, cells were returned to Optimizer full medium for continuous culture until sampled for the genetic mutation assay and functional identification.
Surveyor assay for genome mutagenesis
The genomic DNA was extracted from modified and control cells using a DNeasy Blood and Tissue Kit (Qiagen). After PCR amplification of the CXCR4/CCR5 sgRNA binding sites by Phanta Max Super-Fidelity DNA Polymerase (Vazyme) with the gene-specific primers listed in Supplementary Fig. S1, the Surveyor nuclease (Surveyor mutation detection kit; Transgenomic) was used according to the manufacturer's instructions. The digested DNA was resolved by 12% PAGE on the Gel Imaging System. The ratio of cleaved to un-cleaved products was calculated as a measure of frequency of gene disruption using Image J software (NIH Image-BioLab). 31 For sequence analysis, the purified PCR product was cloned into a BamHI-Hind III dual-digested pUC19 vector using Clone Express II One Step Cloning Kit (Vazyme), and then the recombinants were sequenced using an M13F primer. The assay is sensitive to detecting a single base pair change induced by non-homologous end joining (NHEJ).
Flow cytometry analysis
Single-cell suspensions were prepared from trypsinized cell lines or primary CD4+ T cells. The flow cytometry assay was performed on a C6 flow cytometer (BD Biosciences). All of the antibodies were purchased from BD Biosciences. The following clones were used: CD4 (RPA-T4), CXCR4 (12G5), and CCR5 (3A9). For surface marker measurement, individual antibodies were added to the cell suspension alone or together, with IgG1a-FITC, IgG2a-PE, or IgG2a-APC used as the isotype for anti-CD4-FITC, anti-CXCR4-PE, or anti-CCR5-APC, respectively. Cells were stained with antibodies for 20 min at room temperature followed by washing twice with PBS plus 2% FBS. After resuspension, cells were subjected to fluorescence activated cell sorting (FACS) analysis in <8 h. For the apoptosis assay, cells were first stained with anti-CD4-FITC, washed once with PBS containing 2% FBS, then re-suspended in 100 μL of 1× binding buffer (BD Biosciences). Five microliters of Annexin V-PE and 5 μL of 7-AAD were added and incubated for 15 min in the dark. Finally, cells were diluted with 1× binding buffer and immediately subjected to FACS analysis.
Enzyme-linked immunosorbent assay for p24 protein post HIV-1 infection
Two HIV strains with different co-receptor tropism were used in this assay: HIV-1NL4-3, which uses CXCR4 for entry, and HIV-1YU 2, which uses CCR5 for entry. Both of the strains were prepared and supplied by Prof. Hui Zhang's lab, according to previous research. 30 Approximately 1 × 104 of LV-CRISPR-treated or non-treated GHOST X4R5 cells were exposed to HIV-1NL4-3 and HIV-1YU 2 alone or together at a MOI of 0.8 TCID50 in a humidified 37°C incubator containing 5% CO2. After 6 h, the cells were washed three times with PBS and cultured in fresh medium. At different time points (from day 2 to day 16 post infection), the supernatants were collected, centrifuged to remove cell debris, and frozen. The GFP expression of GHOST X4R5 cells upon infection of HIV-1 was observed and captured by a fluorescence microscope with a digital camera (Nikon). For primary CD4+ T cells nucleofected with Cas9RNPs, the procedure was similar to GHOST X4R5, except for the total dose of HIV-1 (from a MOI of 0.8 TCID50 to a MOI of 0.4 TCID50) and the duration of infection (from 6 h to 3 h) for each infection. If using dual HIV-1 (NL4-3 and YU 2) to infect target cells simultaneously, each strain shared half of the viral dose. All the challenge events occurred in a total volume of 500 μL of serum-free medium. After infection, the medium was removed, and the cells were cultured with fresh complete medium after washing with PBS three times. Once the last sampling was done (day 14 or day 17 post infection), the cell-free supernatant was analyzed for HIV-1 p24 antigen using an HIV-1 p24 Antigen Capture Assay kit (Advanced Bioscience Laboratories) according to the manufacturer's instructions.
Western blot
A fraction of the non-modified or LV-X4/LV-R5/LV-X4R5-treated GHOST X4R5 cells or sgX4 and sgR5 Cas9RNP-transduced primary CD4+ T cells were lysed in a buffer (50 mM of Tris-HCl, pH 7.4, 150 mM of NaCl, 1% NP-40, and 0.25% deoxycholate) containing a protease inhibitor cocktail (Roche Applied Science) for 30 min on ice. The lysates were denatured in 2 × SDS loading buffer by boiling for 10 min and were subjected to a 4–15% continuous SDS-PAGE. After transferring the protein to PVDF membranes (Merck Millipore), the anti-CXCR4 polyclonal antibody (ab1670; Abcam) or anti-CCR5 monoclonal antibody (ab1673, Abcam) and the Immobilon Western chemiluminescent HRP substrate (Merck Millipore) were used to detect the cell surface CXCR4/CCR5 expression. The bands were scanned by the GelDoc XR system (Bio-Rad).
Cell viability and proliferation assays
Freshly isolated human primary CD4+ T cells were seeded onto 96-well plates at a density of 1 × 105 cells per well. After 24 h of pre-stimulation, cells were nucleofected with Cas9RNPs or a PBS control, as described above. Three days later, cytotoxicity assays were conducted using the Cell Counting Kit-8 (Vazyme), according to the manufacturer's instructions. For the cell propagation assay, 1 × 105 primary CD4+ T cells were seeded onto 96-well plates and pre-stimulated for one day. Then, Cas9RNPs or PBS was electroporated to washed CD4+ T cells. Thereafter, the total number of live cells was counted every 2 days based on the trypan blue staining assay.
Off-target analysis
The sgX4-2 and sgR5-H sequences were blasted with an online tool (
Statistical analysis
All statistical analyses were performed with GraphPad Prism software. Statistical significance was determined with Student's t-test, with p < 0.05 considered statistically significant. Increasing levels of confidence are displayed as *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001.
Results
Efficient modification of CXCR4 and/or CCR5 genes via CRISPR/Ca9 in HeLa-CD4 cells
To look for sgRNAs specifically targeting gene loci of interest, the protein-coding region close to the 5′ terminus of the first or second exon was chose to design candidate sgRNAs according to established online guidelines (
Next, the U6 promoter-sgR5-H cassette was inserted into the 3′ end of the sgX4-2 sequence of pLenti-CAS-sgX4, producing a new plasmid named pLenti-CAS-sgX4R5 (Fig. 1C), which contained the two optimal sgRNAs (sgX4-2 and sgR5-H) targeting CXCR4 and CCR5. To assess the influence of the relative order of the two sgRNAs, the other dual-sgRNA plasmid pLenti-CAS-sgR5X4 (sgR5-H is before sgX4-2) was constructed using the same protocol.
The CRISPR/Cas9 lentiviruses (LV-CRISPRs) were produced by transfection of pLenti-CAS-sgX4/-sgR5/-sgX4R5/-sgR5X4, together with psPAX2 and pMD2.G into 293 T cells. The viral supernatant was purified, concentrated, and titered, as described in the Materials and Methods. The produced lentiviruses were named LV-X4, LV-R5, LV-X4R5, and LV-R5X4. As a vector control, the lentivirus expressing Cas9 only (LV-Cas9) was also prepared. Infection of LV-X4R5 or LV-R5X4 into HeLa-CD4 cells at a MOI of 2.5 showed remarkable disruption of both CXCR4 and CCR5 with comparable efficiency in LV-X4 or LV-R5 vectors (Fig. 1F). Furthermore, the relative order of the two tandemly linked sgRNAs targeting CXCR4 and CCR5 had no obvious effect on gene modification efficacy (Fig. 1F). Thus, the LV-X4R5 was selected for subsequent experiments. Collectively, these data proved that lentivirus-vectored CRISPR/Cas9 could efficiently disrupt CXCR4 and CCR5 in mammalian cells.
GHOST (3) CXCR4+CCR5+ cells transduced with LV-X4R5 are resistant to both X4-tropic and R5-tropic HIV-1 infections
To determine whether the established LV-X4R5 could result in the loss of function of both CXCR4 and CCR5 co-receptors, the GHOST (3) CXCR4+CCR5+ cells (GHOST X4R5) were used for a dual gene knockout assay. The GHOST X4R5 cells derived from human osteosarcoma (HOS) cells express CD4, CXCR4, and CCR5 at higher levels than normal HOS cells. The integrated tat-dependent HIV-2 LTR-GFP construct produced GFP in response to HIV/SIV infection, making GHOST X4R5 cells suitable for identifying diverse HIV/SIV viral strains with alternative co-receptor (CXCR4 or CCR5) tropism. GHOST X4R5 cells were transduced with LV-X4, LV-R5, or LV-X4R5 at a MOI of 2.5. After 5 days of culture, some cells were collected for gene mutation analysis based on the Surveyor assay. The CXCR4 mutation efficiencies of bulk GHOST X4R5 cells treated with LV-X4 or LV-X4R5 were 37% and 24%, respectively (Fig. 2A). For CCR5, the gene modification rate was 63% for LV-R5 and 48% for LV-X4R5 (Fig. 2A). Western blot analysis showed that the CCR5 protein was markedly reduced after treatment with LV-R5 or LV-X4R5 compared to untreated or mock-infected controls. For CXCR4, the total protein level post LV-X4 or LV-X4R5 transduction was lower than the controls (Fig. 2B). This may be due to the innate expression of endogenous CXCR4 in GHOST (3) parental cells. 32
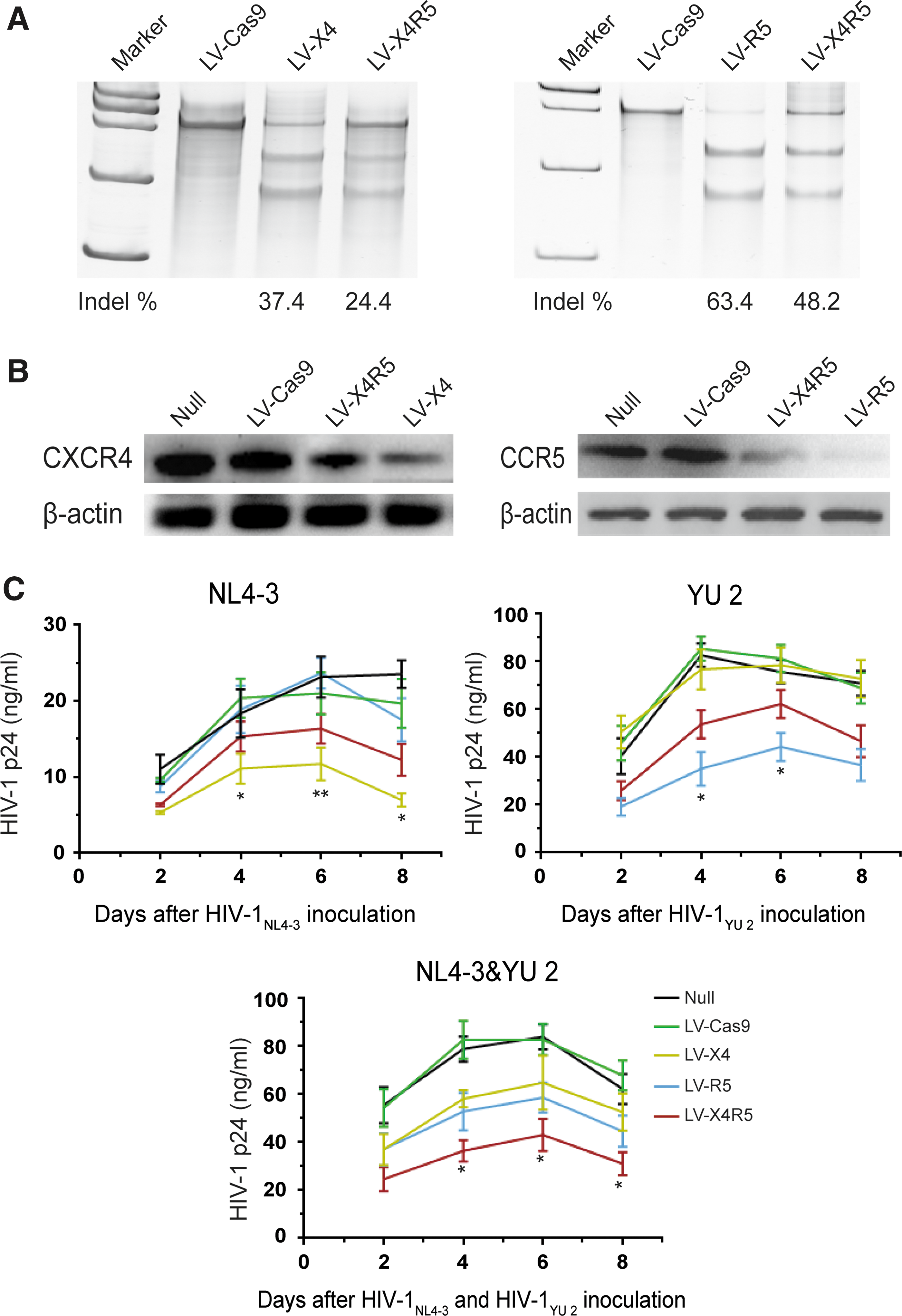
GHOST X4R5 cells transduced with CRISPR/Cas9 lentiviruses are resistant to both X4-tropic and R5-tropic HIV-1 challenges.
Next, the LV-X4/LV-R5/LV-X4R5-transduced GHOST X4R5 cells were infected with two HIV-1 strains, CXCR4-tropic HIV-1NL4-3 and CCR5 tropic HIV-1YU 2. The LV-X4 or LV-R5-transduced GHOST X4R5 cells can significantly resist the X4-tropic or R5-tropic viral infection, respectively, under the challenge of either HIV-1 strain. LV-X4R5-treated cells show partial resistance to either viral infection. There are significant differences between LV-X4 and LV-X4R5 during the period from day 8 to day 12 after HIV-1NL4-3 infection (p < 0.05). Similarly, there are significant differences between LV-R5 and LV-X4R5 during the period from day 4 to day 6 after HIV-1YU 2 infection (p < 0.05). However, when infected with a mixture containing HIV-1NL4-3 and HIV-1YU 2, LV-X4R5 treatment provided more protection to viral infection than LV-X4 or LV-R5, especially during the period from day 4 to day 8 post infection (p < 0.05; Fig. 2C).
To detect a change in the surface expression of CXCR4 and CCR5 after single or dual-gene modification, FACS was used to analyze the superficial co-receptors in GFP-positive GHOST X4R5 cells after treatment with CRISPR/Cas9 lentiviruses. The LV-X4 or LV-R5 single infection efficiently reduced the surface level of CXCR4 or CCR5 co-receptors, respectively, from 60% to 10% for CXCR4 and from 80% to 1% for CCR5 (Fig. 3A). Surprisingly, the LV-X4R5 infection of GHOST X4R5 led to dramatically reduced surface expression of CCR5 (from 80% to 0.7%) but caused lower surface expression of CXCR4 (from 60% to 13.8%). However, the number of CXCR4 and CCR5 double-positive cells was sharply reduced (from 50% to 0.1%), and the number of cells in the double-negative subgroup increased remarkably after transduction of LV-X4R5 compared to LV-X4 or LV-R5 treatments (p < 0.05; Fig. 3B). This implies that approximately half of the GHOST X4R5 cells were resistant to both X4 and R5 tropic HIV-1 challenges.
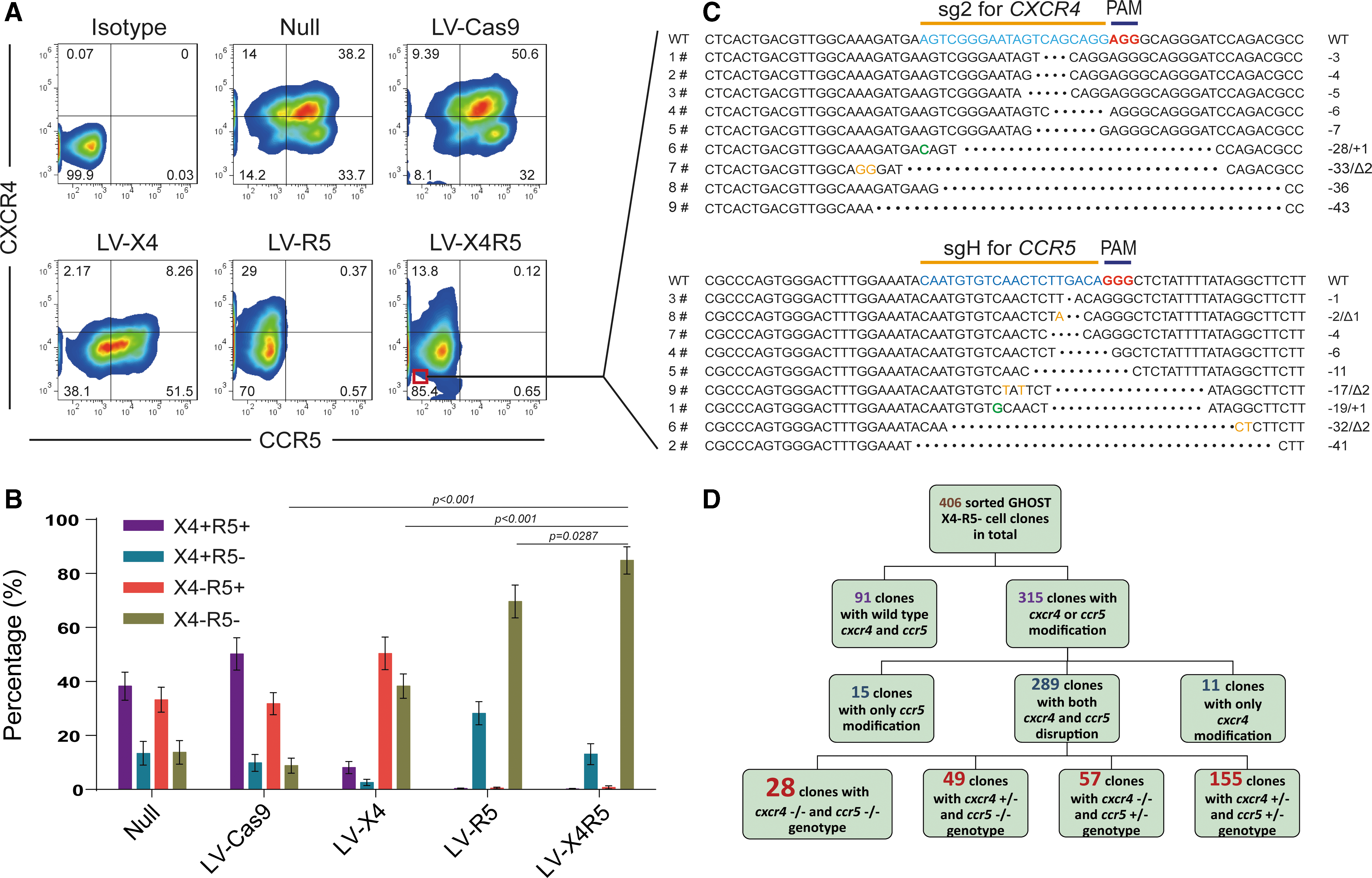
Identification of the genotype for LV-CRISPR-transduced GHOST X4R5 cells and clonal analysis of homozygous gene modification.
To assess the percentage of CXCR4 and/or CCR5 modification after treatment of LV-X4R5 at the single-cell level, the X4 and R5-double negative GHOST X4R5 cells were sorted according to surface expression of the CXCR4/CCR5 co-receptors (Fig. 3C). Next, the sorted cells were subjected to limiting dilution and then seeded into 96-well plates, with each well containing ideally one single cell. After 2–3 weeks of culture, the expanded cell clones were trypsinized separately. All of the cell clones were analyzed with Sanger sequencing with the aim of targeting the CXCR4 and CCR5 region. More than 400 cell clones were analyzed, and 65% of the cell clones (clonal number = 315) had mutations in the CXCR4 and/or CCR5 loci. Eleven clones carried indels only in the CXCR4 locus, while 15 clones carried indels only in the CCR5 locus. Out of the remaining 289 dual gene-mutated cell clones, 155 clones obtained heterozygous mutations in both CXCR4 and CCR5 target sequences, 57 clones obtained homozygous CXCR4 deletions and heterozygous CCR5 deletions, and 49 clones carried homozygous CCR5 deletions and heterozygous CXCR4 deletions. More importantly, there were 28 cell clones with indels in biallelic CXCR4 and CCR5 loci (Fig. 3D). In summary, a tandemly linked CRISPR was successfully constructed in a single LentiCRISPR backbone that could target and modify multiplex genes simultaneously. The double co-receptor-modified GHOST X4R5 cells could resist both X4-tropic and R5-tropic HIV-1 infections.
Human CD4+ T cells modified by Cas9RNPs confer resistance to both X4-tropic and R5-tropic attack
Human CD4+ T cells are the major target for HIV-1 infection. Based on the efficacy of LV-CRISPRs in genetic modifications, an attempt was made to transduce the double-function LV-X4R5 into human CD4+ T cells to disrupt CXCR4 and CCR5. First, two modified CD4+T cell lines were

CD4+ T-cell lines treated with LV-X4R45 are resistant to X4-tropic and R5-tropic HIV-1 challenge.
Next, an attempt was made to modify primary human CD4+T cells via LV-CRISPRs. However, none of the CRISPR/Cas9 lentiviruses could disrupt endogenous CXCR4 or CCR5 in primary CD4+ T cells. Al the possible improvement measures were tried, including purification of viruses with ultrafiltration and anion exchange chromatography, improving pre-stimulation condition, adding more viruses, strengthening the attachment of virions to cell surface, and others (Fig. 5A), but these still failed in deleting genes in primary CD4+ T cells by lentiviral-vectored CRISPR/Cas9.
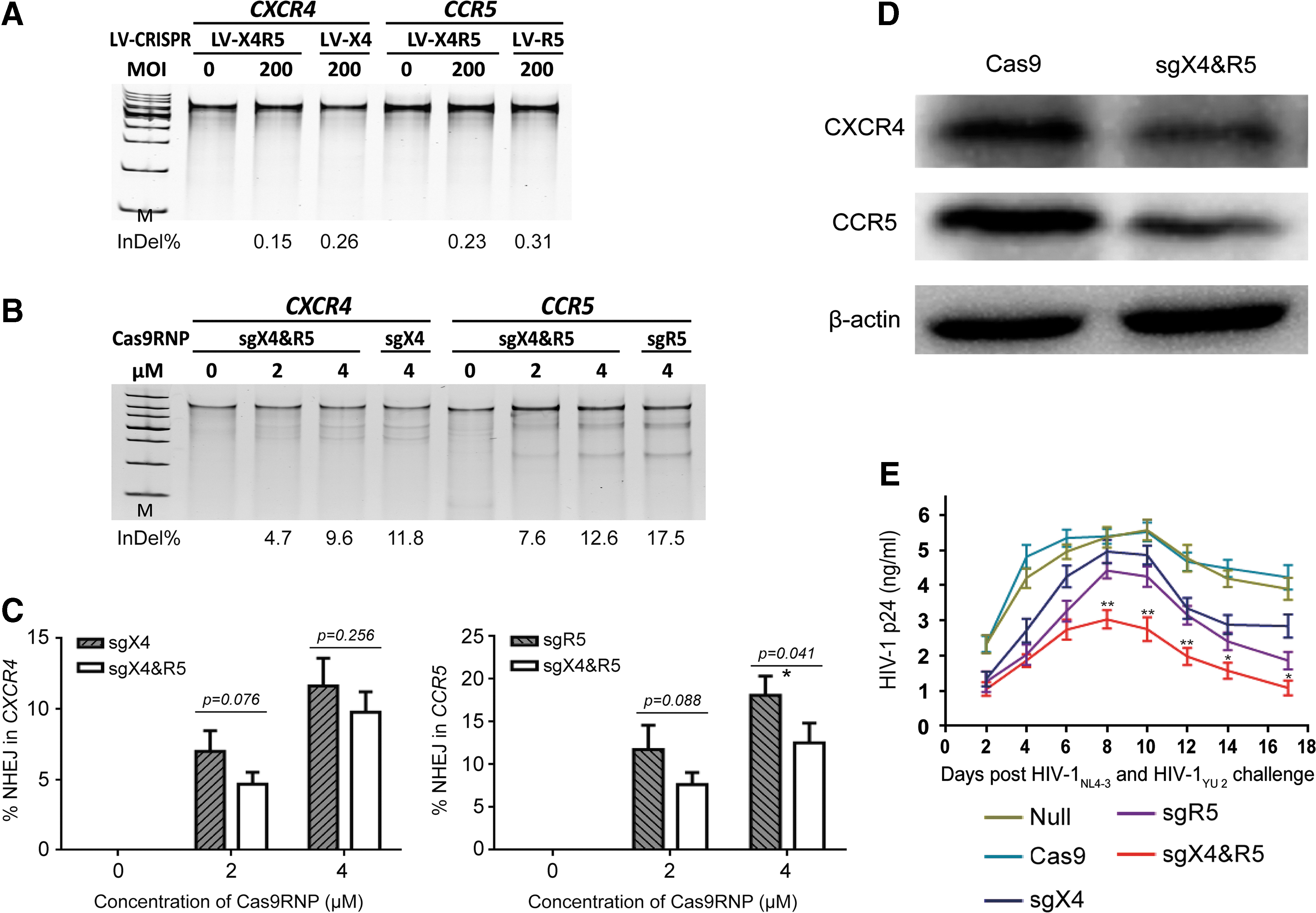
CXCR4/CCR5 disruption by Cas9RNPs protects primary human CD4+ T cells from either X4 or R5-tropic HIV-1 infection.
Next, the focus was on electroporation of the CRISPR/Cas9 system into primary cells, according to recent research. 33,34 For hard-to-transfect cell types, viral vectors hold great promise in obtaining stable transgene expression and in situ infection of certain parts of animal tissue. Accordingly, a lentivirus harboring the CRISPR/Cas9 cassette will randomly integrate the large exogenous DNA sequence into host cells, leading to an unpredictable risk of genetic toxicity. Considering the need for the temporary effect of the RNA-guided Cas9 endonuclease, direct delivery of purified Cas9 protein and gRNAs will function for a limited time and profoundly reduce the potential toxicity of genes. Therefore, a commercial Cas9 nuclease, GeneArt™ Platinum™ Cas9 Nuclease (Thermo Fisher) preassembled with in vitro-transcribed sgX4-2 and sgR5-H alone or together, was delivered to human CD4+ T cells via electroporation, according to the manufacturer's instructions. The molar ratio of total sgRNA to Cas9 protein to form a Cas9RNP complex is approximately 1:1. Three days post electroporation, CD4+ T cells were subjected to a genomic mutation assay. The efficiency of bulk CXCR4 disruption was approximately 12% for sgX4 Cas9RNP, while the efficiency for bulk CCR5 disruption was nearly 18% for sgR5 Cas9RNP (Fig. 5B). Surprisingly, the CXCR4 and CCR5 genome-editing ratios were moderately lower for sgX4 and sgR5. Yet, there were no significant differences between single sgRNA and dual sgRNAs (p = 0.076 with 2 μM of RNP and p = 0.256 with 4 μM of RNP for sgX4 compared to sgX4 and sgR5; p = 0.088 with 2 μM of RNP for sgR5 compared to sgX4 and sgR5), except for sgR5, which was slightly higher than sgX4 and sgR5 with 4 μM of RNP (p = 0.041; Fig. 5C). The protein levels of CXCR4 and CCR5 were also assessed after treatment with different Cas9RNPs. Consistent with gene disruption, sgX4 and sgR5 Cas9RNP treatment yielded a moderately reduced expression of CXCR4 but led to a dramatically lower expression of CCR5 (Fig. 5D). To determine whether modified primary CD4+ T cells obtained a loss-of-function phenotype, Cas9RNPs-treated CD4+ T cells were challenged with HIV-1NL 4-3 and HIV-1YU 2 together at a MOI of 0.4 TCID50, and the HIV-1 p24 core antigen was quantified every other day. During infection, the concentration of p24 increased from 1 ng/mL to nearly 5 ng/mL for CD4+ T cells transduced with Cas9 protein only or PBS. However, the level of p24 in sgX4 Cas9RNP-treated or sgR5 Cas9RNP-treated cell supernatants was moderately lower than null-treated and Cas9 alone-treated cells but remarkably higher than sgX4 and sgR5 Cas9RNP-treated cells, especially from day 8 to day 17 post infection (p < 0.05; Fig. 5E). The CD4+ T cells were not given complete protection from HIV-1 due to the relatively lower efficacy of both CXCR4 and CCR5 gene modifications, indicating the need to improve the gene transfer and gene modification further for better performance. Nevertheless, these results proved that electroporation of Cas9RNPs could modify multiplex endogenous genes concurrently and render primary CD4+ T cells partially resistant to HIV-1 infection.
Viability, propagation, and apoptosis of primary CD4+ T cells after electroporation of Cas9RNPs
There has been concern that electroporation may lead to cell death, especially under high voltage, which is optimal for better transduction. 35 To address this question, Cas9RNPs were delivered into pre-activated human CD4+ T cells according to an optimized protocol of electroporation, and cells were collected at different time points. Cell viability was measured using a CCK8 kit 3 days after transduction, according to the manufacturer's instructions. No apparent differences in cell viability were observed among the different Cas9RNPs (Fig. 6A). Then, cell propagation was assessed by using a trypan blue staining assay every other day for 14 days. The cell growth was identical between non-treated control and Cas9RNPs-treated groups (Fig. 6B), indicating that CRISPR/Cas9 will not affect the propagation and growth of human CD4+ T cells. Moreover, CD4+ T cells were collected for early and late apoptosis assays using AnnexinV and 7AAD (BD Pharmingen). The double-negative subset was regarded as live cells, the AnnexinV- 7AAD+ subgroup was classified into the early phase of apoptosis, and the double positive subpopulation was considered as the late phase of apoptosis or death. After treatment of Cas9RNPs, cells were collected, washed gently, stained sequentially with Annexin V and 7AAD, and subjected to FACS analysis. The majority of the CD4+ T cells were viable post electroporation with each Cas9RNP (Fig. 6C). Importantly, the number of Cas9RNP-transduced cells undergoing apoptosis was comparable to that of non-treated and Cas9 alone-treated controls (Fig. 6D; no significant differences). Collectively, these data indicate that electroporation of Cas9RNP will not influence the viability and proliferation of primary CD4+ T cells.
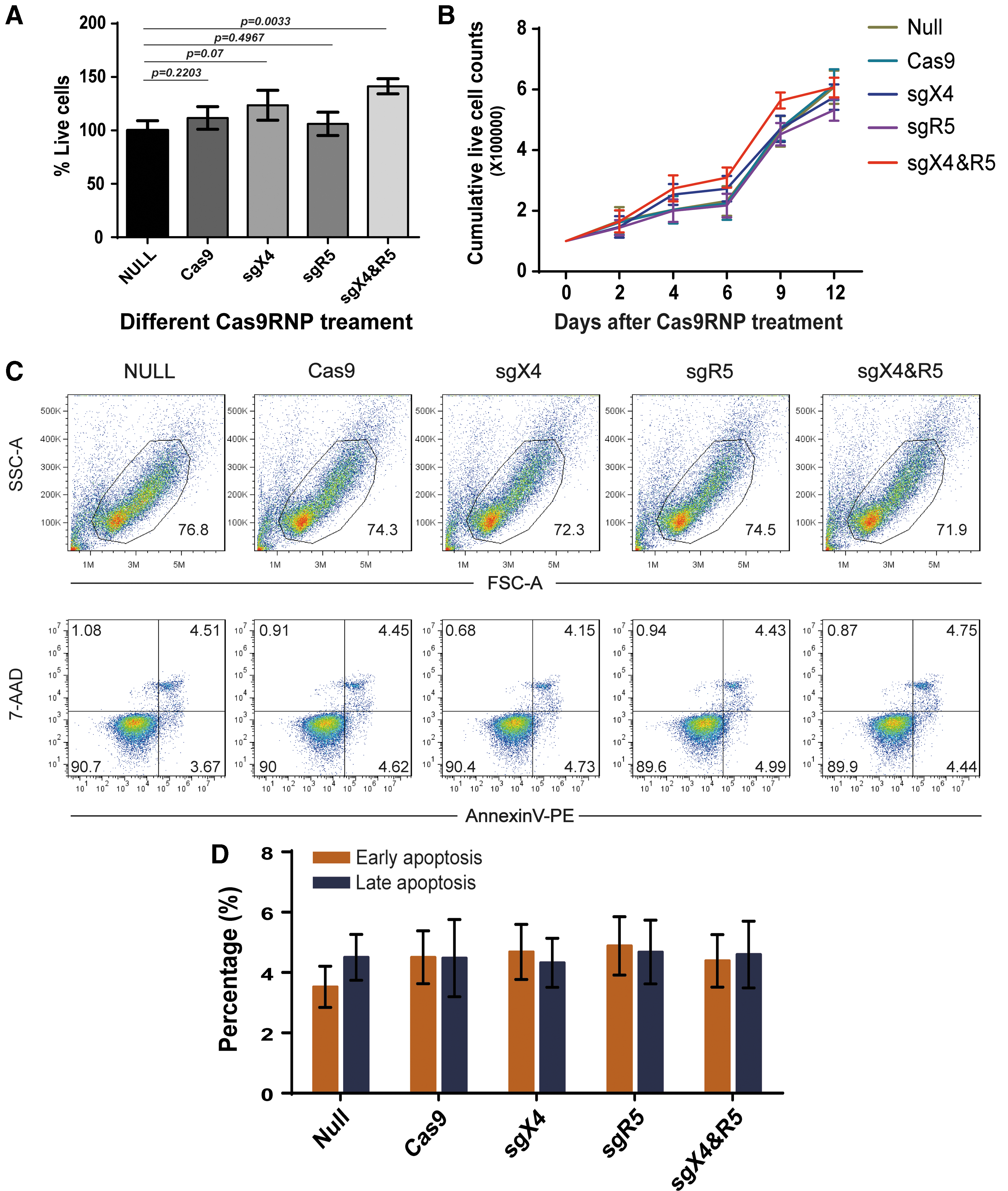
The effect of Cas9RNPs on primary CD4+ T-cell viability, proliferation, and apoptosis.
Off-target mutagenesis in human CD4+ cells treated with CRISPR/Cas9
The CRISPR/Cas9 system serves as a versatile tool for genome manipulation,
36
However, the potential for off-target mutagenesis remains a matter of concern when considering future clinical applications. To determine the off-target effects of the highly efficient CRISPR/Cas9 system, referral was made to the online guidelines (
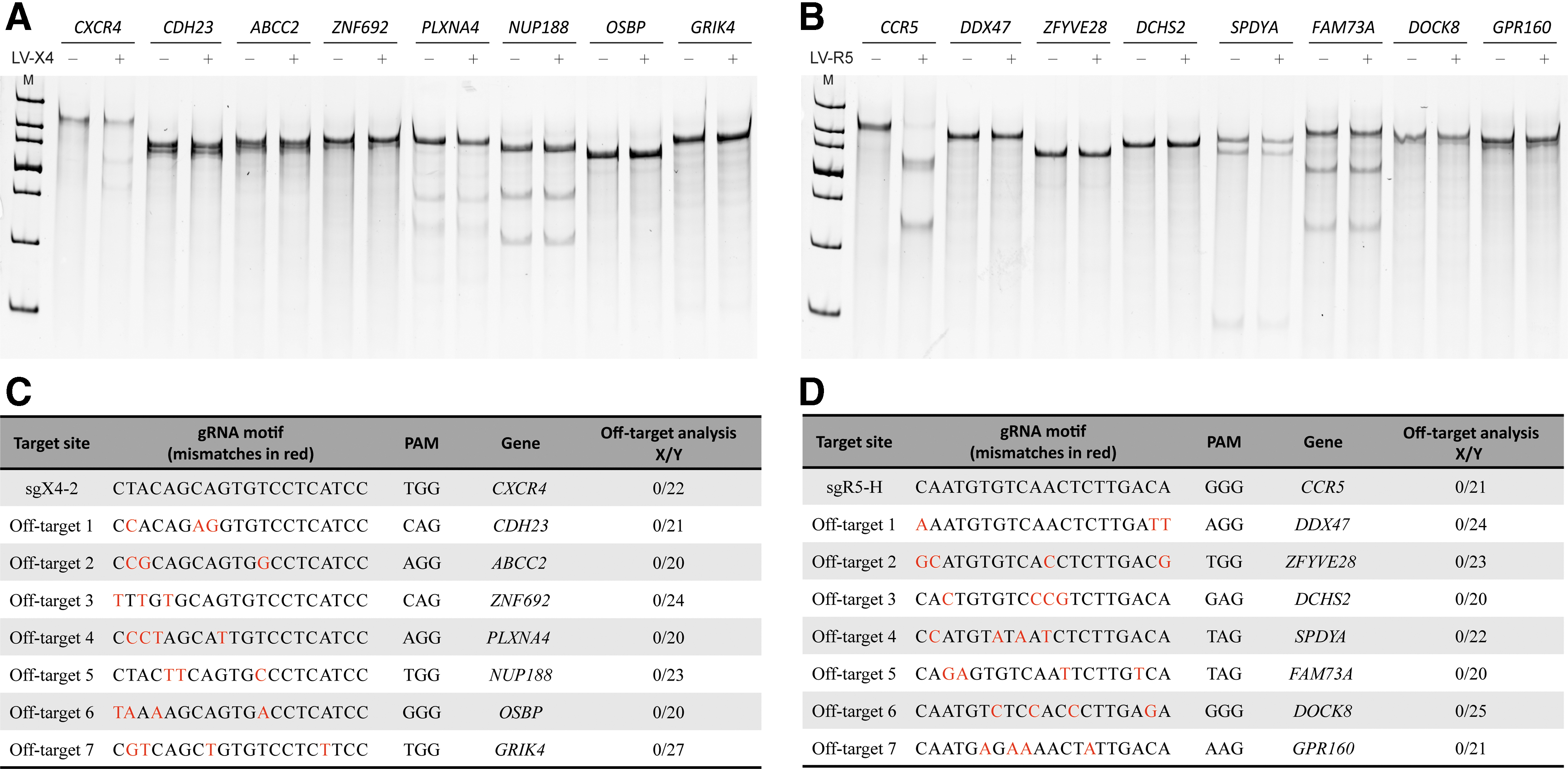
Off-target analysis of CXCR4-sg2 and CCR5-sgH in transduced GHOST X4R5 cells.
Discussion
The successful case of the “Berlin patient” who was cured of HIV-1 infection after transplantation with a homologous CCR5Δ32 donor opened a window to the potential of cell-based gene therapy to control acquired immune deficiency syndrome (AIDS). 37,38 However, there are still obstacles in extending this policy to a broader clinical practice due to the rare percentage of natural CCR5Δ32 homozygous individuals and the limitation of an HLA match. Thus, modification of CCR5 or other co-receptors by manufactured genome-editing tools will provide an alternative choice for a HIV-1 cure. 39
Two potential cellular targets are being tested for CCR5 deletion-based HIV-1 therapy: CD34+ hematopoietic stem cells (HSCs) and CD4+ T cells. 40 The former would give rise to HIV-1-resistant T cells and macrophages to recapitulate the “Berlin patient” case. However, manipulation of HSCs from HIV-1 infected patients is a great challenge. First, HIV-1 has been proven to infect multipotent hematopoietic progenitor cells 41 (possibly involving some primitive HSCs 42 ) and cause irreversible damage to the hematopoietic function of bone marrow. Second, genetic manipulation of primary stem cells, especially HSCs in vitro, holds serious concerns about the balance between improving the efficacy of gene modification and retaining the pluripotency of hematopoietic differentiation. Lastly, the genuine subset of HSCs in peripheral blood is extremely rare, and in vitro proliferation of HSCs is still an obstacle for large-scale therapeutic cell production. 43
In recent years, induced pluripotent stem cells (iPSCs) reprogrammed from somatic cells were utilized to differentiate into multilineage hematopoietic progenitors in vitro. 44 –46 Genome editing and large-scale production of iPSCs are relatively easy to realize. ZFNs/TALENs were previously delivered into human iPSCs by lipofectamine 47 or in the form of penetrating proteins, 12 yielding efficient gene disruption in CCR5 locus. CRISPR/Cas9 was also used to modify CCR5 in iPSCs with high efficiency and conferred progeny cells resistant to R5-tropic HIV-1 infection. 19,48 However, the purity and functionality of genetic-engineered CD34+ cells were suboptimal, and the gene toxicity needs to be further verified in vivo. 12,19,47,48
Compared to HSCs, peripheral CD4+ T cells possess obvious advantages in their availability for genetic manipulation and long-term cultivation in vitro. CD4+ T cells that were transduced with adenovirus-vectored ZFNs targeting the CCR5 locus have been applied in several clinical trials. According to reports by Pablo et al., 14 there was no detectable viral RNA in the peripheral blood of one patient who had been infused with autologous CD4+ T cells modified by CCR5-ZFNs. Moreover, CD4+ T cells contain some stem cell-like subsets, including central memory T cells, effector memory T cells, and stem-cell memory T cells, which hold the potential to self-renewal and maintain the engineered phenotype of resisting HIV-1 challenge. Thus, we applied the CRISPR/Cas9 system to primary CD4+ T cells aiming to confer dual-protection from both X4- and R5-tropic HIV-1 infections.
Cas9s are engineered nucleases that target and cut specific cellular double-stranded DNA sequences under the guidance of the CRISPR array. The treated cells try to repair the double-strand break using NHEJ, an error-prone mismatch repair mechanism that often leads to a nonfunctional gene product. The CRISPR/Cas9 system can be delivered to CD4+ T cells and/or HSCs using nucleofection, 17,33,49 an adenoviral vector delivery system. 50 Some strains of HIV-1 have evolved to use a different cellular co-receptor, CXCR4. These X4-tropic viruses are rarely transmitted but develop within an infected individual over time. They are closely associated with a poor prognosis and rapid disease progression. There is concern that the use of a CRISPR/Cas9 that inactivates CCR5 will lead to the selection of X4-tropic HIV-1 strains. Thus, targeting CCR5 alone could not be used in individuals who harbor dual or mixed-tropic viruses, which is approximately half of the individuals with AIDS. Previous research has demonstrated that independent disruption in CXCR4 or CCR5 will supply resistance to either T-tropic or M-tropic HIV-1, but it is currently unclear whether they could successfully be used synergistically. To protect a cell fully from both R5- and X4-tropic viruses, simultaneous editing of four alleles would be required.
Using ZFNs to target CXCR4 and CCR5 simultaneously has been tested in primary CD4+ T cells isolated from peripheral blood. 19 After transplantation of the modified CD4+ T cells into immune-deficient mice, the animals were resistant to both X4-tropic and R5-tropic HIV-1 infections. ZFNs hold advantages with respect to their smaller molecular size and are suitable for viral vector-loaded delivery into primary cells. For CRISPR/Cas9, the relatively large size of the entire coding sequence brings obstacles in transferring the RNA-guided endogenous nuclease into hard-to-transfect cells. Several improvements have been tested. Staphylococcus aureus Cas9 (SaCas9), with an approximate size of 3 kb, is suitable for adeno-associated virus assembly. 51 Other CRISPR related endonucleases, apart from Cas9, are utilized for genome editing, such as the Cpf1 family, which utilizes a single RNA guide without tracRNA and a T-rich protospacer-adjacent motif for complementary base pairing and cleavage of target DNA. In addition, CRISPR/Cpf1 has also been proven to edit multiplex human genes simultaneously. 52,53 By applying newly developed gene engineering tools, the CXCR4-CCR5 dual modification will likely be optimized to obtain more homozygous gene-deleted CD4+ T cells for better clinical effects.
The present work successfully disrupted both co-receptors in HIV-1 susceptible cell lines and primary CD4+ T cells with undetectable off-target mutagenesis. For GHOST X4R5 cells, the best efficacy for modification was obtained by using a lentivirus delivered CRISPR/Cas9 system in a single construct. It was proven that a single round of transduction of LV-X4R5 can induce sufficiently high levels of both CXCR4 and CCR5 modification, yielding various outputs of NHEJ. The majority of indels are deletions ranging from a loss of a single base pair to >10 bp. The CRISPR/Cas9-treated cells were purified and subjected to monoclonal analysis. Approximately four-fifths of modified cells were given CXCR4-CCR5 dual disruption, and of those, one third were homozygous for both genes. The relatively low rate (∼10%) of editing four alleles simultaneously might be remedied by the addition of X4- and R5-tropic HIV-1 strains to a cell-culture system based on the finding that challenging with HIV-1 will impose a surviving pressure and gradually select the CXCR4 and CCR5-deleted cell progeny. 54 For primary CD4+ T cells, deletion of CXCR4 and CCR5 via electroporation of the Cas9RNP complex confers cell resistance to both tropic HIV-1 challenges, even though this protection is not complete due to the insufficient gene knockout efficacy. Considering the in vivo setting, this strategy is especially intended for curing HIV-1 infected patients in whom the pre-existing HIV-1 viruses will offer a viral pressure and select the CXCR4 and CCR5 modified CD4+ T cells. 11 Finally, the continuous renewal of engineered CD4+ T cells may help the immune system achieve a balance of controlling viral dissemination and maintaining the immune responses resisting opportunistic infections.
Footnotes
Acknowledgments
We thank Prof. Feng Zhang (Broad Institute of MIT and Harvard) for kindly providing pLenti-CRISPRv2 plasmid. We thank Prof. Hui Zhang (Sun Yat-sen Medical School, Sun Yat-sen University) for generously supplying the HIV-1NL4-3 and HIV-1YU 2 viruses and P3-level biosafety facility. The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: GHOST (3) CXCR4+ CCR5+ cells from Drs. Vineet N. KewalRamani and Dan R. Littman. This work was funded by the National Natural Science Foundation of China (no. 81372451, no. 81673003), and Guangzhou Municipal Funds of Science and Technology (no. 201504010016). This work was also supported in part by the National High Technology Research and Development Program of China (no. 2014AA020544) and the Ministry of Sciences and Technology Key Program (no. 2016YFE0107300).
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.