
Abstract
In advanced and metastatic stages of colorectal cancer (CRC), reduced sensitivity to conventional strategies is still a major obstacle to successful treatments. Decorin is an important regulator in the development and progression of various cancers. To examine if CRC patients have altered decorin levels, expression of decorin and its target genes, Met and vascular endothelial growth factor A (VEGFA), were analyzed in their tumors. Compared to normal tissues, decorin expression was reduced in CRC patients' tumors, while there were increased Met and VEGFA levels. To develop a novel therapy for CRC, rAd.DCN.GM, an oncolytic adenovirus encoding decorin and granulocyte macrophage colony stimulating factor (GM-CSF), has been created. Several therapeutic strategies expressing GM-CSF have been employed in clinical trials for treating metastatic colorectal cancer. In this study, infection of CRC cells with rAd.DCN.GM expressed decorin and GM-CSF, and produced cytotoxicity. In murine CT26 xenografts, rAd.DCN.GM and control adenoviruses were administrated intratumorally on days 7 and 10, and tumor volumes were monitored over time. The study showed that rAd.DCN.GM inhibited the tumor growth and lung metastases significantly. rAd.DCN.GM induced apoptosis, inhibited proliferation, and downregulated angiogenesis and epithelial mesenchymal transition markers in the tumors. On day 12 and day 29, the immune-activation in the peripheral blood, tumors, and spleens were analyzed. rAd.DCN.GM increased CD8+ T lymphocytes in the blood, upregulated perforin and granzyme B in the tumors, inhibited transforming growth factor beta expression, and promoted dendritic-cell production in the spleen. In conclusion, rAd.DCN.GM inhibited the tumor growth and metastasis of CT26 tumors, downregulated multiple pro-tumorigenic pathways, and activated antitumor immune responses.
Introduction
C
In addition to conventional strategies, novel technologies, such as nanotechnology, molecular-targeted strategies, and immune-activation approaches, have been developed for the diagnosis and therapy of colorectal cancer. 5 –8 In recent years, oncolytic viruses have also attracted attention for their application as antitumor agents. Oncolytic viruses can not only kill cancer cells through viral replication, but also evoke antitumor immune responses via releasing tumor antigens following the viral-induced tumor cell lysis. 9 Several oncolytic virus–based immunogene therapies have been approved for clinical trials. 10 –12 Among them, an oncolytic herpes simplex virus (HSV) type 1 expressing granulocyte macrophage colony-stimulating factor (GM-CSF; talimogene laherparepvec) has been approved by the U.S. Food and Drug Administration. 10,13,14 Oncolytic adenoviruses are popular vectors for cancer therapy. The safety and efficacy of these viruses have been validated in several clinical studies. The authors' laboratories have been focusing on oncolytic adenovirus-based therapy, including immunogene therapy, and have developed a simplified system for generating oncolytic adenovirus vector carrying one or two transgenes. 15 –18
Decorin, a prototypic member of the small leucine-rich proteoglycans (SLRPs), is an important regulator for various cellular functions, including migration, proliferation, apoptosis, and differentiation. 19,20 Importantly, decorin can block the signaling network of transforming growth factor beta (TGF-β), which functions as a well-known immune suppresser in advanced and metastatic cancers. 21 Decorin is downregulated in tumor lesions of CRC patients, and its restoration suppresses the malignant phenotypes in vitro. 22,23 Previously, Ad.DCN, an oncolytic adenovirus expressing decorin protein, has been developed, which could inhibit the bone metastasis of prostate cancer and breast cancer in mice. 17,18 Moreover, it has been shown that decorin can inhibit the tumor growth and invasion by promoting mitochondrial autophagy and downregulating several metastasis related genes, including c-Met, CTNNB1, and vascular endothelial growth factor (VEGF). 18,20,24 However, the mechanisms of Ad.DCN-mediated antitumor responses in vivo are largely unexplored.
Decorin could effectively block the TGF-β signaling and abolish TGF-β-mediated immune tolerance in the tumor microenvironments. 17,18,25 Data from preclinical experiments and clinical trials has suggested that GM-CSF could evoke an effective antitumor immune response via recruiting natural killer (NK) cells, inducing maturation and differentiation of dendritic cells (DCs), and enhancing activities of macrophage cells. 26,27 Because of these reasons, GM-CSF has been tested in several clinical trials for targeting metastatic colorectal cancers. 28 –30 Therefore, oncolytic adenovirus combining decorin and GM-CSF could be a promising approach to activate an antitumor immune response. Keeping that in mind, a telomerase reverse transcriptase promoter (TERTp) regulated oncolytic adenovirus expressing decorin and GM-CSF, rAd.DCN.GM, has been developed using an established system. 15 In an immunocompetent murine CRC xenograft model, it has been shown that rAd.DCN.GM inhibited the local tumor growth and prevented metastasis to the lungs. As shown previously for Ad.DCN, 17,18 rAd.DCN.GM could also inhibit the expression of decorin target genes. Moreover, rAd.DCN.GM combines the advantages of both decorin and GM-CSF in evoking antitumor immune responses in vivo.
Materials and Methods
Tumor tissues from colorectal cancer patients
Surgical specimens were obtained from 59 CRC patients for clinical pathological examination in Beijing Friendship Hospital (Capital Medical University) from 2010 to 2013. According to the results of pathological examination, the surgical specimens were divided into tumor tissues, para-cancerous tissues, and distal normal tissues. All the procedures were approved by the Ethics Committee of Beijing Friendship Hospital, Capital Medical University. The decorin protein and mRNA expression were detected by immunohistochemistry and real-time reverse transcription polymerase chain reaction (RT-PCR), respectively.
Cell lines
Human colon-cancer cell lines, SW480 and SW620, were purchased from cell bank, Shanghai Institutes for Biological Science, Chinese Academy of Sciences (Shanghai, China). Mouse colon-cancer cell line, CT26, and human embryonic kidney cell line, HEK293, were obtained from American Type Culture Collection (ATCC, Manassas, VA). SW480, SW620, and CT26 cells were maintained in RPMI-1640 media (Gibco, Grand Island, NY) containing 10% fetal calf serum (FCS; Hyclone, Logan, UT). HEK293 cells were cultured in Dulbecco's minimal essential medium (DMEM; Gibco) supplemented with 10% FCS.
Adenoviruses
The oncolytic adenoviruses were constructed by using a simplified system for generating oncolytic adenovirus vector carrying one or two transgenes, as described previously. 15 rAd.DCN.GM is an oncolytic adenovirus, in which the expression of DCN and GM-CSF is regulated by the cytomegalovirus (CMV) promoter and E1B promoter, respectively. The replication of adenoviruses is controlled by TERTp, located upstream of E1A. Oncolytic adenoviruses rAd.DCN and rAd.GM, which expressed DCN and GM-CSF, respectively, were also constructed. rAd.Null, an oncolytic adenovirus without any transgene, was used as control vector in this study. Non-replicating adenovirus Ad (E1-).DCN and Ad (E1-).Null were constructed by the Ad-easy system, as described, 18 and used for biological studies in vitro.
Adenoviral-mediated replication and cytotoxicity in CRC cells
CRC cells, SW480, SW620, and CT26, were seeded into six-well plates (3 × 105 cells per well). The next day, cells were infected with 2.5 × 104 viral particles (vp)/cell of adenoviruses (rAd.DCN.GM, rAd.DCN, rAd.GM, rAd.Null, Ad[E1-].DCN or Ad[E1-].Null). The viral titers of 3 or 48 h crude viral lysates were determined according to Adeno-X Rapid titer kit protocol (Clontech, Mountain view, CA), as previously described. 31 Viral burst size, an indicator of viral replication, was calculated as published. 32
For cytotoxicity assay, colon-cancer cells were plated into 96-well plates (1 × 103 cells per well). The next day, cells were infected with various doses of adenoviruses, as described previously, and the incubation continued for 7 days. The cell survival was determined by the sulforhodamine B staining using uninfected cells as control. 33
Adenoviral-mediated decorin and GM-CSF expression in the colon-cancer cells
CRC cells, SW480, SW620 and CT26, were plated into six-well plates (3 × 105 per well). The next day, cells were exposed to adenoviruses (2.5 × 104 vp/cell). Twenty-four hours post infection, cells were cultured in fresh serum-free media for another 24 h, and the media was collected. The decorin protein in supernatant was detected by enzyme-linked immunosorbent assay (ELISA) using mouse antihuman decorin antibody and botinylated mouse antihuman decorin antibody (R&D Systems, Minneapolis, MB) using previously reported methods. 18 The expression of GM-CSF was detected by using a human GM-CSF ELISA kit (MultiScience Biotech Co. Ltd., Hangzhou, China).
Ad(E1-).DCN-mediated biological activities on human colon-cancer cells
To investigate the effects of Ad(E1-).DCN on human colon-cancer cells, the apoptosis, proliferation (Dye eFluorR 670; eBioscience, San Diego, CA), and migration assays were performed in SW480 cells following infection with replication-deficient adenovirus Ad(E1-).DCN (10,000 vp/cell). 17,18 Moreover, decorin-regulated genes, VEGFA, Met, and CTNNB1, were examined in Ad(E1-).DCN- and Ad(E1-).Null- infected cells (10,000 vp/cell) at 24 and 48 h post infection using RT-PCR.
Decorin and GM-CSF co-expression activated peripheral blood mononuclear cells from colon-cancer patients
Human colon-cancer cells were infected with 25,000 vp/cell of the oncolytic adenoviruses. Cells and media were collected together at 48 h post infection. After three cycles of freezing and thawing, cell lysates were centrifuged, and the conditioned culture media was collected.
Human peripheral blood samples were obtained from CRC patients undergoing diagnostic procedures at Beijing Friendship Hospital. Written informed consent was obtained from each CRC patient. All the procedures were approved by the Ethics Committee of Beijing Friendship Hospital and Beijing Institute of Radiation Medicine. Peripheral blood mononuclear cells (PMBCs) were isolated from heparinized samples by Ficoll-Paque (Amersham Biosciences, Piscataway, NJ) density gradient centrifugation. PBMCs were seeded into 24-well plates at a density of 4 × 105/well/800 μL of complete media, and then 400 μL of conditioned culture media was added to each well, and the incubation continued for 3 days. Total RNA was extracted, and cDNA was synthesized using RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, Wilmington, DE). The mRNA expression of various cytokines and chemokines, interleukin (IL)-2, IL-12, TGF-β, IL-6, interferon gamma (IFN-γ), perforin, and granzyme B, were detected using RT-PCR.
Animal studies
All procedures of animal experiments were approved by the Institutional Animal Care and Use Committee of the Beijing Institute of Radiation Medicine.
CT26 xenograft model and treatment with oncolytic adenoviruses
To establish the colon-cancer xenograft model, 2 × 106 CT26 cells (100 μL) were injected subcutaneously into BALB/c mice (4–6 weeks old). Seven days after the cell injection, when the tumors were visible, tumor volumes were measured by using the following formula: tumor volume = width 2 × length/2. Tumor-bearing mice were divided into five groups without statistical difference (n = 20/group). rAd.DCN.GM, rAd.DCN, rAd.GM, rAd.Null (2.5 × 1010 vp/injection), or 100 μL of phosphate-buffered saline (PBS) was administrated intratumorally. Three days later (day 10), another injection was administered.
Tumor volumes were monitored twice a week. Five mice from each group were euthanized on day 12, and the remaining mice were euthanized on day 29. Samples of the tumor, spleen, liver, lung, and peripheral blood were collected for histopathology, immune activation, and gene expression analyses.
Histopathological analysis and immunohistochemistry
On days 12 and 29, tumor lesions, liver, spleen, and lung were harvested, processed, and stained with hematoxylin and eosin. The apoptosis in tumor lesions was examined by terminal deoxynucleotidyl transferased UTP nick end labeling (TUNEL; Promega, Madison, WI) according to the manufacturer's instructions. The proliferation of tumor cells was detected by proliferating cell nuclear antigen (PCNA) staining using rabbit polyclonal anti-PCNA antibody (Abcam, Cambridge, MA).
Immunophenotype analysis of peripheral blood cells and splenocytes
On days 12 and 29, peripheral blood samples were collected, and the subtypes of T lymphocytes were analyzed by flow cytometry. Briefly, heparin-anticoagulated peripheral blood was stained with APC-conjugated rat anti-mouse CD3e antibody (APC-CD3e), PE-conjugated rat anti-mouse CD4 antibody (PE-CD4), and FITC-conjugated rat anti-mouse CD8 antibody (FITC-CD8a; eBioscience). After incubation for 30 min at room temperature, the erythrocytes were lysed by red blood cell (RBC) lysis buffer (BD Biosciences, San Jose, CA; 1 × ). Then, the immune-phenotypes were analyzed by flow cytometry.
On days 12 and 29, the spleens were isolated, sliced into small pieces, and pressed through a 70 μm cell strainer (BD Falcon, Franklin Lakes, NJ). Single-cell suspension was collected and lysed by RBC lysis buffer. The immune-phenotypes of T lymphocytes were detected as described above. Moreover, single-cell suspensions were also stained with FITC rat anti-mouse CD11c antibody (FITC-CD11c) and PE rat anti-mouse CD86 antibody (PE-CD86; eBioscience). The percentage and number of DCs were analyzed and calculated by flow cytometry.
Gene expression analysis of GM-CSF, decorin, decorin target genes, cytokines, and chemokines in the tumor lesions
On days 12 and 29, the mice were euthanized, and the tumors were removed. The expression of human GM-CSF, human decorin, decorin-regulated genes, epithelial mesenchymal transition (EMT) markers (E-cadherin, N-cadherin, and Vimentin), cytokines, and chemokines in the tumors were analyzed using RT-PCR. Cytokines and chemokines examined were mouse IL-2, IL-4, IL-6, IL-10, IL-12, tumor necrosis factor alpha (TNF-α), TGF-β, IFN-γ, perforin, and granzyme B.
RT-PCR
The mRNA expression of various genes were quantified by using SYBR® Premix Ex Taq™ (Tli RNaseH Plus; Takara, Shiga, Japan) on a 7500 Fast Real-Time PCR System (Applied Biosystems/Life Technologies, Foster City, CA). The relative expression levels were calculated by 2–ΔΔCT using β-actin as the control.
Statistical analysis
Data are presented as mean ± standard error of the mean and were statistically analyzed using GraphPad Prism v5 (GraphPad Software, San Diego, CA). Longitudinal data were analyzed using a two-way repeated measure analysis of variance (ANOVA) followed by Bonferroni post hoc tests for all the data over the time course. Paired t-tests were used to analyze decorin, Met, and VEGFA expression in the tumor tissues of CRC patients, and unpaired t-tests were used to analyze other data with two groups. One-way ANOVA followed by Bonferroni post hoc tests were performed to analyze multiple groups. Differences were considered significant at two-sided p < 0.05.
Results
Reduced decorin expression in the colorectal cancer patients
Lower levels of decorin expression in the tumor microenvironment are generally poor prognostic marker and are associated with aggressive tumors. 34 –36 In this study, tumor samples and corresponding distal normal tissues were collected from 59 CRC patients, and decorin expression was analyzed. As shown in Fig. 1A, compared to distal normal tissues, decorin protein was downregulated in the tumor lesions. Reduced mRNA expression of decorin were observed in 25/31 and 26/28 cases from colon- and rectal-cancer patients, respectively (Fig. 1B).

Decorin expression in tumor lesions of colorectal cancer (CRC) patients. Surgical specimens were obtained from 59 CRC patients for clinical pathological examination.
It has been suggested that decorin inhibits the angiogenic network through suppression of Met and VEGFA.
37,38
Therefore, this study investigated Met and VEGFA expression in the samples of 51 patients who had reduced decorin expression. The data showed that Met and VEGF expressions were upregulated significantly in most of the cases. Compared to the rectal-cancer patients, the upregulation in colon-cancer patients was much more noticeable (Fig. 1C–E). However, in rectal-cancer patients, lower decorin levels and upregulation of Met and VEGF indicated a higher risk of metastasis and recurrence (Supplementary Fig. S1; Supplementary Data are available online at
rAd.DCN.GM replication, viral-induced cytotoxicity, and decorin and GM-CSF production in the colon-cancer cell lines
It has been previously shown that decorin could block TGF-β signaling, which plays pivotal role in tumor growth and metastasis. 17,18,20 It was hypothesized that oncolytic adenovirus expressing decorin could offer a potential therapeutic approach for CRC, as suggested earlier for breast and prostate cancers. 17,18 To enhance the antitumor responses further, GM-CSF and decorin were co-expressed in the recombinant oncolytic adenovirus, as described in the Materials and Methods. rAd.DCN.GM is an oncolytic adenovirus that contains both decorin and GM-CSF. The two genes are regulated by CMV and E1B promoter, respectively. rAd.DCN, an oncolytic adenovirus containing only decorin gene; rAd.GM, an oncolytic adenovirus containing only GM-CSF gene; and rAd.Null, an oncolytic adenovirus without any transgene (Fig. 2A) were used as the controls in these studies. These oncolytic adenoviruses could infect both tumor and normal tissues, but would not spread among normal tissues.
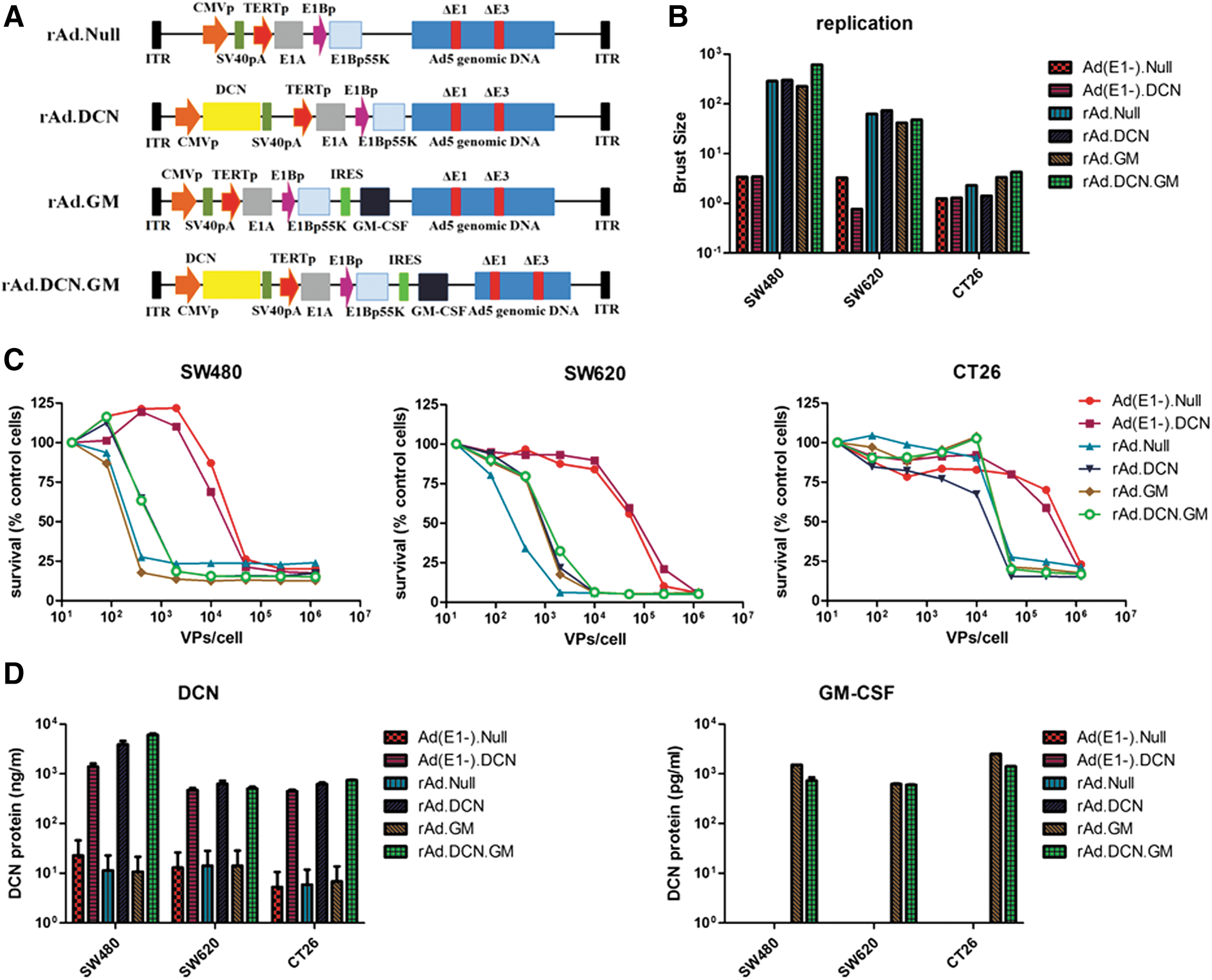
Construction of oncolytic adenovirus rAd.DCN.GM, viral replication, viral-induced cytotoxicity, and viral-mediated production of decorin and granulocyte macrophage colony stimulating factor (GM-CSF) in colon-cancer cell lines.
In human CRC cells, the viral titers increased significantly from 3 to 48 h post infection with oncolytic adenoviruses (rAd.DCN.GM, rAd.DCN, rAd.GM, and rAd.Null), indicating effective viral replication. However, no significant replication was detected in mouse CT26 cells (Fig. 2B). Interestingly, oncolytic adenoviruses killed colon-cancer cells, including the mouse CT26 cells, and the cell killing was viral dose dependent (Fig. 2C). These results suggest that besides viral replication, other unknown mechanisms might be involved in the oncolytic adenoviral-mediated cytotoxicity in the mouse cells. Next, it was shown that Ad(E1-).DCN-, rAd.DCN-, and rAd.DCN.GM-infected cells expressed and secreted decorin protein into the extracellular media (Fig. 2D). rAd.GM and rAd.DCN.GM also produced GM-CSF protein in CRC cells (Fig. 2D).
In SW480 cells, replication-deficient adenovirus, Ad(E1-).DCN, mediated decorin expression inhibited migration, downregulated target genes, Met, CTNNB1 (catenin beta 1), and VEGF, and reduced TGF-β expression (Supplementary Fig. S2), suggesting that decorin overexpression is a potential viable approach to inhibit tumor cell migration and invasion.
rAd.DCN.GM inhibits tumor growth and lung metastasis in CT26 xenograft model
Murine colon-cancer CT26 xenografts were established subcutaneously in BALB/c mice. The oncolytic adenoviruses, rAd.DCN.GM, rAd.DCN, rAd.GM, or rAd.Null, were injected intratumorally, as described in the Material and Methods. Compared to the buffer group, all the oncolytic adenoviruses inhibited tumor growth significantly. Moreover, rAd.GM.DCN, rAd.DCN, and rAd.GM produced much stronger inhibitory effects than rAd.Null, indicating that decorin and GM-CSF alone or in combination could enhance oncolytic adenoviral-mediated antitumor responses (Fig. 3A and B).
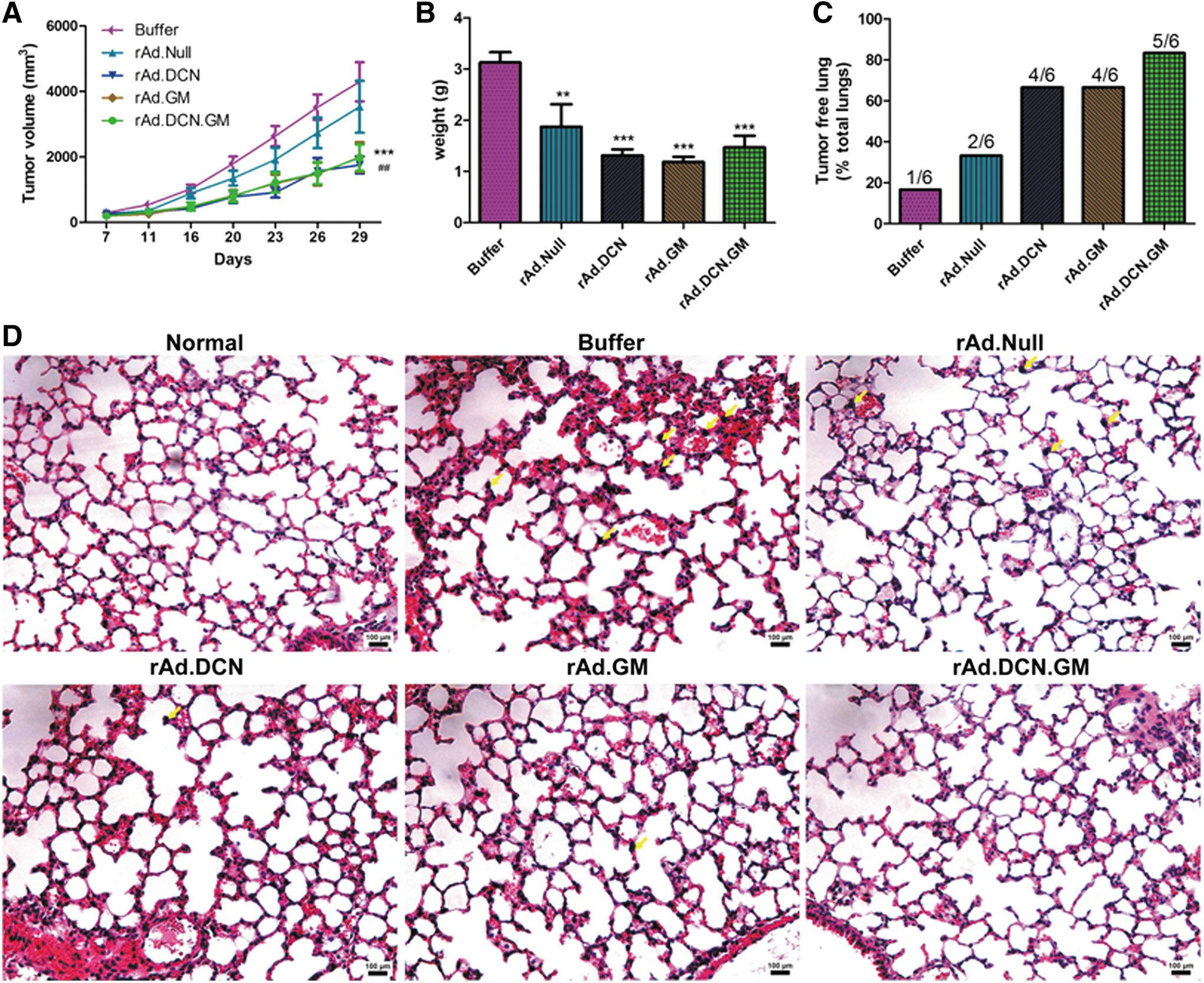
rAd.DCN.GM-mediated antitumor responses in a murine colon-cancer cell, CT26, xenograft model. 2 × 106 CT26 cells (100 μL) were injected subcutaneously into BALB/c mice (4–6 weeks old). On day 7, tumor-bearing mice were divided into five groups (n = 20/group) without statistical differences in tumor volumes (width
2
× length/2). rAd.DCN.GM, rAd.DCN, rAd.GM, rAd.Null (2.5 × 1010 vp/injection of each virus) or 100 μL of phosphate-buffered saline was administrated intratumorally. A repeat injection (2.5 × 1010 vp) was administered 3 days later (day 10). Tumor volumes were monitored twice a week and are presented in
In addition to the local tumor growth, tumor metastasis was also investigated in this study. The data showed that tumor metastatic lesions could be detected in the lungs of CT26 bearing mice, but not in the liver or spleen (Fig. 3C and D and Supplementary Fig. S3A and B). On day 29, the lungs of six mice randomly collected from each group were analyzed. Lung metastasis was detected in five mice from the buffer group (5/6), and reduced metastasis could be found in rAd.DCN- (2/6), rAd.GM- (2/6), and rAd.DCN.GM-treated (1/6) groups (Fig. 3C and D). These results suggest that combining oncolytic adenovirus with decorin and GM-CSF not only reduced local tumor burden, but also prevented tumor metastasis to the lungs.
rAd.DCN.GM induces cell death and inhibits angiogenesis and EMT in tumor lesions
Next, viral-induced cell apoptosis/death of the tumor cells within the tumors was examined by TUNEL. The data showed that all the oncolytic adenoviruses induced cell apoptosis/death in the tumor lesions. However, rAd.DCN.GM, rAd.GM, and rAd.DCN treatments were all more effective than Ad.Null in inducing cell apoptosis/death, indicating that decorin- and GM-CSF-enhanced oncolytic adenoviruses induced cell apoptosis/death (Fig. 4A). Moreover, PCNA staining suggested that the proliferation of tumor cells was inhibited by the oncolytic adenoviruses. However, no obvious difference could be detected among various oncolytic adenoviral treatments (Fig. 4A).
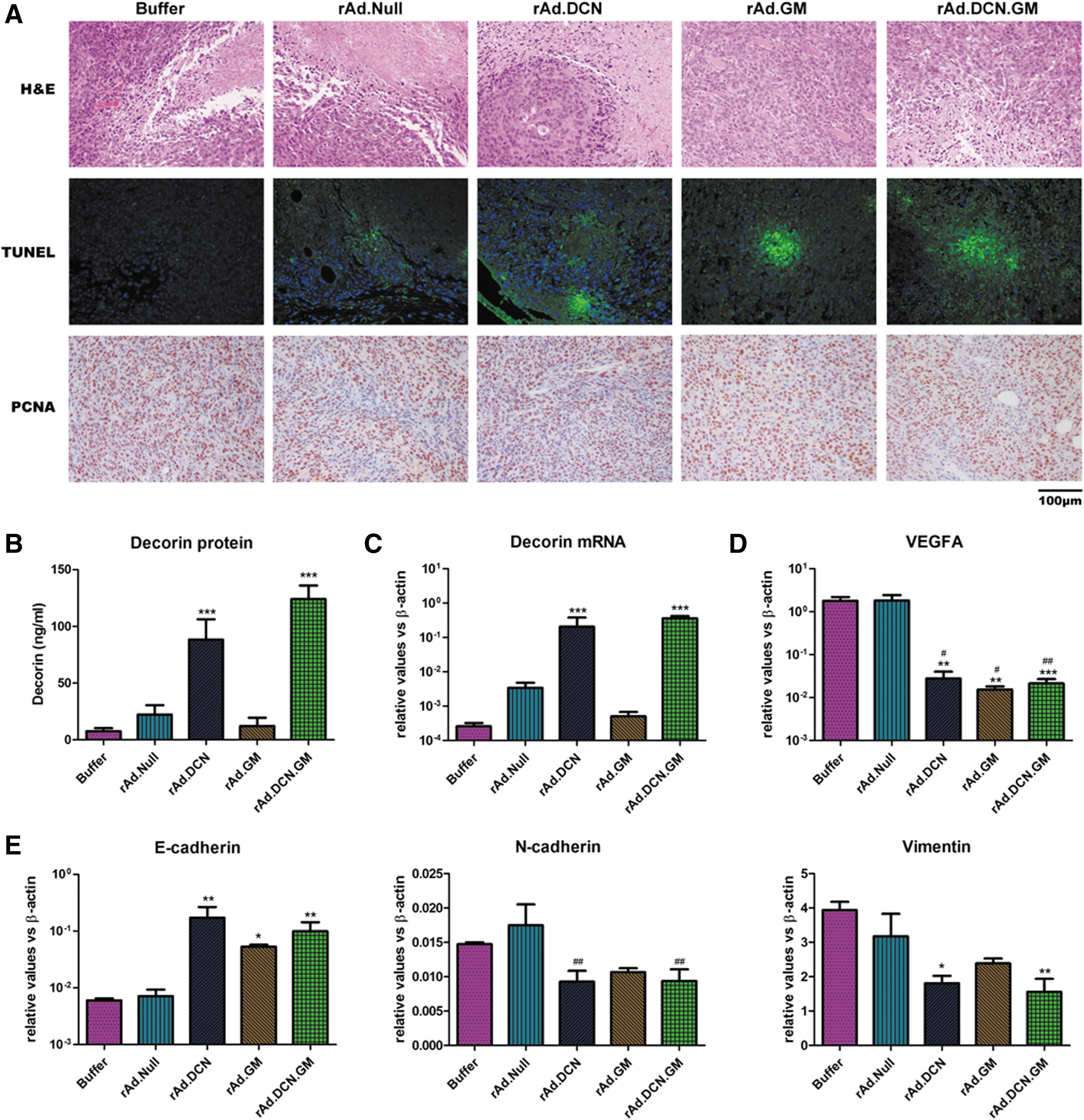
rAd.DCN.GM induced apoptosis/death of tumor cells in vivo, and produced decorin protein to inhibit angiogenesis and epithelial mesenchymal transition (EMT) in the tumor lesions. Two days after the last injection of oncolytic adenovirus (day 12), five mice from each group were euthanized. The tumors were removed and processed, as described in the Materials and Methods. Oncolytic adenovirus-induced apoptosis was evaluated by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), and the proliferation of tumor cells was examined using proliferating cell nuclear antigen (PCNA) staining
Both rAd.DCN.GM and rAd.DCN treatments produced high levels of decorin in the tumor lesions. Decorin was also subsequently secreted into the sera (Fig. 4B and C). VEGFA, a well-known target gene of decorin, was downregulated in the tumor tissues of rAd.DCN.GM- and rAd.DCN-treated groups. Interestingly, rAd.GM also inhibited VEGFA expression (Fig. 4D), suggesting that GM-CSF could inhibit the expression of VEGFA through other mechanisms. Moreover, rAd.DCN.GM and rAd.DCN treatments reduced the Vimentin and N-cadherin expression significantly, while E-cadherin expression was elevated. rAd.GM treatment has much weaker effects on these genes (Fig. 4E), suggesting decorin protein could play an important role in mediating EMT of the tumor cells in vivo. Thus, rAd.DCN.GM can inhibit tumor metastasis possibly via regulating angiogenesis as well as EMT in the tumor lesions.
rAd.DCN.GM produces antitumor immune responses in vivo
rAd.DCN.GM rapidly increases CD8+ T cells both in peripheral blood and the spleen
Several studies conducted in animal models and in clinical trials have demonstrated that oncolytic viruses expressing GM-CSF can evoke strong antitumor immune responses. 10,11,27 It has been shown that inoculation of CT26 tumors with rAd.DCN.GM produces GM-CSF expression in the tumors (Fig. 5A). It was also found that perforin and granzyme B expression increased following oncolytic adenoviruses treatment. Moreover, gene-armed oncolytic viruses (rAd.DCN.GM, rAd.DCN, and rAd.GM) induced stronger effects on perforin and granzyme B expression (Fig. 5B and C). Similar results were found in patient-derived PBMCs after stimulation with oncolytic adenoviruses infected cell lysates (Supplementary Fig. S4A and B).

rAd.DCN.GM-mediated GM-CSF expression in the tumors, and activation of CD8+ T lymphocytes in peripheral blood. On day 12, five mice from each group were euthanized. The tumors were removed, and total RNA was isolated. GM-CSF
Next, the immuno-phenotypes of T lymphocytes were analyzed in peripheral blood. Oncolytic adenoviruses increased the percentage of CD8+ T cells in peripheral blood, on both days 12 and 29, which could potentially activate both viral- and tumor-specific CD8+ T cells. Importantly, rAd.DCN.GM treatment resulted in the strongest increase in the CD8+ T lymphocytes on day 12 (p < 0.05 vs. buffer group), which was consistent with the increased expression of perforin and granzyme B in the tumors (Fig. 5D). Interestingly, compared to other oncolytic adenoviruses, rAd.DCN.GM treatment slightly downregulated the CD8+ T lymphocytes and upregulated CD4+ T lymphocytes on day 29 (Supplementary Fig. S5). This might be due to the initial activation of CD4+ T lymphocytes, which are then converted to CD4+ T memory cells for long-term immune surveillance. The results suggest that GM-CSF expression could enhance the immune responses of oncolytic adenoviruses through activation of both CD8+ and CD4+ T lymphocytes.
rAd.DCN.GM downregulates Th2 cytokines and increases DCs in the spleen
It was found that the expression of IFN-γ in the spleen was upregulated after treatment with oncolytic adenoviruses on day 12, perhaps due to the activation of antiviral immune responses. Compared to rAd.Null, gene-modified oncolytic adenoviruses (rAd.DCN.GM, rAd.DCN, and rAd.GM) could slightly downregulate IFN-γ expression (Fig. 6A). It has also been reported previously that TGF-β is a strong immune suppressor. This study therefore analyzed TGF-β expression in the spleen. TGF-β levels were reduced in the rAd.DCN- and rAd.DCN.GM-treated groups. The strongest inhibition was observed in the rAd.DCN.GM group. Two important target genes of TGF-β—IL-6 and TNF-α—were also downregulated in the rAd.DCN.GM-treated group (Fig. 6B and D). Importantly, rAd.DCN.GM also regulated these cytokines in patient-derived PBMCs, which is consistent with the in vivo data (Supplementary Fig. S4C and D).
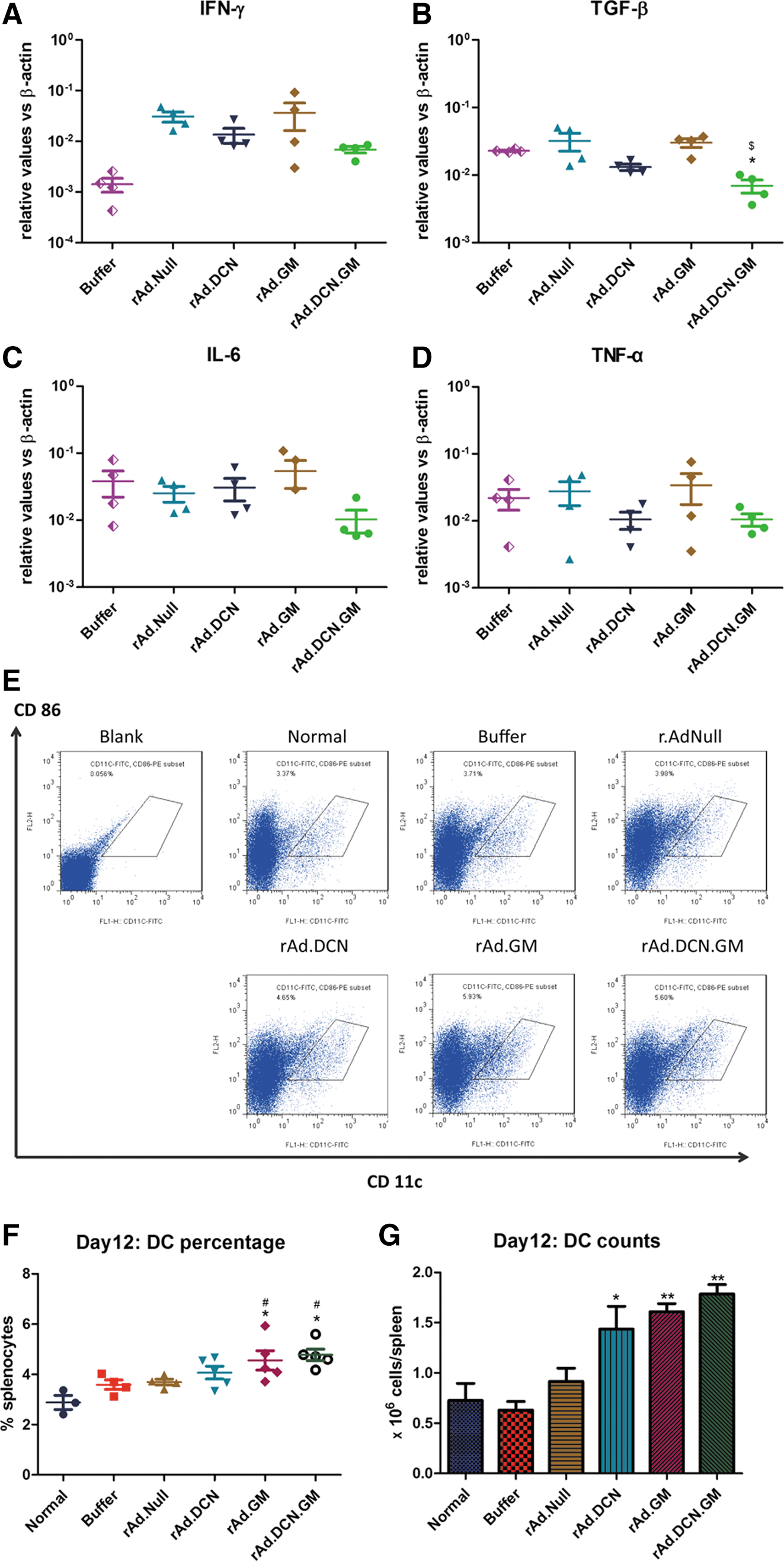
rAd.DCN.GM produces GM-CSF and decorin, inhibits the expression of transforming growth factor beta (TGF-β), and increases dendritic cells (DCs) in the spleen. On day 12, five mice from each group were euthanized. The spleens were isolated, and a single-cell suspension was obtained, as described above. After lyses by red blood cell lysis buffer, cells were counted.
DCs are important immune cells involved in mediating antitumor activities, and several DC-based tumor vaccines have shown strong clinical responses. 39 Since GM-CSF plays an important role in inducing the maturation and differentiation of DCs, this study examined DC production in the spleen. rAd.DCN.GM, rAd.DCN, and rAd.GM treatment resulted in an increased percentage and numbers of DCs in the spleen, while a control adenovirus Ad.Null did not produce such an effect. However, the upregulation of DCs in rAd.GM- and rAd.DCN.GM-treated groups was quite significant, and superior to that observed by Ad.DCN (Fig. 6E–G). These results suggest that oncolytic adenoviral-mediated GM-CSF expression could promote the amplification of DCs in the spleen. It is possible that rAd.DCN.GM could promote the amplification of DCs, not only by GM-CSF-mediated proliferation and maturation of DCs, but also via decorin-mediated inhibition of TGF-β signaling.
Discussion
Immunotherapy has emerged as one of the promising approaches for cancer therapy in the past decades. 40 –43 Although great breakthroughs have been achieved in immunotherapy, there are still several obstacles to overcome, such as the induction of a cytokine storm and local immune suppression. 44 Recently, oncolytic virus-mediated immunotherapy has provided an alternative approach for cancer immunotherapy. 10,12,14,44 Oncolytic viruses modulate antitumor immune responses by releasing tumor specific antigens and promoting immune cell infiltration in tumor lesions. 45,46 Even though oncolytic serotype 5 adenoviruses have several shortcomings, including the high prevalence of neutralizing antibodies, hepatotoxicity, and low expression of its receptor in tumor cells, they are suitable vectors for immunotherapy due to their safety and high immunogenicity. 47 This study developed an oncolytic adenovirus, rAd.DCN.GM, encoding decorin and GM-CSF. In an immune-competent murine CRC model, it was shown that rAd.DCN.GM could activate the antitumor immune responses significantly and inhibit spontaneous metastasis.
Downregulation of decorin, a well-known tumor suppressor, has been reported in several cancers, including prostate cancer, breast cancer, and CRC. 34 –36 Consistent with previous reports, the present study showed that decorin was significantly downregulated in tumor lesions of CRC patients. Moreover, lower decorin expression was detected in patients with metastatic lesions. Previous reports have shown that decorin can target multiple tyrosine kinase receptors, such as Met, EGFR, and IGFR, known to support tumor growth. 25,48 The present study showed that Met was upregulated in most of the decorin downregulated CRC patients, and decorin restoration reduced Met expression in human CRC cells. VEGFA, a well-known pro-angiogenic factor, is one of the most important target genes of decorin. 17,18,20 In the present study, VEGFA upregulation was frequently detected in CRC patients, and oncolytic adenoviral-mediated decorin expression reduced VEGFA expression in CT26 xenograft. Besides VEGFA, decorin can also downregulate pro-angiogenic proteases, such as matrix metalloprotease (MMP)-9 and MMP-2, and inhibit tumor angiogenesis. 37,38 It has been demonstrated that decorin inhibits migration of tumor cells via stability of E-cadherin protein, an important marker of EMT. 49,50 This study has also shown that decorin overexpression significantly inhibited the migration of CRC cells in vitro and prevented lung metastasis in vivo. In the CT26 xenograft model, rAd.DCN and rAd.DCN.GM potentially inhibited EMT by upregulating E-cadherin and downregulating N-cadherin and Vimentin. Therefore, oncolytic adenoviruses armed with decorin could inhibit tumor growth and metastasis via regulating multiple decorin target genes.
Previous studies have demonstrated that decorin could effectively block TGF-β signaling. 17,18,20 In advanced cancers, TGF-β promotes tumor growth, invasion, and metastases via enhancing proliferation of tumor cells, promoting angiogenesis and EMT, and inducing immunosuppression in the tumor microenvironment. 51 –53 Many studies have demonstrated that TGF-β plays a pivotal role in regulating immune tolerance in the tumor microenvironment, mainly through suppressing the maturation of T helper cells, DCs, and NK cells, inhibiting cytotoxicity of CD8+ T cells, and inducing M2 polarization of macrophage cells. Moreover, TGF-β could regulate cytokines expression and drive Th1/Th2 balance toward Th2 immune phenotype. 54,55 Therefore, blocking aberrant TGF-β signaling is one of the potential strategies for the treatment of advanced and metastatic cancers. Several TGF-β inhibitors have been developed and investigated in animal tumor models. 16,32,56,57 In the CT26 xenograft model, it was shown that rAd.DCN and rAd.DCN.GM treatments reduced the expression of TGF-β and its target genes, IL-6 and TNF-α, both in the tumors and in the spleens. To improve the antitumor response, decorin was expressed along with GM-CSF in oncolytic adenovirus rAd.DCN.GM. GM-CSF has been widely used to arm oncolytic viruses, and virally expressed GM-CSF can recruit NK cells, induce maturation and differentiation of DCs, and promote the differentiation and enhance the activities of macrophage cells. 26,27 However, clinical studies have shown that GM-CSF alone induced only limited CD8+ T-cell mediated immunity against the tumors. This study demonstrated that a combination of decorin with GM-CSF in rAd.DCN.GM not only enhanced decorin-mediated downregulation of TGF-β, but also enhanced the GM-CSF-induced antitumor response, possibly by increasing CD8+ T lymphocyte population in the peripheral blood and DCs in the spleen.
In conclusion, a novel oncolytic adenovirus rAd.DCN.GM encoding decorin and GM-CSF has been developed. rAd.DCN.GM expressed decorin and GM-CSF proteins in the CRC cells, and inhibited the migration of CRC cells. In an immune-competent murine colon-cancer xenograft model, rAd.DCN.GM produced significant inhibition of tumor growth and distant metastasis. This is associated with viral-induced oncolysis, and simultaneous production of decorin and GM-CSF. Both decorin and GM-CSF proteins are released into the systemic circulation and thus produce local antitumor as well as distant anti-metastasis effects. The biological effects of decorin and GM-CSF include the inhibition of tyrosine kinase growth factor receptors, inhibitions of EMT, tumor cell migration and angiogenesis, downregulation of TGF-β, blockade of the immune suppression, and the immune activation including the promotion of maturation and differentiation of DCs.
Taken together, the in vitro and in vivo studies presented here suggest that in addition to rAd.DCN and rAd.GM oncolytic adenoviruses, rAdDCN.GM can also be developed as a potential treatment for CRCs. As shown in this study, many CRC patients have reduced levels of decorin protein in the tumors, particularly in metastases and during tumor recurrence. Therefore, the potential of rAdDCN.GM to be developed as a novel gene therapy drug for treating CRC patients can be envisioned.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No.81402558&81472396) and the National High Technology Research and Development Program of China (863 Program; SS2014AA020515). Studies at the North Shore Research Institute were funded by an anonymous donor and a Clinical and Translational Science Award (P.S.).
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.