
Abstract
Oncolytic virotherapy is a novel and intriguing treatment strategy for cancer therapy. However, the clinical potential of oncolytic virus as single agent is limited. M1 virus is a promising oncolytic virus that has been tested in preclinical studies. In this study, we investigated the effect of the combination use of M1 virus and Bcl-2 family inhibitors. A chemical compounds screening including ten Bcl-2 family inhibitors demonstrated that pan-Bcl-2 inhibitors selectively augmented M1 virus oncolysis in cancer cells at very low doses. The mechanism of the enhanced antitumor effect of pan-Bcl-2 inhibitors with M1 virus is mainly due to the inhibition of Bcl-xL, which synergizes with M1-induced upregulation of Bak to trigger apoptosis. In xenograft mouse models and patient-derived tumor tissues, the combination of M1 and pan-Bcl-2 inhibitors significantly inhibited tumor growth and prolonged survival, suggesting the potential therapeutic value of this strategy. These findings offer insights into the synergy between Bcl-xL inhibition and oncolytic virus M1 as a combination anticancer treatment modality.
Introduction
C
Although oncolytic viruses specifically kill tumor cells, the outcome of oncolytic virus infections may not be as expected due to the complicated interactions between the viruses and the host defense system. 14 Therefore, researchers aimed to increase effect of oncolytic viruses by genetically modifying them or combing them with chemical sensitizers. It is reported that small molecular inhibitors, such as histone deacetylase inhibitors or microtubule-destabilizing agents can specifically enhance oncolytic effect of oncolytic viruses in cancer cells. 15 –18 Our group has reported that activation of cyclic adenosine monophosphate signaling pathway or a classical protein kinase A inhibitor, H89, inhibit M1-induced expression of antiviral factors, thus enhancing the replication of M1 and the subsequent virus-mediated oncolysis. 19,20 Hence, combining chemical compounds with M1 would be a rational strategy to increase M1 oncolysis.
Bcl-2 family members are the major regulators of the apoptotic pathway at the decision phase. The Bcl-2–related anti-apoptotic proteins include Bcl-2, Bcl-xL, Bcl-w, A1 (Bfl-1), and Mcl-1. 21 These proteins block cell death by sequestering the pro-apoptotic proteins, Bax and Bak, preventing them from oligomerizing and forming pores in the mitochondrial outer membrane. 22 Bcl-2-related anti-apoptotic proteins are frequently expressed at high levels in a variety of cancers. 23 Thus, inhibition of Bcl-2 and Bcl-xL might be expected to potentiate the effect of cytotoxic therapy by enhancing the apoptotic response to cellular stress. Bcl-2 family members have preclinical activity as single agents and in combination with other antineoplastic agents. 24,25 ABT-199, a potent and selective Bcl-2 inhibitor developed by AbbVie Inc., has become the first United States Food and Drug Administration–approved Bcl-2 family inhibitor in 2016. 26 Clinical trials of several investigational drugs targeting the Bcl-2 family (ABT-263, ABT-737, Oblimersen sodium) are ongoing. 27
Here, we reported that small molecular anti-apoptotic Bcl-2 family inhibitors cooperated with M1 virus to kill cancer cells by potentiating mitochondrial apoptosis. Importantly, we demonstrated that Bcl-xL inhibition is critical for this augmented M1 oncolysis. These findings support that the strategy of combing M1 virus with Bcl-xL specific inhibitors may possess the potential to be further developed into targeted enhanced oncolytic virotherapy.
Materials and Methods
Cell culture and viruses
Cell lines were purchased from American Type Culture Collection. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (vol/vol) Fetal bovine serum and 1% penicillin/streptomycin (Life Technologies). Primary cancer and normal cell were purchased from ScienCell Research Laboratories and cultured according to instructions. M1 was grown in Vero cells. Virus titer was determined by 50% tissue culture infective dose assay using BHK-21 cells and converted to plaque-forming units (PFU). The variant of M1 in this study was described previously.
Cell viability assay
Cells were seeded in 96-well plates at 3,000 cells per well in 0.1 mL media. After treatment, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added to cells (1 mg/mL final concentration), and cells were allowed to grow at 37°C for another 3 h. MTT containing media were removed, and MTT precipitate was dissolved in 100 μL DMSO. The optical absorbance was determined at 570 nm using a microplate reader (iMark; Bio-Rad).
RNA interference
Specific and nontargeting small interfering RNAs (siRNAs) were synthesized by Sigma and Ribobio (Guangzhou, China). Cells were replaced with 10% fetal bovine serum DMEM (without penicillin/streptomycin). SiRNAs were transfected using Lipofectamine RNAiMAX (13778-150, Thermo Fisher) with OPTI-MEM (31985070, Thermo Fisher).
The sequence of siRNA was listed as follows: siBcl-2-sense: 5′-GGUGUUACUUCACUUGCAUdTdT-3′ siBcl-2-antisense:5′-AUGCAAGUGAAUUGAACACC dTdT-3′ siBcl-xL-sense: 5′-GGAUACAGCUGGAGUCAGUdTdT-3′ siBcl-xL-antisense: 5′-ACUGACUCCAGCUGUAUCCdTdT-3′ siBcl-w-sense: 5′-CAGCUGUAUUCCAUUACAUdTdT-3′ siBcl-w-antisense: 5′-AUCUAAUGGAAUACAGCUGdTdT-3′ siBak-sense: 5′-AUGAGUAUCCGUAAGAGACdTdT-3′ siBak-antisense: 5′- GUCUCUUACGGAUACUCAUdTdT-3′
Antibodies and reagents
Antibodies used in this study are listed as follows: Bcl-2 (50E3, Cell Signaling Technology); Bcl-xL (2762, Cell Signaling Technology); Bcl-w (31H4, Cell Signaling Technology); Bax (2272, Cell Signaling Technology); Bak (D4E4, Cell Signaling Technology); glyceraldehyde 3-phosphate dehydrogenase (AP0060, Bioworld); M1, E1, and NS3 (produced by Beijing Protein Innovation, Beijing, China); and Ki-67 (9449s, Cell Signaling Technology). ABT-263, ABT-737, obatoclax mesylate (GX15-070), TW-37, AT101, HA14-1, UMI-77, Gambogic Acid, and ABT-199 were purchased from Selleckchem. WEHI-539 was purchased from Apex Bio Technology (Houston, TX).
Western blotting
Cells were lysed using M-PER Mammalian Protein Extraction Reagent (Thermo Scientific), and sodium dodecyl sulfate–polyacrylamide gel electrophoresis was performed. Membranes were visualized on a ChemiDoc XRS+ System (Bio-Rad) using Immobilon Western Chemiluminescent HRP Substrate (Millipore).
Caspase activity analyses
Cells were cultured in 96-well plates and infected with M1 virus (0.001 PFU/cell) in the presence or absence of Bcl-2 family inhibitors. Caspase-3/7 and Caspase-9 activities were determined by Caspase-Glo Assay Systems (Promega, Madison, WI) according to manufacturer's protocols. The results were normalized to cellular viability (MTT assay).
Animal models
This study was approved by the Animal Ethical and Welfare Committee of Sun Yat-sen University. For the subcutaneous xenograft model, Hep3B (5 × 106 cells/mouse) or LoVo (1 × 107 cells/mouse) cells were inoculated subcutaneously into the hind flank of the 4-week-old female BALB/c-nu/nu mice. After 6 days, palpable tumors developed (50 mm3), and mice were divided into four groups by random. M1 virus (2 × 106 PFU/day) was intravenously injected and ABT-263 (10 mg/kg/day) was intraperitoneal injected three times, tumor length and width were measured every other day, and the volume was calculated according to the formula (length × width2)/2. For the orthotopic xenograft model, Hep3B cells (1 × 106 cells/mouse) were inoculated into the liver parenchyma of nude mice. After 6 days, mice were randomized into four groups. M1 virus (2 × 106 PFU/day) was intravenously injected and ABT-263 (10 mg/kg/day) was intraperitoneal injected three times.
Immunohistochemistry assay
The expressions of Ki-67 and Cl-caspase3 in tumors were characterized by immunohistochemistry using specific antibodies. Briefly, tumor sections (4 μm) were dewaxed in xylene, hydrated in descending concentrations of ethanol, immersed in 0.3% H2O2–methanol for 30 minutes, washed with phosphate-buffered saline, and probed with monoclonal antibodies or isotype control at 4°C overnight. After washing, the sections were incubated with biotinylated goat anti-rabbit or anti-mouse Immunoglobulin G at room temperature for 2 hours. Immunostaining was visualized with streptavidin/peroxidase complex and diaminobenzidine, and sections were then counterstained with hematoxylin.
Ex vivo
Tissue culture-end point staining-computer image analysis (TECIA) was used to evaluate the ex vivo anticancer activity. TECIA is an improved histoculture drug response assay and is described elsewhere. 28 Primary tumor tissue specimens were obtained from consenting patients who underwent tumor resection. Informed consent was obtained from the patients before tissue collection. The work was approved by an ethics review committee at Sun Yat-sen University (Guangzhou, China). The institutional review board of Sun Yat-sen University Cancer Center has approved all human studies. Tumor samples were received in cell culture medium and processed within 2–6 hours. Samples were manually divided using a scalpel blade into approximately 1 mm3 blocks using sterile techniques. The explants were placed on moist but not submerged filter-paper inserted into single wells of 24-well plates with 1 mL DMEM containing 15% fetal bovine serum and cultured at 37°C with 5% CO2 for 24 hours. The A-score was recorded by volumetric integral of samples using the Image Analysis System. Samples were then exposed to saline (negative control), M1, ABT-263, and ABT-263 plus M1 for 4 days. After treatment, 100 μL of MTT (5 mg/mL) was added and cultured for 4 hours. The B-score was determined by the area and intensity of staining using the Image Analysis System. Every treatment on each sample was tested in quadruplicate. The efficacy of different treatments was presented as percentage inhibition that was calculated using the following formula: Inhibition (%) = [1−(mean of B-scores of treated sample/mean of A-scores of treated sample) / (mean of B-scores of control)/(mean of A-scores of control)] × 100%. Neither negative control samples with low MTT staining nor positive control samples with low inhibition (<80%) were accepted for analysis.
Statistical analysis
All statistical analyses were performed using SPSS 13.0 software (SPSS, IBM, Armonk, NY). Most of the data were subjected to Student's t-test or one-way analysis of variance followed by Dunnett's multiple post-hoc tests. Values of tumor volume were analyzed by repeated measures one-way analysis of variance. Unless otherwise indicated, all error bars indicate standard deviation. Significance was defined as p < 0.05.
Results
Screen of Bcl-2 family inhibitors identifies M1 enhancer
Most alphavirus-induced cell death includes loss of mitochondrial membrane integrity. 29 To investigate the mechanism of alphavirus M1-induced cell death, we performed JC-1 staining to examine the potential of mitochondrial membrane. As expected, M1 virus infection induced loss of mitochondrial membrane potential (Fig. 1A). Considering that the balance of proteins of Bcl-2 family play key roles in cell fate, we attempted to determine whether inhibition of anti-apoptotic Bcl-2 family members can potentiate M1-induced mitochondrial apoptosis. To test this, we conducted a Bcl-2 family inhibitors screening to identify enhancer of M1 virus. We used difference of area under curve to assess the synergy effect of the inhibitors (Fig. 1B–D). We identified ABT-263 and ABT-737 as the top two hits in our screening. Selective Bcl-xL inhibitor WEHI-539 also performed better in this screen. However, the selective Bcl-2 inhibitors, ABT-199, GX15-070, and HA14-1, seemed no synergy effect with M1 virus (Fig. 1E).

Bcl-2 family inhibitor screening to identify M1 enhancer.
ABT-263/ABT-737 sensitizes cancer cells to M1 oncolysis but spares normal cells
To further confirm the synergy effect of ABT-263 and ABT-737, which inhibit Bcl-2, Bcl-xL, and Bcl-w. We examined the cell viability of the combination use of ABT-263 or ABT-737 and M1 in different cancer cells. The presence of ABT-263 or ABT-737 significantly decreased cell viability of Hep3B, T24, and LoVo cells in different multiplicity of infection of M1 virus (Fig. 2A–C). The fact that Bcl-2 family inhibitors can enhance the M1 oncolysis even at very low titer suggests that the mechanism-based combination strategy can be utilized to overcome the limited viral replication efficiency within tumors, especially through systemic delivery.

Bcl-2 family inhibitors ABT-263/ABT-737 potentiate oncolysis of M1 virus but spare normal cells.
Another critical issue is the safety of this combined strategy. Normal colorectal cell line NCM460 and normal hepatic cell line L-02 were treated with either ABT-263 or ABT-737 plus M1 virus. Neither M1 virus nor combination treatment reduced cell viability (Fig. 2D and E). Furthermore, the same experiments were detected in three human primary cells: HH (human hepatocytes), HCEpic (human corneal epithelial cells), and HAEC (human aortic endothelial cells) (Supplementary Fig. S1A–C; Supplementary Data are available online at
The combination of M1 and ABT-263 or ABT-737 induces apoptosis through mitochondrial pathway
We next sought to determine whether ABT-263 or ABT-737 lead to increased replication of M1 virus. To this end, we detected the expression of M1 virus protein E1 as well as the titer of M1 virus. The expressions of M1 virus protein E1 did not increase upon treatment with ABT-263 in Hep3B tumor cells (Supplementary Fig. S2A). Accordingly, both inhibitors did not affect the titer of M1 virus upon a long infection period in these cells (Supplementary Fig. 2B). These results indicate that virus replication does not contribute to the enhanced cell death mediated by combined treatment.
Based on the evidence that both M1 virus and Bcl-2 family inhibitors can induce mitochondrial apoptosis, we postulated that the combination treatment may potentiate intrinsic apoptosis pathway. We first detected cell apoptosis via Annexin V–fluorescein isothiocyanate/propidium iodide staining and observed that combing M1 with either ABT-263 or ABT-737 significantly induced cell apoptosis, while single treatments did not (Fig. 3A). We next examined the executive stages of apoptosis by measuring the activation of caspase 3/7 and caspase 9. Consistently, cancer cells exposed to M1 plus either Bcl-2 family inhibitor showed much stronger activation of caspase 3/7 and caspase 9 than did single treatment (Fig. 3B–D), indicating that the combination of M1 and Bcl-2 family inhibitors exerts stronger mitochondrial apoptosis-inducing effect on cancer cells.
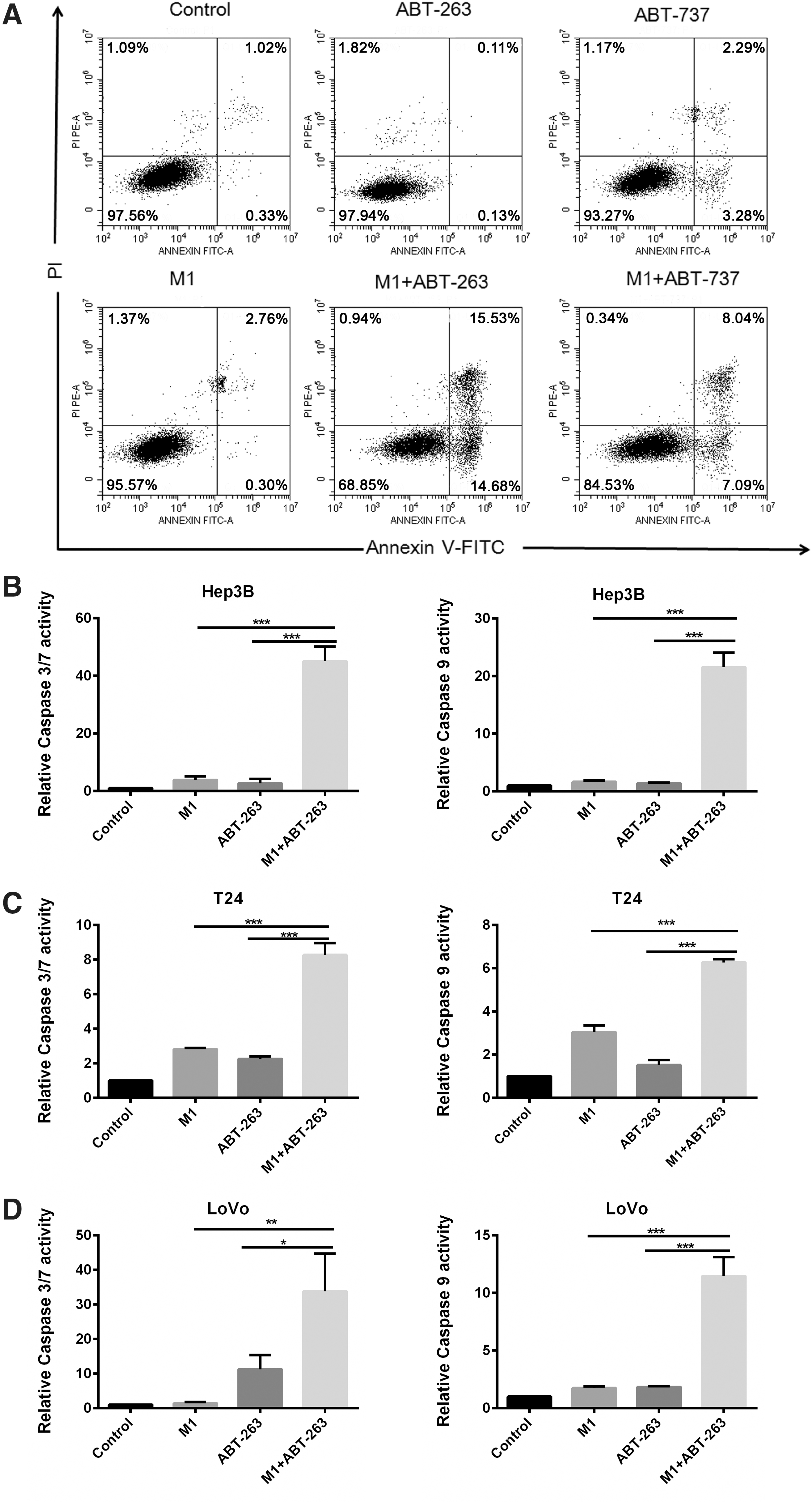
The combination of M1 and ABT-263/ABT-737 induces apoptosis through the intrinsic mitochondrial pathway.
Selective inhibition of Bcl-xL mediates the enhanced cell apoptosis driven by M1 plus ABT-263 or ABT-737
Given that both ABT-263 and ABT-737 target Bcl-2, Bcl-xL, and Bcl-w, we next studied the contribution of the three targets on the increased M1 oncolysis by using siRNA to silence Bcl-2, Bcl-xL, or Bcl-w respectively. Morphology studies showed that silencing Bcl-xL obviously enhanced the M1-induced oncolysis (Fig. 4A and B). Nevertheless, silencing Bcl-2 or Bcl-w plus M1 did not show any significant difference in cell viability (Fig. 4C and D), suggesting that Bcl-2 or Bcl-w was not the critical target. These findings suggest that selective inhibition of Bcl-xL exerts notable effects on oncolytic effect of M1 virus. To validate this conclusion, we measured cell viability by MTT assay and determined apoptosis via Annexin V–fluorescein isothiocyanate/propidium iodide staining (Fig. 4E). Consistent with the reported observations, silence of Bcl-xL combined with M1 infection markedly suppressed viable cells and induced apoptosis in different cancer cells. The observations above indicate that specific suppression of Bcl-xL drives the M1 plus Bcl-2 family inhibitors–induced enhancement of apoptosis.
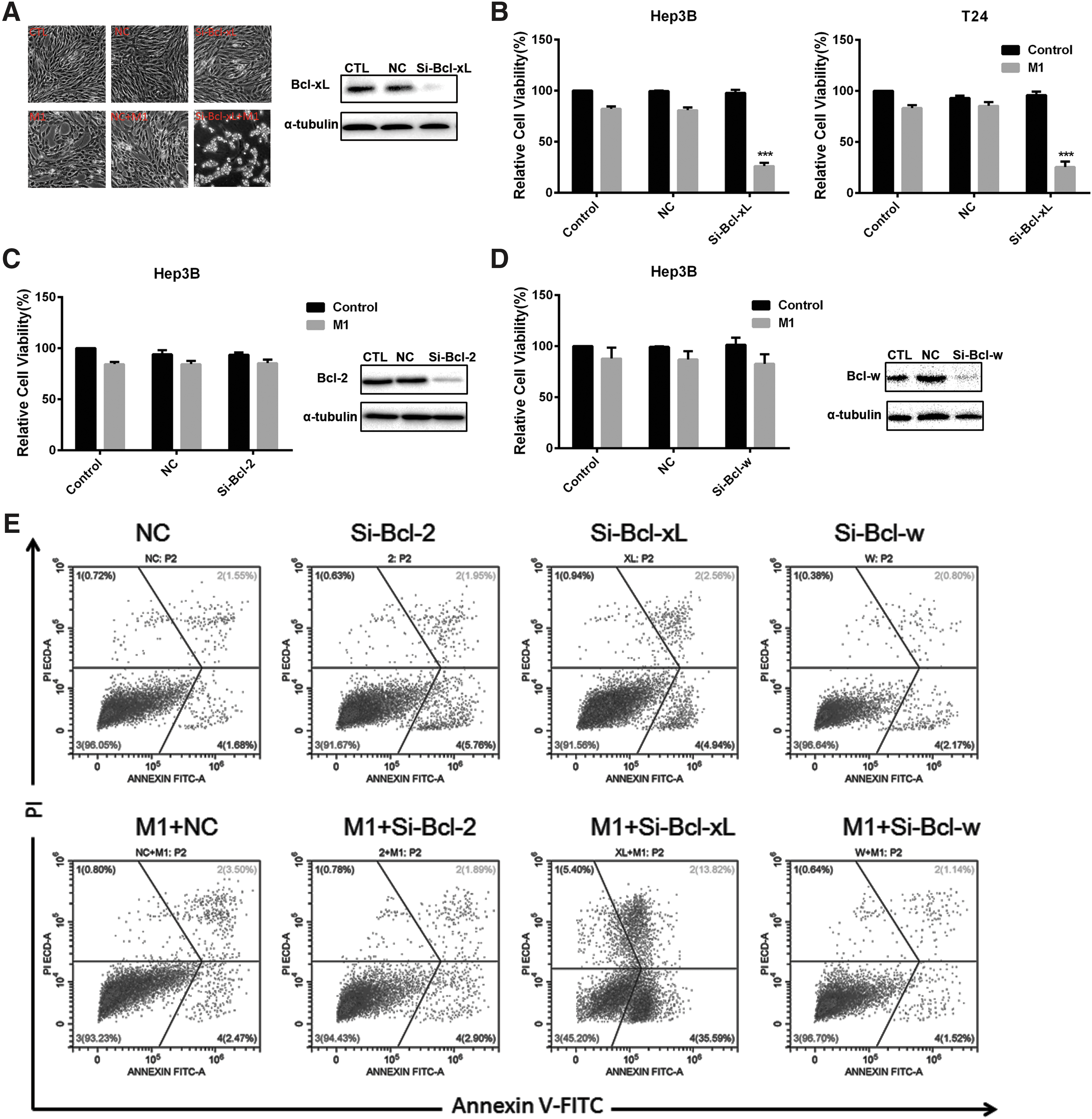
The antitumor effect of M1 and ABT-263/ABT-737 on cancer cells is mainly due to inhibition of Bcl-xL.
Inhibition of Bcl-xL synergizes with M1-induced upregulation of Bak to promote cancer cell apoptosis
We next investigate why targeting Bcl-xL can enhance M1 oncolysis. Thus, we examined the protein of Bcl-2 family after M1 virus (multiplicity of infection = 0.01) infection. The anti-apoptotic protein Bcl-2, Bcl-xL, Bcl-w and pro-apoptotic protein Bax did not alter, while pro-apoptotic protein Bak was upregulated after M1 infection (Fig. 5A). Bcl-xL tends to bind with Bak to prevent apoptosis. We next performed siRNA targeting Bak and found that knockdown of Bak could abrogate the increased oncolytic activity by ABT-263 and ABT-737 (Fig. 5B–D). Inhibition of Bcl-xL can thus cooperate with M1-induced Bak upregulation to activate apoptosis. This finding indicates that inhibition of Bcl-xL synergizes with M1-induced upregulation of Bak to promote cancer cells apoptosis.
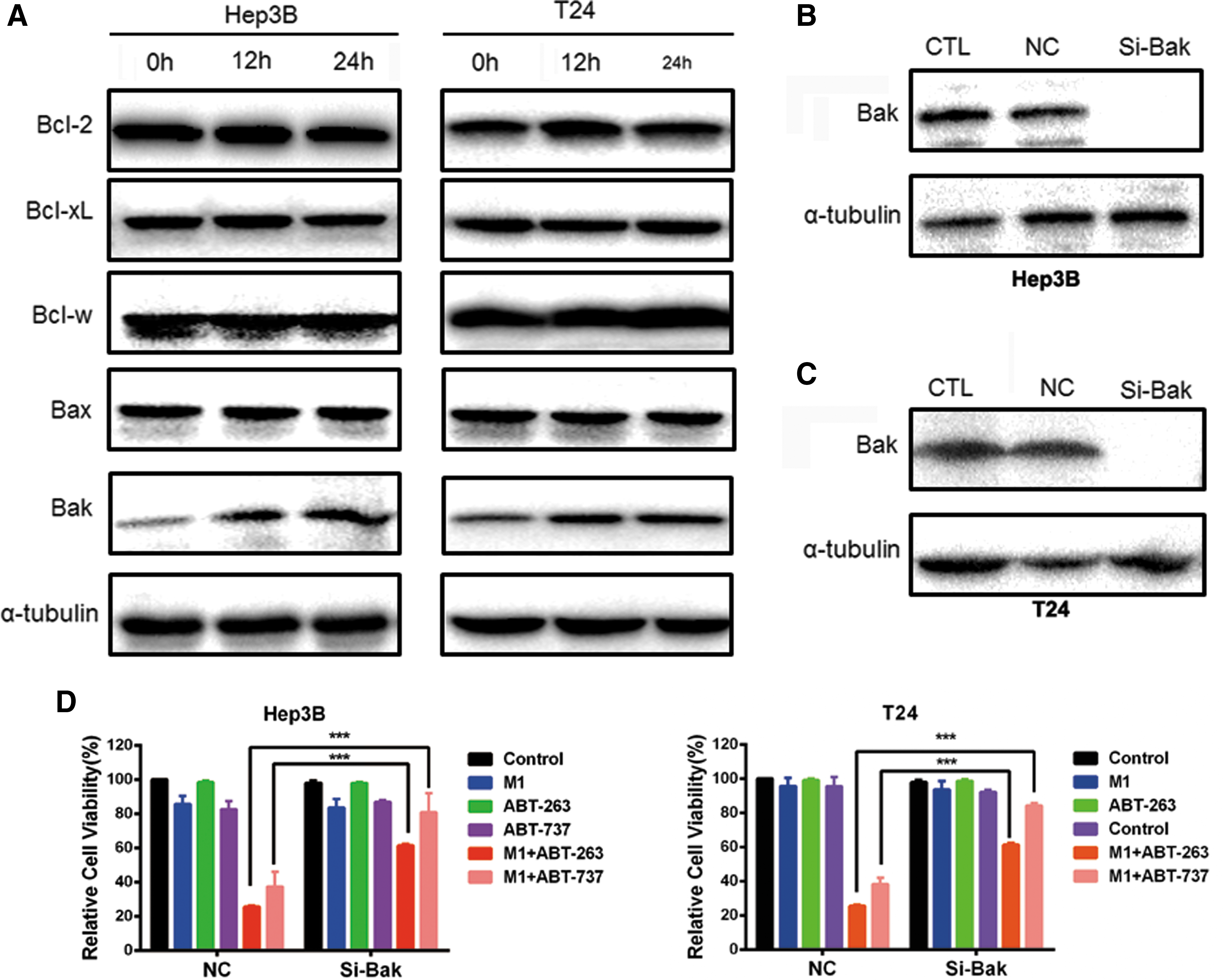
Inhibition of Bcl-xL synergizes with M1-induced up-regulation of Bak to promote cancer cells apoptosis.
ABT-263 potentiates M1-induced in vivo and ex vivo antitumor efficacy
The significantly enhanced M1-induced oncolysis in cancer cells prompted us to further investigate the in vivo antitumor effect of this combination strategy. To monitor tumor growth directly, we first established subcutaneous xenograft models using Hep3B or LoVo cells in nude mice. When palpable tumors formed, mice were treated with or without intravenously inoculated M1 virus and/or intraperitoneally injected ABT-263. Consistent with in vitro observations, combined treatment led to a significant inhibition of tumor growth in xenografts compared with control group or the groups treated with either drug alone (Fig. 6A–F). Body weight was also measured and no difference was found in all groups, indicating the safety of the combination treatment (Supplementary Fig. S3A).
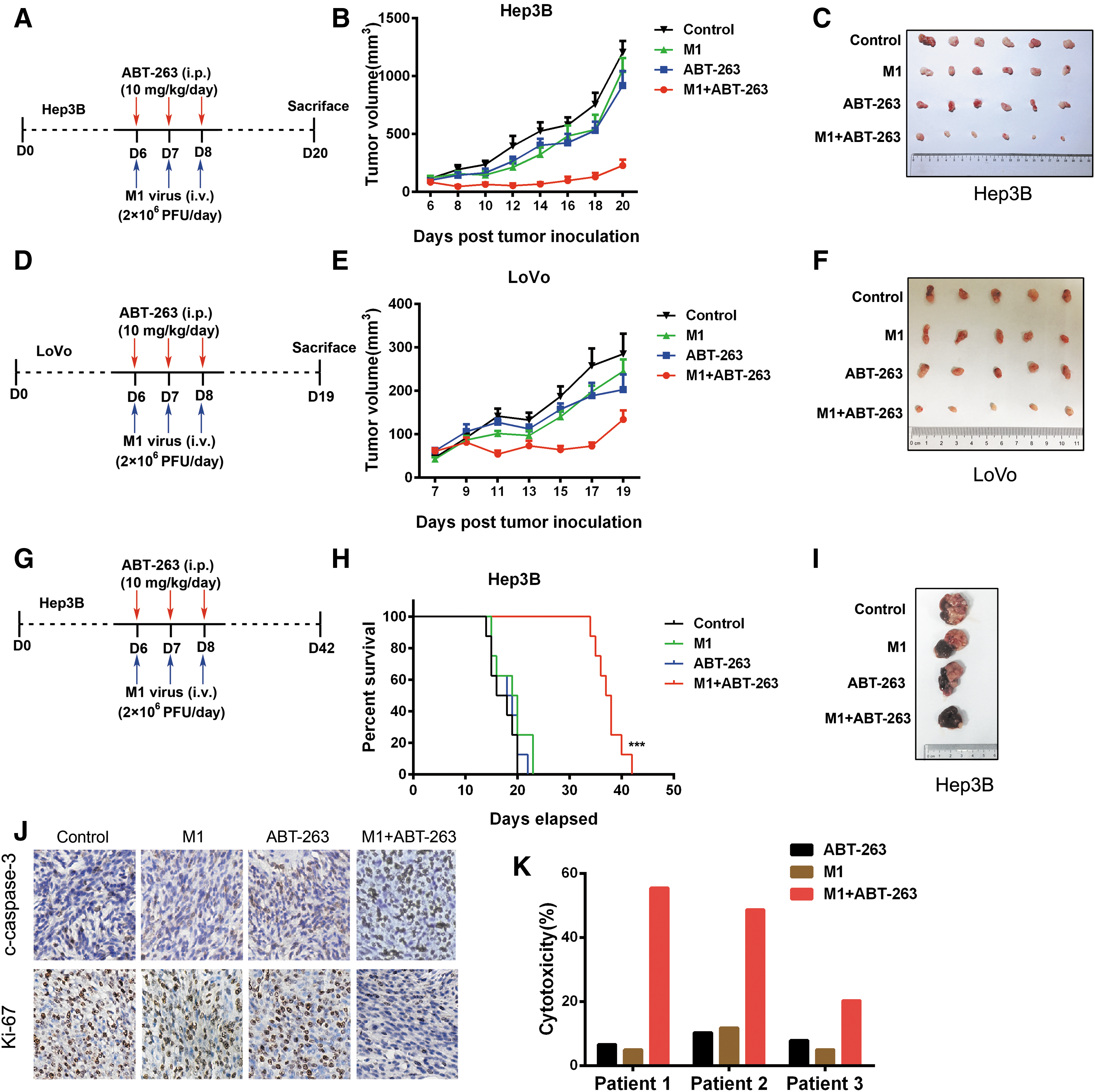
In vivo antitumor effect of M1 and ABT-263.
We next developed an orthotopic Hep3B xenograft model to further investigate the antitumor efficacy of M1 plus ABT-263 combination therapy. Accordingly, M1 virus or ABT-263 alone had minimal effect on tumor growth, whereas combination therapy significantly reduced tumor burden in Hep3B xenograft model (Fig. 6G–I). Moreover, mice received M1 plus ABT-263 showed a statistically significant increase in overall survival. Immunohistochemistry analyses were further performed on subcutaneous xenograft tumor sections. Ki-67, a proliferative marker, was obviously downregulated by M1 virus plus ABT-263 treatment. Meanwhile, higher level of cleaved-caspase 3 was observed in combination group, indicating a stronger apoptosis-inducing effect of M1 plus ABT-263 versus single treatments (Fig. 5J).
To validate the antitumor efficacy of M1 and ABT-263, we carried out ex vivo experiments on primary human colon tumor surgical samples by tumor histoculture end-point staining computer image analysis (TECIA). Consistent with the in vitro and in vivo oncolytic effects, three colorectal cancer patients also showed promising antitumor prospect of this combination therapy, supporting the therapeutic potential of M1 and ABT-263. (Fig. 6K).
These findings demonstrate that Bcl-2 family inhibitor ABT-263 can potently increase the in vivo oncolytic efficiency of M1 in a safe manner.
Discussion
In this study, we design a mechanism-based combination therapeutic strategy to enhance M1-induced oncolysis with Bcl-2 family inhibitors. In vitro and in vivo studies demonstrate this potentiated anticancer effect of the combination strategy on human cancers. Importantly, the combination treatment retains the intrinsic tumor selectivity of M1 virus, thus indicating great safety in clinical use in future. Mechanism studies suggest that targeting Bcl-xL, rather than Bcl-2 or Bcl-w, can significantly increase M1-induced mitochondrial apoptosis.
Interactions between pro-apoptotic and pro-survival members of the Bcl-2 family proteins are decisive in the initiation of pore opening, thus operating the mitochondrial gateway to cell death. 30 Although higher multiplicity of M1 infection leads mitochondrial apoptosis, low doses of virus infection may also initiate apoptotic pathways. However, the overexpressed pro-survival Bcl-2 family proteins in cancer cells may block mild apoptosis. Bcl-2 family inhibitors seem to remove these blockers thus allowing the activation of M1-induced intrinsic pathway of apoptosis.
One current key shortage of oncolytic virotherapy is the limited viral spread within tumors. Researchers' attempt to screen and discover small molecules to inhibit host antiviral immunity thus promoting viral replication. Indeed, some chemical drugs, such as histone deacetylase inhibitors, are identified according to this strategy. However, some other researchers also concern about the potential safety problem of burst of viral replication. Increased viral replication is often accompanied by inhibition of interferon-related antiviral response, which may be harmful for tumor patients' anti-infection ability. Instead of increasing viral replication, Bcl-2 family inhibitors augment the M1-induced apoptosis even at extremely low titer of M1, hence overcoming the limitation of viral replication efficiency on virus-induced oncolysis.
The finding that inhibition of Bcl-xL, but not Bcl-2 or Bcl-w, is mainly responsible for the augmented M1-induced oncolysis in cancer cells suggests that Bcl-xL selective inhibitors may present greater performance in potentiating the oncolytic effect of M1. We postulate that the expression of Bcl-xL can be used to predict the antitumor efficacy of combination therapy with Bcl-xL selective inhibitors plus M1 virus. Unlike other members of Bcl-2 family, elevated expression of Bcl-xL was found in many solid tumors, including neuronal tumors, adenocarcinoma, bladder cancer, gastric cancer, and so on, 31 suggesting a broad spectrum of solid tumors would respond to this combination therapeutic strategy.
Our study does present two limitations. First, we discovered that selective antagonism of Bcl-xL potentiates M1 oncolysis by enhancing mitochondrial apoptosis. However, the inhibitor we used in animal experiments was ABT-263, which is a pan-Bcl-2 family inhibitor. Thus, more specific Bcl-xL inhibitors should be tested in animal experiment in future. Second, inhibition of Bcl-xL has been reported to induce thrombopenia in clinical trials. In this study, we used low dose of ABT-263, thus, we concluded that the side effect of Bcl-xL inhibition could be limited. That being said, whether the thrombopenia occurred or not after M1/Bcl-xL-inhibition treatment should be further investigated in the future.
In summary, we demonstrate that Bcl-2 family inhibitors potentiate the M1-induced in vitro and in vivo oncolysis by inhibiting the anti-apoptotic effect of Bcl-xL in human cancers. We predict that oncolytic virus M1 combines with Bcl-2 family inhibitors, especially Bcl-xL selective ones, will be further developed into potentially personalized therapy against human cancers.
Footnotes
Acknowledgments
This work was funded by National Natural Science Foundation of China (81503088 and 81603127), China Postdoctoral Science Foundation (2015M580762 and 2015M580761), Natural Science Foundation of Guangdong Province (2016A030310160 and 2016A030310146), the research and development project of applied science and technology of Guangdong Province, China (No. 2016B020237004), and Medical Scientific Research Foundation of Guangdong Province, China (No. A2015102).
Author Disclosure
All authors state that they have no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.