
Abstract
Primary immunodeficiencies, including Wiskott-Aldrich syndrome (WAS), are a main target for genome-editing strategies using specific nucleases (SNs) because a small number of corrected hematopoietic stem cells could cure patients. In this work, we have designed various WAS gene-specific CRISPR/Cas9 systems and compared their efficiency and specificity with homodimeric and heterodimeric WAS-specific zinc finger nucleases (ZFNs), using K-562 cells as a cellular model and plasmid nucleofection or integration-deficient lentiviral vectors (IDLVs) for delivery. The various CRISPR/Cas9 and ZFN SNs showed similar efficiency when using plasmid nucleofection for delivery. However, dual IDLVs expressing ZFNs were more efficient than dual IDLVs expressing Cas9 and guide RNA or all-in-one IDLVs, expressing Cas9 and guide RNA in the same vector. The specificity of heterodimeric ZFNs and CRISPR/Cas9, measured by increments in γ-H2AX focus formation in WAS-edited cells, was similar for both, and both outperformed homodimeric ZFNs independently of the delivery system used. Interestingly, we show that delivery of SNs, using IDLVs, is more efficient and less genotoxic than plasmid nucleofection. We also show the similar behavior of heterodimeric ZFNs and CRISPR/Cas9 for homology-directed gene knock-in strategies, with 88 and 83% of the donors inserted in the WAS locus, respectively, whereas when using homodimeric ZFNs only 45% of the insertions were on target. In summary, our data indicate that CRISPR/Cas9 and heterodimeric ZFNs are both good alternatives to further develop SN-based gene therapy strategies for WAS. However, IDLV delivery of WAS-specific heterodimeric ZFNs was the best option of all systems compared in this study.
Introduction
W
Advances in targeted genome-editing (GE) tools have brought gene therapy the opportunity to specifically modify the affected locus with no or minimal alterations in the target cells. 12 The revolution started with the possibility to generate nucleases that specifically cut in the selected locus in the human genome. 13 –16 These specific nucleases (SNs) recognize specific nucleotide sequences, generating double-strand breaks (DSBs) that the cell repairs by homologous recombination (HR) or by nonhomologous end-joining (NHEJ). Depending on the cell's mechanism used to repair the DSBs and the materials delivered into the cell, we can repair, insert, or delete DNA fragments in the targeted genome. The specificity of SNs can rely on protein–DNA recognition, such as zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), or on RNA–DNA recognition clustered regularly interspaced short palindromic repeats (CRISPR/Cas system).
The CRISPR/Cas system derives from bacterial adaptive immune systems, 17,18 and it is one of the most powerful GE technologies at the moment. 19 The guide RNA (gRNA) hybridizes with an ∼20-nucleotide sequence next to a protospacer-adjacent motif (PAM) and enables the Cas9 endonuclease to cleave the target site, creating DSBs. 16 Various Cas9 nucleases derived from various bacterial species have been used to develop efficient GE tools. 19 Each CRISPR/Cas9 system differs in terms of gRNA design and PAM requirements in order to achieve specific DSBs, and all of them have been shown to efficiently edit the genome; nevertheless, Cas9 from Streptococcus pyogenes (SpCas9) has been the most widely used. 20 However, CRISPR/Cas9 systems generate DSBs outside of the target sequences (off-target sites) that can differ by several nucleotides from on-target sites and can generate different types of mutations. 21,22
ZFNs are chimeric proteins composed of an endonuclease (the bacterial FokI restriction enzyme) fused with several zinc finger domains that bind specifically to the target sequence. These zinc fingers consist of 30 amino acids in a ββα configuration and recognize 3 or 4 base pairs of DNA. Because the cleavage domain must dimerize, the system requires two different ZFNs (left and right) binding to opposite strands of DNA at a certain distance (5–7 bp). 23 However, it has been reported that the ZFNs can also form homodimers (ZFNleft–ZFNleft and ZFNright–ZFNright), generating DSBs in genome locations different from the intended target (off-target sites). 24,25 To increase ZFN specificity, Miller and colleagues developed ZFNs harboring a mutated FokI enzyme that is active only when forming heterodimers. 13 The specificity of ZFNs can also be increased by decreasing the intracellular concentration of ZFNs. 26
Efficiency and safety are the two main aspects of any therapeutic strategy. Successful GE therapeutic approaches must be able to deliver the SNs (and the donor DNA for gene correction) into a large number of target cells in order to have a clinical benefit. In addition, the delivery system must be innocuous to the target cells. This is a relatively easy task for immortalized cell lines, but primary human cells are more difficult and require special delivery systems. HSCs, the final target for WAS gene therapy, are particularly resistant to the various delivery technologies. Nevertheless, various SNs as well as donor DNA can be efficiently delivered to HSCs by nucleofection, 27,28 adeno-associated viruses (AAVs), 29,30 and integration-deficient lentiviral vectors (IDLVs). 31,32 IDLVs appeared initially to be promising delivery tools for HSC GE because of their low toxicity and high efficiency. However, ZFNs have been delivered efficiently to HSCs, using IDLV systems, 31,33 –35 whereas the delivery of other SNs such as TALENs and CRISPRs has been more difficult. 36
The efficiency of SNs can be easily measured by analyzing the percentage of cells harboring insertions and deletions (indels) or incorporating the desired modifications in the targeted sequence. However, safety is more complicated to study, due to the difficulty in detecting off-target cleavage of the SNs. Most studies determine specificity by in silico predictions of potential off-target sites followed by experimental analysis with CEL (celery) nuclease (commercialized under the brand name Surveyor) or deep sequencing. However, these biased systems cannot detect unpredicted sites 37 as demonstrated by several studies that detect off-target cleavage sites in live cells without any kind of previous restriction. 21,22,38 –40= However, unbiased off-target analysis (GUIDE-seq [genome-wide, unbiased identification of DSBs enabled by sequencing], IDLVs, deep sequencing, etc.) is generally costly and technically challenging. An alternative way to measure safety is to quantify the amount of DSBs generated by the SNs. One of the best markers by which to quantify DSBs is H2AX phosphorylation. H2AX is a variant of H2A that becomes the center of the cellular response when DNA damage occurs and is phosphorylated, leading to γ-H2AX focus formation. 41 γ-H2AX staining has been used to quantify the DNA damage produced by specific site endonucleases, such as ZFNs. 13,42 This quantification can be used to analyze the specificity of the various SNs, as the number of foci can be correlated with off-target cleavage.
In this paper we compare the efficiency and safety of various WAS-specific SNs (ZFNs_wt, ZFNs_mt, and CRISPR/Cas9_gRNA9) targeting the same locus and using two different delivery methods (plasmid nucleofection and IDLVs). We use the human leukemia cell line K-562 as target, which is considered to be an adequate model for studying GE because the results can be translated to HSCs, 27,32 the target for WAS GE therapeutic strategies. We study the efficiency and safety of gene disruption, using plasmid nucleofection or IDLVs to deliver the various SNs. We also use nucleofection of the various SNs together with donor DNA to study the efficiency and safety of GE of the WAS locus by HR. Our data indicate that ZFNs_mt and CRISPR SNs achieve similar efficiencies and specificities by nucleofection, but delivery of ZFNs_mt with IDLVs renders higher efficiencies than with CRISPR/Cas9 in K-562 cells.
Materials and Methods
Plasmids
Plasmids encoding original homodimeric ZFNs (ZFNs_wt) targeting intron 1 of the WAS gene (pVAX-N2A-3FN-15724fok and pVAX-N2A-3FN-15755fok) were designed by Sangamo BioSciences (Richmond, CA) as previously described. 43 Plasmids encoding optimized ZFNs (ZFNs_mt) (pVAX-N2A-3FN-15724fokEL and pVAX-N2A-3FN-15755fokKK) were described previously 44 and have a mutated version of the FokI nuclease (EL and KK) that increases the affinity for heterodimerization. 13,45 The ZFN cDNAs were included in the SEWP plasmid 46 (a self-inactivated LV expressing enhanced green fluorescent protein [eGFP] under the control of the spleen focus-forming virus [SFFV] promoter) by standard molecular techniques, replacing eGFP for the ZFNs to generate pSZFN24, pSZFN55, pSZFN24EL, and pSZFN55KK.
The Cas9 nuclease cDNA was obtained through gene synthesis (plasmid pUC-coCas9nls2x; GenScript, Piscataway, NJ) of a human codon-optimized cDNA version of the Streptococcus pyogenes M1 Cas9 sequence (ref AE004092.2; from nt 854751 to nt 858857) further modified to eliminate the restriction sites for EcoRI, ClaI, BamHI, NotI, MluI, BmgBI, AscI, AarI, AsiI, PstI, SbfI, XhoI, SciI, KpnI, and XbaI and to introduce a BstXI site at the +43 position and two BlpI sites at the +2500 and +2710 positions. In addition, a sequence containing a hemagglutinin (HA) tag and two nuclear localization signals (NLSs) from the simian virus 40 T antigen (SV40T) was included at the 3′ end. Finally, the coCas9-HA-nls2x sequence was flanked by a BamHI–AscI site at the 5′ end and a PstI–AarI site at the 3′ end to facilitate cloning into a lentiviral plasmid backbone.
Guide RNAs (gRNAs) were designed by searching for protospacer adjacent motif (PAM) sequences near the cutting site of the WAS-specific ZFNs. Basic Local Alignment Search Tool (BLAST) searches of the various gRNAs came up with only one gRNA (gRNA9: 5′-GAGGCAGGAAGGACCAGGTC-3′) with the desired characteristics (targeting the same region as the ZFNs and having no homology with other regions in the genome) (Supplementary Fig. S1; Supplementary Data are available online at
SELg9 and SELg9WP plasmids were obtained as follows: a XhoI–XbaI fragment of the SEWP plasmid containing the 3′ long terminal repeat (LTR) was subcloned into a pUC plasmid to obtain pUC-LTR. The gRNA9scaffold was introduced into the 3′ LTR BbsI site of pUC by standard molecular techniques to obtain pUC-LTR-gRNA9. Finally, a XhoI–XbaI fragment of pUC-LTR-gRNA9 containing LTR-gRNA9 was used to replace the XhoI–XbaI fragment of the SE and SEWP lentiviral vectors to obtain the SELg9 and SELg9WP plasmids.
To obtain SCas9Lg9 and SCas9Lg9WP, the eGFP cDNA from SELg9 and SELg9WP were removed by BamHI and PstI and replaced with a BamHI–PstI fragment from the pUC-coCas9nls2x plasmid containing the codon-optimized Cas9 cDNA.
To obtain the SCas9Xg9WP and the SCas9Eg9WP plasmids, gRNA9 was amplified by PCR with primers containing the XhoI or EcoRI sites (see Supplementary Table S1) and inserted into the XhoI and EcoRI sites of SCas9WP, respectively.
Cell lines and culture media
293T cells (CRL-11268; American Type Culture Collection, Manassas, VA) were grown in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Edinburgh, Scotland) with GlutaMAX and supplemented with 10% fetal bovine serum (FBS) (PAA Laboratories, Pasching, Austria). The K-562 cell line (chronic myelogenous leukemia) was purchased from the ATCC (CCL-243) and cultured in RPMI 1640 (Gibco-BRL, Middlesex, UK) supplemented with 10% FBS (PAA Laboratories).
Vector production
Lentiviral vectors were produced by cotransfection of 293T cells with three plasmids: (1) a vector plasmid (SEWP, SCas9WP, SELg9W, SCas9Lg9, SCas9Lg9W, SCas9Xg9W, SCas9Eg9W, SZFN24W, SZFN55W, SZFN24ELW, or SZFN55KKW), (2) the human immunodeficiency virus (HIV) packaging plasmid pCMvdR8.91 for integration-competent LVs or pCMvdR8.91D64V for IDLVs, and (3) the envelope plasmid pMD2.G. 47 Vector production was performed as previously described. 48 Briefly, 293T cells were plated on amine-coated petri dishes (Sarstedt, Newton, NC), in order to ensure 80–90% confluence for transfection. The three plasmids were resuspended in LipoD293 (SignaGen, Gaithersburg, MD) as described previously. 49 The plasmid–LipoD293 mixture was added to prewashed cells and incubated for 6–8 hr, when new medium was added. After 48 hr viral supernatants were collected, filtered through a 0.45-μm (pore size) filter (Nalgene, Rochester, NY), and concentrated either by ultrafiltration at 2000 × g at 4°C, using 100KD centrifugal filter devices (Amicon Ultra-15; Millipore, Billerica, MA) or by ultracentrifugation (23,000 rpm, 2 hr, at 4°C; Beckman Coulter, Brea, CA). Concentrated vectors were either directly used or aliquot and frozen at −80°C.
Viral titers of eGFP-expressing LVs and IDLVs were determined by transduction of 1 × 105 K-562 cells per well in 48-well tissue culture plates (BD Biosciences, San Jose, CA), using a 4-fold serial dilution of the supernatants and then assaying the percentage of eGFP-positive cells by fluorescence-activated cell-sorting (FACS) analysis 5 days (LVs) or 48 hr (IDLVs) after transduction. LV and IDLV particle numbers per milliliter were determined with a lentivirus titration kit (qPCR lentivirus titration kit; Applied Biological Materials [ABM], Richmond, BC, Canada) according to the manufacturer's protocol. Briefly, samples were diluted 1:100 and 1:1000 in 100 μl of phosphate-buffered saline (PBS) before lysis. Viral lysis was performed with 2 μl of diluted viral supernatant added to 18 μl of the virus lysis buffer provided. The qRT-PCR program was performed with an Applied Biosystems 7300 real-time PCR system (Thermo Fisher Scientific, Waltham, MA), using the following qRT-PCR parameters: 1 cycle of reverse transcription (42°C for 20 min), 1 cycle of enzymatic activation (95°C for 10 min), 40 cycles of denaturation (95°C for 15 sec), and 40 cycles of annealing/extension (60°C for 1 min).
Cell transduction
293T and K-562 cells were washed with Dulbecco's PBS (1 × ) (PAA Laboratories) and dissociated with Invitrogen TrypLE Express (Thermo Fisher Scientific) diluted one-third in PBS (293T cells) or directly centrifuged at 1250 rpm for 5 min and counted. Cells (1 × 105) were plated in 24- or 48-well plates (BD Biosciences) and incubated for 5 hr with the various LVs or IDLVs. Serial dilutions were made to determine titers. Medium was changed after 5 hr.
Cell nucleofection
For gene disruption, K-562 cells (1 × 106) were nucleofected with the various pairs of ZFN-expressing plasmids (3 μg each; SZFN24W/pSZFN55W [ZFNs_wt] or SZFN24ELW/SZFN55KKW [ZFNs_mt]), with two plasmids expressing Cas9 and gRNA9 (SCas9WP/SELg9W), with 6 μg of the all-in-one CRISPR plasmid SCas9Lg9W, or with 6 μg of control (irrelevant) plasmid (SδEWP, expressing mutated eGFP). For the homology directed (HD) gene insertion assay, the donor plasmid (12–18 μg) was also included together with the selected SN plasmids. Nucleofection was performed with an Amaxa Nucleofector II and solution V (Lonza, Basel, Switzerland), applying program T16 and following the nucleofection protocol for K-562 cells.
Flow cytometry
Controls and transduced K-562 cells (1–4 × 105) were collected and washed with cold FACS buffer (PBS containing 2% FBS and 0.5% bovine serum albumin [BSA]). 293T cells were dissociated with TrypLE Express, washed with PBS containing 2% FBS, and centrifuged at 1250 rpm for 5 min and washed with cold FACS buffer. Cells were stained with 7-aminoactinomycin D (7-AAD) viability dye (eBioscience, San Diego, CA) and viable cells (7-AAD-negative cells) were gated for the subsequent analysis. Finally, cells were analyzed for GFP expression, using a FACSCanto II flow cytometer (Becton Dickinson, Franklin Lakes, NJ) equipped with FACSDiva analysis software (BD Biosciences).
Quantification of cleavage efficiency of WAS target site
Genomic DNA extraction from K-562 cells was performed with a QIAamp DNA mini kit (Qiagen, Chatsworth, CA), in accordance with the protocol for cells in suspension. The genomic regions flanking the CRISPR and ZFNs target site for each treatment were amplified by PCR using hWASP5Fw/hWASP5Rev primers (primers are listed in Supplementary Table S1). The 2-kb fragments were purified with a QIAquick spin column (Qiagen), in accordance with the manufacturer's protocols. Total purified PCR product (400 ng) was mixed with 2 μl of 10 × Taq DNA polymerase PCR buffer and ultrapure water to a final volume of 20 μl, and subjected to a reannealing process to enable heteroduplex formation. After reannealing, products were treated with Surveyor nuclease (Integrated DNA Technologies, San Diego, CA), in accordance with the manufacturer's recommended protocols, and analyzed on 2% agarose gels. Two fragments of 1.2 and 0.8 kb will appear if the SNs have generated a DSB at the target site. The percentage of cleavage was determined by densitometry of the 2.0-, 1.2-, and 0.8-kb bands, using the following formula:
DSB analysis: analysis of H2AX phosphorylation
K-562 cells were nucleofected or transduced with the various SNs and, 48 hr later, 2.5 × 105 cells were collected and washed with cold FACS buffer (PBS containing 2% FBS and 0.5% BSA). Cells were then fixed (80% methanol for 5 min), permeabilized (0.1% PBS–Tween for 20 min), and incubated with PBS–10% goat serum for 10 min to block nonspecific protein–protein interaction, followed by incubation with anti-histone γ-H2AX (phosphorylated at Ser-139) antibody (diluted 1:100; Abcam, Cambridge, UK) for 30 min at room temperature. Subsequent to washing with PBS, cells were incubated with goat anti-rabbit IgG H + L Alexa Fluor 647 antibody (diluted 1:10,000; Abcam) for 30 min at room temperature. Isotype control antibody was rabbit IgG monoclonal (Abcam) (0.1 μg), also incubated for 30 min at room temperature. After washing with PBS, cells were analyzed for γ-H2AX expression, using the FACSCanto II flow cytometer (Becton Dickinson) equipped with FACSDiva analysis software (BD Biosciences).
Measurement of γ-H2AX by image cytometry
K-562 cells were nucleofected or transduced with the various SNs as described above. Formation of γ-H2AX in response to DSBs was analyzed by image cytometry, using a FlowCellect histone H2A.X phosphorylation assay kit (Merck Millipore) according to the manufacturer's instructions. γ-H2AX expression was measured with an ImageStream X Mark II imaging flow cytometer (Merck Millipore) and analyzed with IDEAS software, using the Spot Wizard. To determine the number of γ-H2AX foci in each cell, the Peak Mask was applied, which gives the number of foci per nucleus according to an internal algorithm. The number of individual masks in a cell was enumerated with the Spot Count feature and plotted for every cell in histograms.
Off-target detection
Possible off-target sites for the CRISPR/Cas9_gRNA system were identified through BLAST 51 and Bowtie 52 algorithms, using the entire sequence of gRNA9 (GAGGCAGGAAGGACCAGGTCTGG) or just the first 16 bp including the PAM sequence (GAAGGACCAGGTCTGG). For ZFNs we used the genome-wide tag scanner for nuclease off-sites, 53 using the binding sites of both ZFNs as target. We selected the eight best potential off-targets for each nuclease (see Supplementary Tables S2 and S3) and analyzed indel generation by Surveyor as described previously and using the primer pairs described in Supplementary Tables S2 and S3.
Efficiency and fidelity of HD donor insertion into the WAS locus
K-562 cells were nucleofected with plasmids expressing the various SNs (pSZFN24/pSZFN55, pSZFN24EL/pSZFN55KK, or pSCas9WP/SELg9) and donor DNA expressing the eGFP and neomycin resistance (NeoR) genes. Cells transfected with the nucleases and donor DNA were selected with neomycin for 20 days, and GFP-positive cells were sorted with a FACSAria II (BD Biosciences) and seeded into a 96-well plate to obtain single cells (1 cell/5 wells). Clones were grown in the presence of neomycin, and genomic DNA was extracted from more than 50 clones from each condition. The DNAs were used as templates for PCRs, using (1) a primer pair (hWASP5Fw/hWASP5Rev) flanking the WAS target sequence, outside the donor homology domains; (2) two primer pairs that amplify the endogenous WAS and donor DNA at the 5′ junction (hWASP5Fw/eGFPRev) and 3′ junction (eGFPFw/hWASP5Rev); and (3) a primer pair (SFFV1aFw/NeoRRev) that amplifies an internal fragment from the donor DNA (see Supplementary Fig. S2 and Supplementary Table S1). All the PCRs were carried out with Kappa Taq polymerase (Kapa Biosystems, Wilmington, MA).
Statistical analysis
All data are expressed as means ± SEM. Statistical comparisons were performed by Student t test, with the assumption of normal distribution. Statistical significance was defined as p < 0.05.
Results
Construction of WAS-specific ZFNs and CRISPR/Cas9 systems
WAS-specific homodimeric ZFNs (ZFNs_wt) and heterodimeric ZFNs (ZFNs_mt) were designed by Sangamo as described in Materials and Methods. A WAS-specific CRISPR/Cas9 system was designed and home-made, based on previously published systems. 16,54 The Cas9 nuclease cDNA was obtained through gene synthesis (GenScript) of a codon-optimized cDNA version of the Streptococcus pyogenes M1 Cas9 sequence incorporating an HA tag and two NLSs from SV40T at the 5′ end (see Materials and Methods for details). We searched for WAS-specific guide RNAs (gRNAs) targeting the same sequence as the ZFNs and without potential off-target sites (see Materials and Methods for details and Supplementary Fig. S1). Only one gRNA (gRNA9) fulfilled these requirements and was synthesized by GenScript together with the scaffold and the U6 promoter.
The efficiency and safety of specific nucleases can vary depending on expression levels, which are highly influenced by the delivery system used. We have therefore constructed various plasmids (Fig. 1; see Materials and Methods for details) that can be used to deliver the SNs by plasmid nucleofection and by IDLVs. Each ZFN (ZFN24, ZFN55, ZFN24EL, and ZFN55KK) is expressed by a plasmid harboring the SFFV promoter and all the elements required to be packed into lentiviral particles. The same plasmids are used to express Cas9 and gRNA9 separately. K-562 cells were nucleofected with the plasmids or transduced with IDLV particles, and the various samples were analyzed for the expression of ZFNs, Cas9, and gRNA 2 days later to verify expression of the various SNs and gRNA (data not shown).
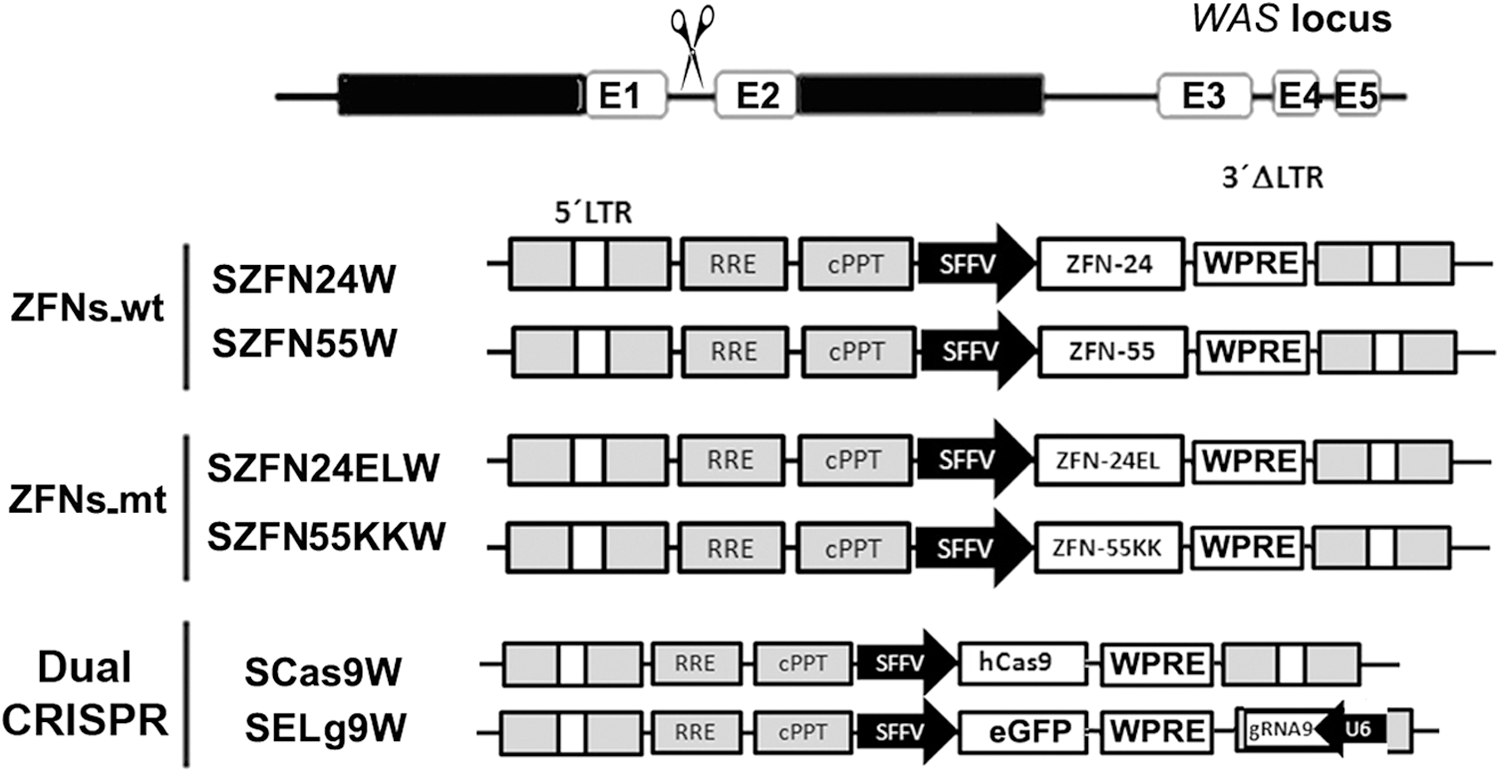
Plasmid constructs used to express the various WAS-specific nucleases. Top: A drawing of the WAS target site. Each zinc finger nuclease (ZFN) pair (ZFNs_wt and ZFNs_mt) is expressed by two lentiviral vector plasmids expressing each ZFN (24 and 55 or 24EL and 55KK) through the spleen focus-forming virus (SFFV) promoter and containing the posttranscriptional regulatory element of woodchuck hepatitis virus (WPRE). For comparison, the CRISPR system (Cas9/gRNA, bottom) is also expressed by two different plasmids: one expressing human codon-optimized Cas9 cDNA, using the same backbone as the ZFNs (SCas9W), and the other expressing gRNA9 through the U6 promoter and eGFP through the SFFV promoter (SELg9W).
Delivery of SNs by nucleofection showed similar efficiency and specificity of ZFNs and CRISPR/Cas9
We first compared the cutting efficiency and specificity of both ZFNs versus CRISPR/Cas9 systems, using nucleofection as the delivery method. Plasmids expressing a ZFNs_wt pair (SZFN24W, SZFN55W), a ZFNs_mt pair (SZFN24ELW and SZFN55KKW), and dual CRISPR (Scas9W and SELg9W) were nucleofected into K-562 cells, using the Amaxa Nucleofector, and analyzed for specific (Surveyor) and unspecific (γ-H2AX-staining) DSB generation (Fig. 2). Although highly variable, we observed similar WAS gene-targeting efficiencies of all systems (Fig. 2A, right). Nonspecific DSB generation by the various SNs was analyzed first by measuring the increments in γ-H2AX staining by flow cytometry (FACS) and using K-562 cells nucleofected with irrelevant plasmid as controls (Mock) (Fig. 2A). The CRISPR/Cas9_gRNA9 and ZFNs_mt systems showed similar γ-H2AX staining compared with K-562 cells nucleofected with irrelevant plasmid, indicative of the low off-target activity of these SNs (Fig. 2A). However, at similar cutting efficiencies, ZFNs_wt presented two to four times higher percentages of γ-H2AX-positive cells compared with Mock (Fig. 2A).
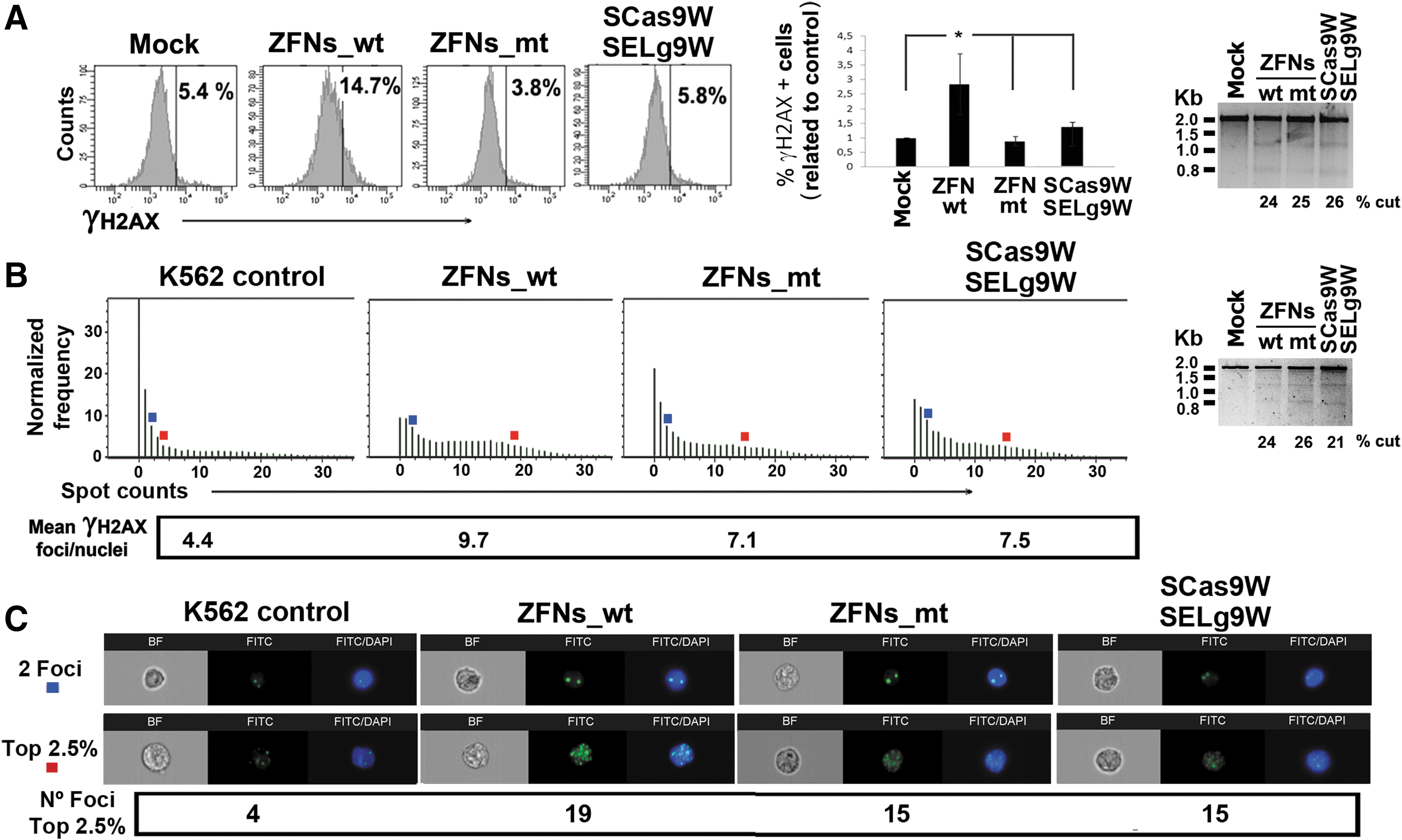
Comparison of WAS-specific zinc finger nucleases and CRISPR systems delivered by nucleofection.
We next investigated whether γ-H2AX staining on FACS correlates with γ-H2AX focus formation (indicative of DSB repair) and whether plasmid nucleofection could also affect DSB generation on K-562 cells. K-562 cells were therefore nucleofected with ZFNs_wt, ZFNs_mt, CRISPR, irrelevant plasmids, or without plasmid DNA. Forty-eight hours later the various samples were analyzed for γ-H2AX focus formation, using the ImageStream and IDEAS analysis software (Fig. 2B and C) (see Materials and Methods for details). This system allowed us to verify that γ-H2AX staining indeed identified γ-H2AX spots (see Fig. 2B and C, blue and red squares). The IDEA software was used to quantify the amount of spots per nucleus, plotted in histograms, and the mean γ-H2AX foci per nucleus calculated in each sample (Fig. 2B, left). The results showed that, as observed by FACS, K-562 cells nucleofected with ZFNs_mt, CRISPR, and irrelevant plasmids had similar DSB levels when measured as mean γ-H2AX foci per nucleus (Fig. 2B: 7.1 and 7.5; and data not shown), whereas ZFNs_wt rendered the highest DSB levels (9.7). We also observed the same number of foci in populations that have a normalized frequency of 2.5% (top 2.5%) in K-562 cells nucleofected with the ZFNs_mt or CRISPR plasmids (Fig. 2B and C, red squares; 15 foci per nucleus), whereas K-562 cells nucleofected with the ZFNs_wt plasmid showed up to 19 foci per nucleus. Of note, K-562 cells nucleofected with no DNA showed lower DSB levels than K-562 cells nucleofected with ZFNs_mt, CRISPR, or irrelevant plasmid (Fig. 2B and C, K-562 control vs. ZFNs_mt and CRISPR; and data not shown).
All together, these data showed that nucleofection with plasmids expressing ZFNs_mt and CRISPR had similar low off-target activity that was below the limit of detection by γ-H2AX staining, in part due to the genotoxicity of plasmid nucleofection. However, nucleofection with plasmids expressing ZFNs_wt increased DSB generation compared with nucleofection with irrelevant plasmid and must therefore be due to the off-target activity of ZFNs_wt.
We also analyzed the specificity of the various nucleases by in silico analysis of potential off-target sites. For CRISPR/Cas9_gRNA9, we used BLAST 51 and Bowtie 52 algorithms to search for potential gRNA9 off-target sites. For ZFNs we used the genome-wide tag scanner for nuclease off-sites. 53 We selected the eight highest scoring off-targets for each nuclease (see Supplementary Tables S2 and S3) and analyzed indel generation by Surveyor (≈2% sensitivity). However, no detectable off-target activity was observed for any SNs, using this technology (data not shown), probably due to the low sensitivity of this technique.
IDLVs-ZFNs are more efficient that IDLVs-CRISPR but have similar specificity
Delivery of SNs by IDLVs is an interesting alternative for SN delivery because of their wide tropism, their ability to incorporate various elements (SNs, donor, miRNA), and the potential to be used in vivo. We therefore generated IDLVs expressing each of the CRISPR components (Cas9 and gRNA9) and each of the ZFNs_mt, using the plasmids showed in Fig. 1. K-562 cells were transduced with equivalent particle numbers per cell (PNC, 400) of each IDLV pair (SCas9W/SELg9W or SZFN24ELW/SZFN55KKW) and the cutting efficiencies and mRNA expression levels were determined (Fig. 3). Our data showed higher cutting efficacies of IDLVs-ZFNs_mt (35.6%) compared with the dual IDLVs-CRISPR system (25.8%) (Fig. 3A); this could be due, at least in part, to the lower mRNA expression levels of the latter (Fig. 3B). Indeed, our analysis showed that IDLVs-CRISPR expressed 3–10 times lower mRNA levels compared with IDLVs-ZFNs_mt, probably due to a longer messenger (Cas9, 4.2 kb vs. ZFNs, 1.1 kb).
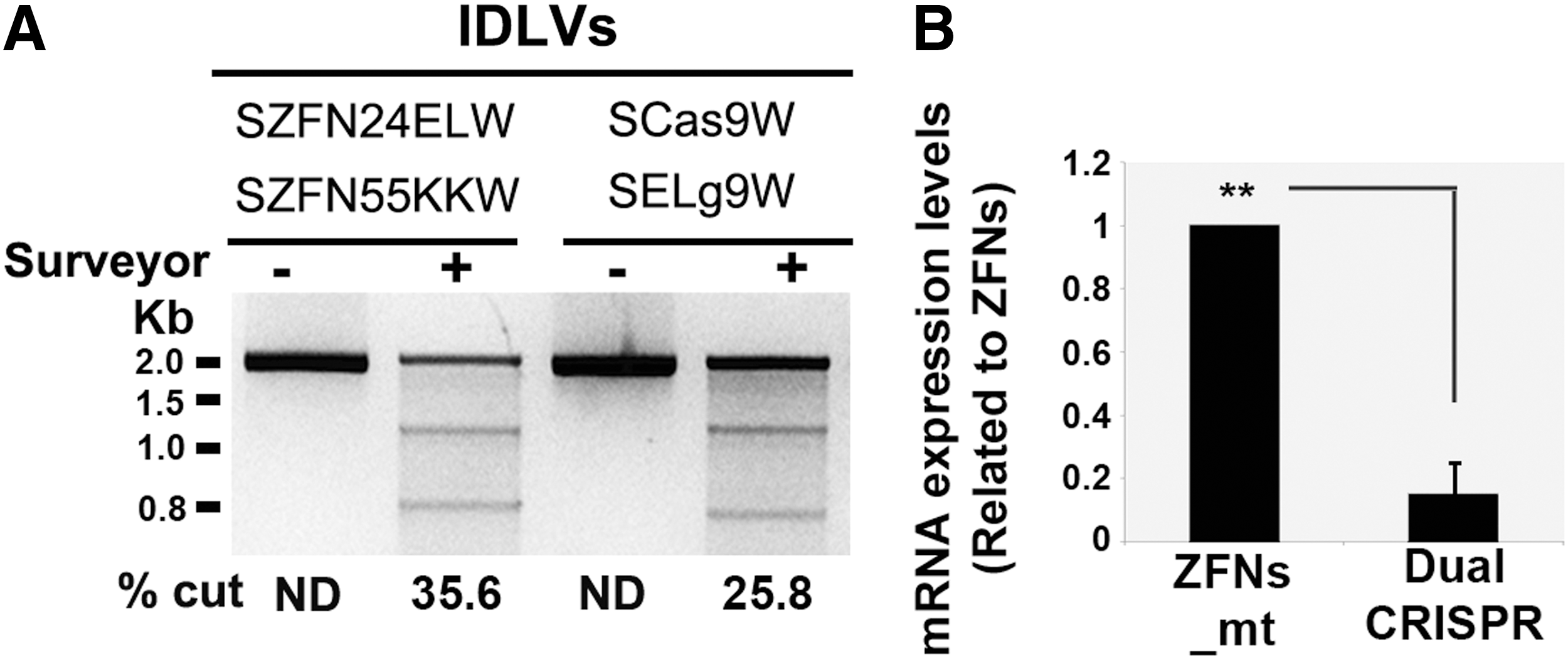
Comparative analysis of the efficiency of IDLVs ZFNs_mt and dual CRISPR systems to edit the WAS locus.
To increase the efficiency of the CRISPR system, we constructed various all-in-one IDLVs to express both Cas9 and gRNA9 (Fig. 4A, top) and analyzed their efficiency in K-562 cells, using similar volumes (Fig. 4B, bottom) or PNC (Supplementary Fig. S3B). Optimized IDLVs achieved cutting efficacies ranging from 21.2% to 35.5% (PNC, 250–350), showing that these all-in-one IDLVs-CRISPR can be used for WAS GE. However, although the CRISPR all-in-one IDLVs outperformed the dual IDLVs, their efficacy was still two to four times lower compared with the dual IDLVs-ZFNs (Fig. 4B and Supplementary Figs. S3B and S4). Interestingly, WAS gene disruption using any of the IDLV systems did not cause any harm to the treated cells (Supplementary Fig. S5). Because the SCas9Lg9W IDLVs showed the best efficiencies we used this construct for further studies.

Development of all-in-one IDLVs expressing Cas9 and gRNA9.
We next generated SCas9Lg9W IDLV particles as well as IDLVs expressing each ZFN and compared their efficiency and specificity to disrupt the WAS locus. It is important to note that the specificity of the various SNs must be compared in target cells edited at similar levels because higher cutting efficiencies at the WAS locus will result in higher γ-H2AX staining and could also render higher off-target activity. Therefore, K-562 cells were cotransduced with IDLV-ZFN24W/IDLV-ZFN55W (IDLVs-ZFNs_wt), IDLV-ZFN24ELW/IDLV-ZFN55KKW (IDLVs-ZFNs_mt), or all-in-one SCas9Lg9W IDLVs at various PNCs in order to reach similar cutting efficiencies. Forty-eight hours later we analyzed DSBs by γ-H2AX and indel generation at the target site (WAS locus) by Surveyor (Fig. 5A, right and Supplementary Fig. S6). We required two to four times higher PNCs for IDLVs-CRISPR compared with IDLVs-ZFNs in order to reach similar efficiencies. Interestingly, as observed with nucleofection, FACS analysis showed similar levels of γ-H2AX-positive cells in CRISPR- and ZFNs_mt-treated K-562 cells, whereas cells treated with ZFNs_wt showed higher γ-H2AX staining, indicating higher off-target activity (Fig. 5A and Supplementary Fig. S6).
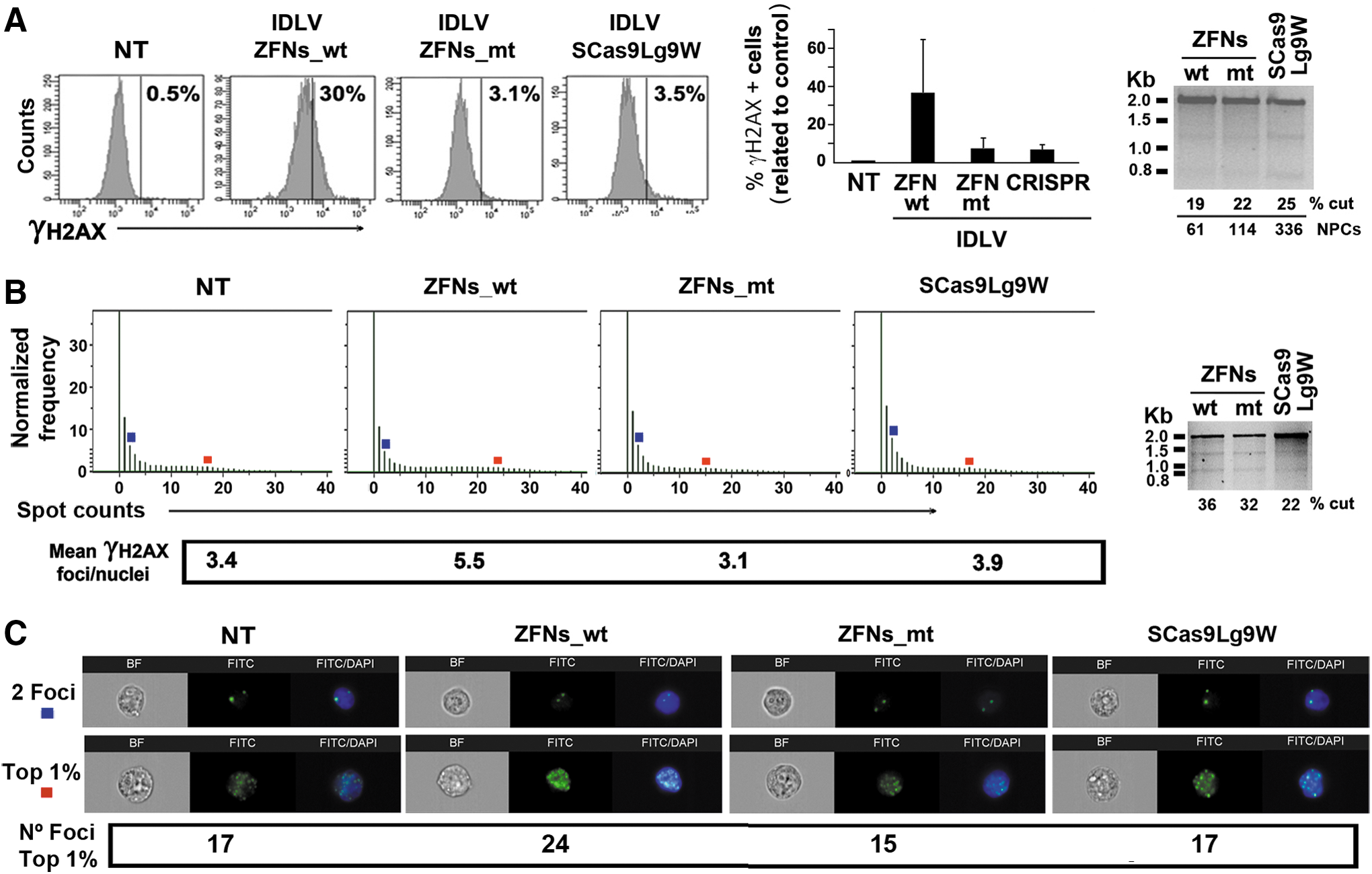
Comparison of WAS-specific zinc finger nucleases and all-in-one CRISPR system delivered by IDLVs.
We further compared efficacy and specificity of the dual IDLVs-ZFNs and the all-in-one IDLVs-CRISPR by incubating 106 K-562 cells with equal volumes of concentrated IDLVs and analyzing the cutting efficiency and DSB generation (Fig. 5B and C). As observed previously, we detected lower efficiency of the all-in-one IDLVs-CRISPR compared with the dual IDLVs-ZFNs_mt or IDLVs_wt (Fig. 5B, right; 22% vs. 32 and 36%, respectively). We also confirmed that γ-H2AX staining and FACS correlated (at least partially) with the amount of γ-H2AX foci observed with the ImageStream cytometer (Fig. 5B, left). Indeed, K-562 cells transduced with IDLVs-ZFNs_wt showed a mean number of γ-H2AX foci per nucleus higher than that of K-562 cells transduced with IDLVs-ZFNs_mt (Fig. 5B, left; mean γ-H2AX foci per nucleus of 5.5 vs. 3.1, respectively). We cannot compared ZFNs_mt with CRISPR in this experiment because the WAS cutting efficacy was lower in the latter (32 vs. 22%). However, we could show that the average amounts of DSBs in control cells (NT; nontransduced) were similar to those found in K-562 cells edited with IDLVs-ZFNs_mt and IDLVs-CRISPR (3.4, 3.1, and 3.9, respectively; Fig. 5B). In the same direction, we also observed a similar number of foci in populations that have a normalized frequency of 1% (top 1%) in K-562 control cells and in K-562 cells transduced with IDLVs-ZFNs_mt or IDLVs-CRISPR (Fig. 5B and C, red squares; 17, 15, and 17 foci per nucleus, respectively), whereas K-562 cell transduced with ZFNs_wt IDLVs showed up to 24 foci per nucleus. These data indicate that delivery of SNs by IDLV had a low impact on the target cells, something that was also observed in Fig. 3.
Interestingly, we could also observed that IDLVs showed lower γ-H2AX focus formation compared with nucleofection in spite of achieving higher cutting efficacies of the WAS gene (compare Fig. 2B with Fig. 5B and Supplementary Fig. S7). Indeed, K-562 WAS GE using IDLVs-ZFNs_mt and IDLVs-CRISPR achieved 32 and 22% cuts and mean γ-H2AX foci per nucleus of 3.1 and 3.9, respectively, whereas the same cells edited using nucleofection of plasmid ZFNs_mt and CRISPR achieved efficiencies of 26 and 21% cuts and mean γ-H2AX foci per nucleus of 7.1 and 7.5, respectively.
All together, these data indicate that the WAS-specific ZFNs_mt have similar specificity compared with CRISPR-gRNA9, and that delivery of SNs by IDLVs has lower impact on DSB generation in target cells compared with nucleofection of plasmid DNA.
Efficiency and specificity of ZFNs and CRISPR for HD GE of WAS locus
We next investigated the efficiency and specificity of WAS-specific ZFNs_wt, ZFNs_mt, and CRISPR systems to insert donor DNA into the WAS locus. We designed a donor containing eGFP and neomycin resistance gene expression cassettes flanked by 1-kb homologous arms to the WAS locus (Fig. 6A). Plasmids expressing the various SNs were nucleofected together with the donor DNA, and eGFP expression was analyzed on days 3 and 20 (Fig. 6B). We achieved transfection of about 30–45% of the K-562 cells (Fig. 6B, top plots), which renders between 0.3 and 6% of K-562 cells stably expressing eGFP (Fig. 6B, bottom plots) when cotransfected with the SNs. After 20 days, most of these cells stably expressing eGFP have integrated the donor DNA into the chromosome. To study only those cells in which donor integration has occurred, we sorted the eGFP+ cells and generated clones in the presence of neomycin. These clones (GFP+NeoR+) must have integrated the complete donor DNA. The various clones were analyzed by PCR as depicted in Fig. 6C to differentiate between donor integrations in the WAS locus (In-target) and integrations outside the WAS locus (Out-target) (see also Supplementary Fig. S2 for details). The results showed that ZFNs_mt and CRISPR achieved 88 and 83% of the insertions in the targeted locus, respectively (Fig. 6C and D). On the other hand, cells transfected with donor DNA and ZFNs_wt had 55% of the insertions landing outside the WAS locus (Fig. 6D).
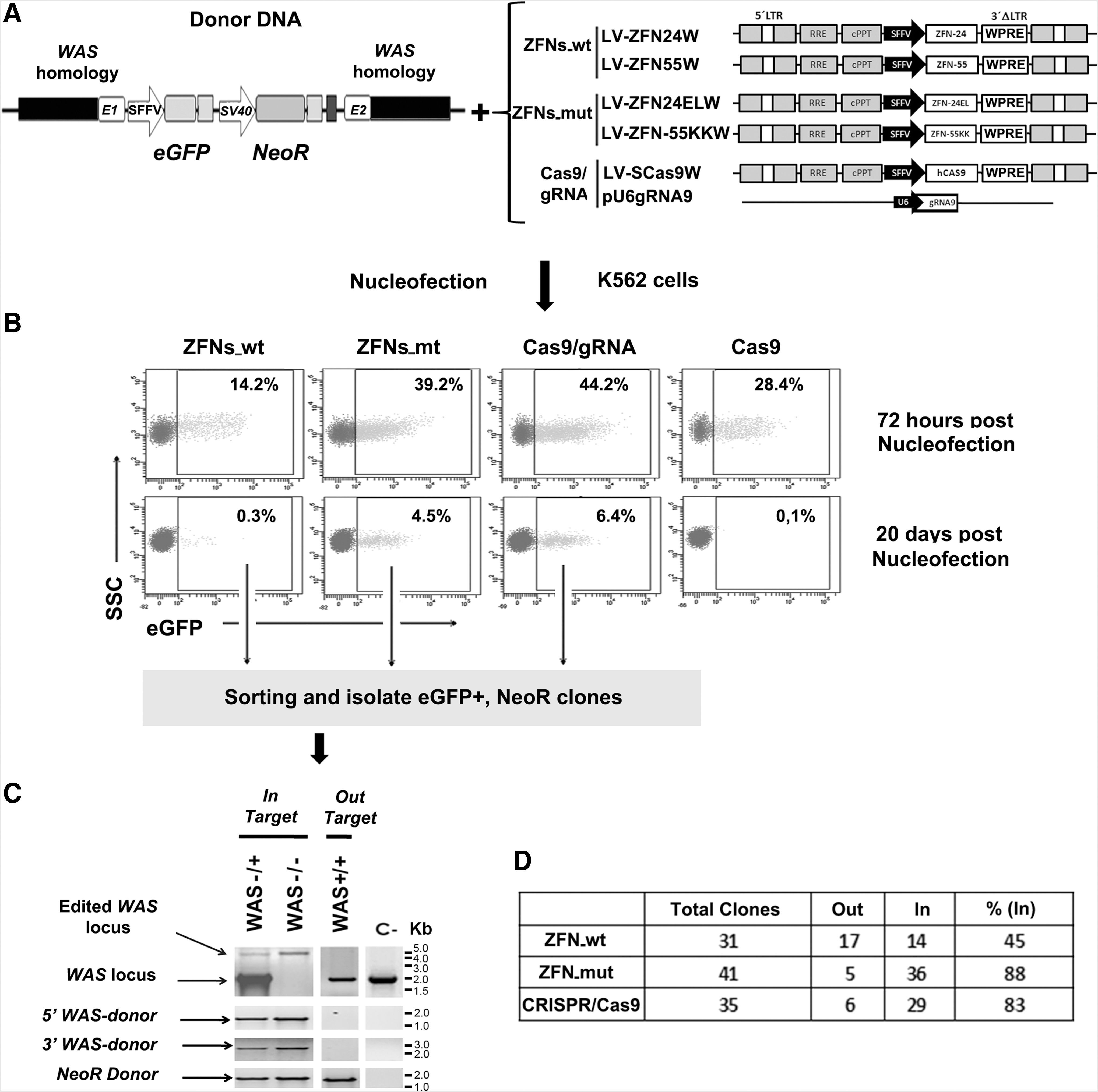
Analysis of the efficiency and safety of ZFNs and CRISPR/Cas9 for homologous recombination (HR)-directed WAS genome editing.
These data are in line with the cutting specificity data of the various SNs obtained previously. To further confirm the integration of donor DNA in the WAS locus, we analyzed WASP expression levels in homozygous and heterozygous clones by FACS analysis. Supplementary Figure S8 shows the absence or reduction of WASP expression in homozygous and heterozygous clones, respectively.
Discussion
In this study we have compared the efficiency and safety of various WAS-specific nucleases (ZFNs_wt, ZFNs_mt, and CRISPR/Cas9_gRNA9) for gene disruption as well as for HD gene insertion into the WAS locus, using K-562 cells as a cellular model. We used K-562 cells as target because they are considered an adequate model for studying GE in hematopoietic cells and the results can be translated to HSCs, 27,32 the final target for WAS gene therapy. Because the efficiency and safety of SNs depend on the efficiency of the delivery system as well as the expression levels achieved on target cells, we generated a set of plasmids that can be used for delivery of ZFNs and the CRISPR/Cas9 system either by nucleofection or IDLVs.
We first showed that the method used to deliver the SNs affected differently the efficiency of ZFNs and CRISPR/Cas9 systems. Indeed, both SNs showed similar WAS cleavage when delivered by plasmid nucleofection. However, delivery of the CRISPR system using two IDLVs showed lower cutting activity compared with ZFNs, even when the CRISPR system was delivered as all-in-one IDLVs. Our data also indicate that the lower efficiency of the IDLVs-CRISPR system was in part due to low expression levels of Cas9 mRNA. Indeed, we have hypothesized that the Cas9 levels achieved by the IDLVs-CRISPR system must be close to the threshold required to achieve GE. This could explain why IDLVs-CRISPR systems require higher NPCs than IDLVs-ZFNs systems and also the observed differences in efficacy of various experiments using IDLVs-CRISPR, even when similar NPCs are used. This hypothesis is partially supported by the fact that integrative LVs-CRISPR are always more robust and achieve similar or even higher cutting efficacies than integrative LV-ZFNs (integrative LVs express higher mRNA levels that their IDLV counterparts) (data not shown).
Our desire to improve IDLV delivery of the CRISPR system prompted us to design IDLVs expressing both Cas9 and gRNA in the same vector and to optimize their expression levels by inserting the gRNA-expressing cassette into the LTR and including the WPRE. The all-in-one IDLVs-WAS-specific CRISPR systems showed good WAS gene cleavage, although efficacy was still two to four times lower compared with the dual system expressing each ZFN under two different IDLVs. In addition, we could not detect any differences in cell viability when comparing similar cutting efficiencies, indicating that IDVLs-ZFNs could be a better option than IDLVs-CRISPR-gRNA9.
In a clinical setting it is fundamental that SNs cut only in the target site because off-target cleavage could generate a wide set of mutations that could, in the worst scenario, cause cellular transformation. We therefore analyzed the specificities of the WAS-targeted homodimeric and heterodimeric ZFNs versus CRISPR/Cas9_gRNA9 by Surveyor analysis of potential off-target sites (see Materials and Methods and data not shown) as well as for γ-H2AX staining. γ-H2AX immunostaining allows for identification of DSB foci that can be generated by SNs at the target locus but also at the off-target loci, and can therefore be used to measure them. 13,41,42 However, DSBs are also generated in the DNA of cells growing under normal conditions, and this noise can sometimes interfere with the off-target analysis. In addition, higher cutting efficiencies of the SNs will also render higher γ-H2AX staining, and therefore this methodology can be used only to compare DSB generation in cells edited at similar levels. Indeed, we verified in our system that higher cutting efficacies at the WAS locus rendered higher γ-H2AX signal (Supplementary Fig. S6). Another technical difficulty in off-target assessment based on the γ-H2AX signal is uncertainty concerning whether it correlates with focus formation due to DSB generation. Because FACS cannot discriminate between signals coming from spots (DSBs) or from uniform nuclear staining (unspecific), we proceeded to analyze off-target generation using γ-H2AX staining and ImageStream cytometry, which allows the identification, localization, and quantification of DSB foci. Using this technology we showed that, independent of the delivery method used, heterodimeric ZFNs and CRISPR/Cas9-gRNA9 behaved similarly and both showed lower levels of nonspecific DSB generation compared with homodimeric ZFNs. These data indicate that the CRISPR/Cas9 system can be designed to reach similar specificities compared with heterodimeric ZFNs. Interestingly, we also showed that IDLVs showed lower γ-H2AX focus formation compared with plasmid nucleofection in spite of achieving even higher levels of WAS gene disruption. In this direction, we also observed that control plasmid nucleofection increased the amount of DSBs in target cells compared with nucleofection without DNA or with untreated cells. Therefore, we must take into account the nonspecific DSBs generated by nucleofection rather than by the off-target activity of the SNs.
Another important aspect to take into account before translating gene repair strategies into the clinic is the fidelity of GE. Indeed, although SNs enhanced the efficiency and fidelity of HD recombination, 55,56 the administered donor DNAs can be integrated into DSBs generated by SN off-targets, cellular stress, or by the presence of homologous microdomains. 57,58 These undesired insertions can be highly deleterious for the targeted cells. We therefore analyzed whether the efficiency and fidelity of donor insertion into the WAS locus was affected by the SN used. Our system allowed the identification and isolation of those K-562 clones having the donor integrated in their genome, facilitating the analysis of the integration sites on individual cells. PCR analysis of the various clones showed again a similar behavior of heterodimeric ZFNs and CRISPR/Cas9_gRNA9, with 88 and 83% of the donors inserted in the WAS locus, respectively. Interestingly, when using homodimeric ZFNs only 45% of the insertions landed in the WAS locus, which corroborates the lower specificity of these ZFNs compared with heterodimeric ZFNs and with CRISPR/Cas9_gRNA. These data are in line with previously published data analyzing targeted integration of donor DNAs. 32 However, considering that DSBs can be generated by the plasmid nucleofection process, a good proportion of the donor insertions outside of the WAS locus must be due to the delivery process. However, the differences observed between ZFNs_mt and CRISPR versus ZFNs_wt must be due to the higher off-target activity of the latter.
Our studies have been performed in K-562 cells, which are easy to grow and with similar resistance for gene modification compared with many primary human cells, including HSCs. 27 The possibility to generate clones for K-562 cells allowed us to investigate the specificity of HD gene insertion in single cells. K-562 cells have been widely used as hematopoietic cellular models for GE of a wide range of target genes including WAS, 44 AAVS1, 59 –61 ILR2, the HHB locus, 62 CCR5, ABCC11, OPN1SW, CHI3L1, CEL, VEGFA, and C4BPB. 63 Results observed in K-562 cells can be translated to HSCs, although the efficiency of HD gene repair is much lower in HSCs compared with K-562 cells. 32
Taken together, our data indicate that both CRISPR/Cas9_gRNA9 and heterodimeric ZFNs rendered good efficiencies and specificities for WAS GE and that IDLVs outperformed plasmid nucleofection as delivery vehicle. We also showed that ZFNs delivered by IDLVs render higher efficiencies than CRISPR-IDLVs, probably due to lower expression levels.
Footnotes
Acknowledgments
This work has been financed by Fondo de Investigaciones Sanitarias ISCIII (Spain) and Fondo Europeo de Desarrollo Regional (FEDER) from the European Union, through research grants PI12/01097, PI15/02015 (F.M.), and ISCIII Red de Terapia Celular (TerCel: RD12/0019/0006) (F.M.); by the CICE and CS de la Junta de Andalucía FEDER/Fondo de Cohesion Europeo (FSE) de Andalucía 2007–2013 through research grants P09-CTS-04532, PI-57069, PI-0001/2009, and PAIDI-Bio-326 (F.M.); PI-0160/2012, PI-0014-2016 (K.B.), and PI-0407/2012, PI-0318/2014 (M.C.). A.G.-G. and S.S.-H. are doctoral students from the Biomedicine Doctorate Program from Granada University (Spain). S.S.-H. is funded by Plan de Empleo Juvenil (MINECO).
Author Disclosure
All of the authors declare no competing interests and no personal financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.