
Abstract
Porphobilinogen deaminase (PBGD) gene therapy represents a promising therapeutic option for acute intermittent porphyria (AIP) patients suffering recurrent acute attacks. A first-in-human Phase I clinical trial confirmed the safety and tolerability of adeno-associated virus (AAV)-AAT-PBGD gene therapy, but higher doses and/or more efficient vectors are needed to achieve therapeutic expression of the transgene. This study assayed the insertion into the promoter of a short enhancer element able to induce transgene expression during exposure to endogenous and exogenous stimuli related to the pathology of the disease. The inclusion in tandem of two elements of the minimal functional sequence of human δ-aminolevulinic acid synthase drug-responsive enhancing sequence (ADRES) positioned upstream of the promoter strongly induced transgene expression in the presence of estrogens, starvation, and certain drugs known to trigger attacks in porphyria patients. The inclusion of two ADRES motives in an AAV vector improved therapeutic efficacy, reducing 10-fold the effective dose in AIP mice. In conclusion, the inclusion of specific enhancer elements in the promoter of gene therapy vectors for AIP was able to overexpress the therapeutic transgene when it is most needed, at the time when porphyrinogenic factors increase the demand for hepatic heme and precipitate acute porphyria attacks.
Introduction
A
Severe and recurrent porphyria attacks can be cured only by allogeneic liver transplantation. As a clinical alternative to transplantation, PBGD liver gene therapy mediated by recombinant viral vectors might restore hepatic PBGD activity and prevent the occurrence of acute attacks. The proof of concept was obtained in deficient AIP mice using adeno-associated virus (AAV) vectors encoding PBGD under the control of a liver-specific promoter. 7,8 Recently, a first-in-human Phase I clinical trial confirmed the safety and tolerability of AAV2/5-EalpAAT-PBGD gene therapy, but higher doses and/or more efficient vectors are needed to obtain therapeutic expression of the transgene. 9
It was speculated that the efficacy of gene therapy vectors for AIP might be improved by enhancing the expression of the therapeutic transgene when it is most needed—when porphyrinogenic factors increase the demand for hepatic heme synthesis and precipitate the acute porphyria attacks. Hypothetically, the insertion upstream of the promoter of the therapeutic vector of the minimal active sequence of the drug-responsive sequence that enhances hepatic ALAS1 (acid synthase drug-responsive enhancing sequence [ADRES]) gene expression might induce an overexpression of the transgene during the onset of the acute attack, thus expanding the therapeutic efficacy of the vector.
Materials and Methods
Murine model of AIP
Compound heterozygote T1 (C57BL/6-pbgd tm1(neo)Uam) and T2 (C57BL/6-pbgd tm2(neo)Uam) strains, as described by Lindberg et al., 10 exhibit a hepatic PBGD activity that is only 33% of normal. To imitate a human porphyria attack biochemically, mice were intraperitoneally injected with increasing doses of phenobarbital (75, 80, 85, and 90 mg/kg of body weight) for four consecutive days. After the last phenobarbital dose, animals were housed for 24 h in metabolic cages (reference 3600M021; Biosis Biologic Systems SL, Barcelona, Spain) in order to collect 24 h urine samples. Experimental protocols were approved by the Ethics Committee of the University of Navarra (CEEA022-06) according to European Council Guidelines.
Construction of recombinant expression vector
Plasmids for hydrodynamic transfer experiments
A set of different plasmids derived from a pTRE2 vector (Clontech Laboratories, Inc., Heidelberg, Germany) were designed and constructed to conduct several comparative hydrodynamic transfection assays. The pTRE-EalpAAT-Luciferase plasmid (pLuc) was used as the starting product to construct other plasmids, and was also included in the assays as a control. The reporter gene luciferase (CDS sequence: 304 … 429, 487 … 694, 746 … 1079, 1128 … 1448, 1498 … 1852, 1896 … 2061, and 2109 … 2251; GenBank accession number M15077) was cloned downstream of the EalpAAT, which is a potent chimeric promoter in directing stable gene expression in liver cells. 11 A panel of plasmids that carried one or several ADRES elements was also constructed. The nucleotide sequence of an ADRES element (174 bp) spans from −20.918 bp to −20.745 bp of the 5′-flanking region of human ALAS1 gene (GenBank accession no. AC006252). Different plasmid clones were obtained and were analyzed by sequencing.
Production of AAV2/8 vectors
Single-stranded AAV2/8-EalpAAT-PBGD and AAV2/8-2 × ADRES-EalpAAT-PBGD vectors were produced for evaluation in an AIP mouse model. First, the plasmids pro-AAV-PBGD and pro-AAV-2 × ADRES were generated by cloning expression cassettes of the vectors in a pro-AAV cloning vector. 12,13 The expression cassette inserted into pro-AAV-PBGD had the polyA insulator sequence, the liver-specific promoter EalAAT, the codon optimized synthetic sequence encoding human housekeeping PBGD, and the 3′-UTR and polyadenylation sequences of human PBGD described by Unzu et al. 7 The expression cassette inserted into pro-AAV-2 × ADRES had these same elements plus two tandem repeats of ADRES inserted after the polyA insulator at the 5′ end of EalAAT promoter. Second, recombinant AAV serotype 8 (rAAV8) vectors with wild-type AAV2 inverted terminal repeats (ITRs) were then produced in HEK 293T cells. For each production, a mixture of plasmids, 20 μg of corresponding pro-AAV-based plasmid, and 55 μg of pDP8.ape (PlasmidFactory, KG, Bielefeld, Germany) was transfected into 293T cells 15 cm2 plate using linear polyethylenimine 25 kDa (Polysciences, Warrington, PA) as described. 14 AAVs were purified by ultracentrifugation in Optiprep Density Gradient Medium-Iodixanol (Sigma–Aldrich, St Louis, MO), following the manufacturer's instructions. The purified batches were then concentrated and filtered by passage through Centricon tubes (YM-100; Millipore, Bedford, MA). After concentration, the viral batches were stored at −80°C. To titer the AAV productions, viral DNA was isolated using the High Pure Viral Nucleic Acid kit (Roche Applied Science, Mannheim, Germany). The concentration of viral particles was subsequently determined by real-time quantitative polymerase chain reaction (RT-PCR) using primers specific to the PBGD polyadenylation sequence: pPBGD fw: 5′-gctagcctttgaatgtaacca-3′; and pPBGD rv: 5′-ccttcagaactggtttattagtagg-3′.
Experimental procedures
Effects of the incorporation of ADRES elements in a plasmid expressing the luciferase gene marker
First, luciferase transgene expression depending on the number and disposition of ADRES elements were tested in vivo by hydrodynamic transfer into the livers of mice of different plasmids (pLuc, 1 × ADRES, 2 × ADRES, 3 × ADRES, 2 × ADRES + invADRES, or 2 × invADRES–EalpAAT-luciferase). Fifty micrograms of DNA plasmid re-suspended in 2 mL of 0.9% NaCl saline was hydrodynamically delivered by the tail vein to BALB/c mice (n = 5/group) as described by Unzu et al. 15 The day after the hydrodynamic procedure, transfected animals received 90 mg/kg of phenobarbital for two consecutive days. Liver transgene expression levels were determined 2 h after the second phenobarbital doses by bioluminescence measurements performed in anesthetized mice intraperitoneally injected with 100 μL of D-luciferin (30 mg/mL in 150 mM of NaCl). Five minutes after D-luciferin injection, animals were placed in the imaging chamber of a Xenogen IVIS system (Xenogen, Alameda, CA), which includes a cooled charge-coupled device (CCD) camera. Gray-scale photographs of the animals were acquired, followed by bioluminescence image acquisition. Regions of interest (ROIs) were traced over the positions of greatest signal intensity on the animal, as well as over regions of “no signal,” which were used as background readings. The grayscale photograph and data images from all studies were superimposed using Living Image software (Xenogen). Light intensity was quantified as photons/s/cm2/steradian. Luciferase expression was also expressed as fold change relative to bioluminescence values measured in the control group that received the pLuc plasmid.
Second, other male BALB/c mice (n = 10) were also hydrodynamically transfected with 2 × ADRES-AAT-luciferase (50 μg of DNA plasmid re-suspended in 2 mL of 0.9% NaCl saline) in order to assess the performance of plasmid vectors modified with ADRES elements in response to different porphyrinogenic stimuli. Half of the animals received a dose of 90 mg of phenobarbital/kg, and luciferase expression was daily monitored by luminescence measurement, as previously described.
Third, cohorts of male BALB/c mice (n = 6) were also hydrodynamically transfected with 2 × ADRES-EalpAAT-luciferase. Transfected animals were exposed to a porphyrinogenic stimulus selected from 24 h fasting, 17α-ethinyl-estradiol injection (estrogen, 1 μg two times a day), and cardiotrophin 1 injection (CT-1; 10 μg/kg/day). Luciferase expression was measured at baseline and after 24 h fasting or 2 days after continuous exposure to estrogen or CT-1. The effect of CT-1 as a porphyrinogenic stimulus was identified using whole-transcript arrays to study the hepatic gene expression in wild-type mice injected with recombinant CT-1 (Dr. Prieto and Dr. Bustos, pers. commun.).
Fourth, male BALB/c wild-type mice were hydrodynamically transfected with 50 μg of pLuc (n = 6), 2 × ADRES-EalpAAT-luc (n = 6), or a construction including five estrogen response element (ERE) copies cloned at the 5′ end of the EalAAT promoter (5xERE-EalpAAT-luc; n = 10). Half of the animals in each of the three groups received 1 μg of porphyrinogenic estrogen 17α-ethinyl-estradiol twice a day for 6 days after transfection. Luciferase expression was measured daily and reported as the ratio between estradiol-treated and untreated animals (fold change).
Effects of the incorporation of ADRES elements into a rAAV2/8 vector carrying the therapeutic PBGD gene
The AAV2/8-2 × ADRES-EalpAAT-PBGD was tested at treatment doses of 7.7 × 109, 1.5 × 1010, and 2.3 × 1010 genome copies/kg body weight. The control AAV2/8-EalpAAT-PBGD vector was tested at treatment doses of 7.7 × 109, 5 × 1010, 1 × 1011, 2 × 1011, and 3 × 1011 gc/kg. Compound heterozygous AIP mice aged 12–25 weeks (four female mice per group) were injected intravenously via the tail vein with a total volume of 200 μL of the corresponding viral vector. Extra groups of wild-type and AIP mice were included as non-treated controls. Thirty days after AAV administration, groups of mice were injected for 4 days with increasing doses of phenobarbital. On day 4, 24 h urine samples were collected, and porphyrin precursors, PBG and ALA, were quantified in urine using a quantitative ion exchange column method (BioSystems SA, Barcelona, Spain) and were measured by spectrophotometry (Ultrospc 3000; Pharmacia Biotech, Little Chalfont, United Kingdom) at 555 nm. On day 35, 24 h after the last dose of phenobarbital, mice were administered with one supplementary dose of phenobarbital and sacrificed after 20 min. The livers were harvested and divided in different pieces, and PBGD expression was measured by enzymatic activity and immunohistochemistry, as reported by Unzu et al. 7 Other liver samples were frozen at −80°C until DNA or RNA extraction. The steady state mRNA level of the ALAS1 was analyzed by quantitative RT-PCR using iQ SYBR green supermix in an iQ5 real-time PCR detection system (Bio-Rad, Hercules, CA). PCR amplification was performed under the following conditions: one cycle of 3 min at 95°C; followed by 35 cycles of 15 s at 95°C, 30 s at 60°C, 25 s at 72°C, and 10 s at 70°C, 10 s at 75°C, and 30 s at 80°C; followed by a single final extension cycle of 72°C for 4 min. The relative transcript level was determined using primers annealing specific cDNA sequences of heat shock protein 70 (HSP70) (fw: 5′-tggtgctcacgaagatgaag-3′, rv: 5′-aggtcgaagatgagcacgtt −3′, product length: 235 bp), total XBP1 (tXBP1s: 5′-gcaggtgcaggcccagttgtcac-3′; tXBP1as: 5′-ccccactgacagagaaagggagg-3′, product length: 228 bp), and spliced XBP1 (sXBP1s: 5′-cgggtctgctgagtccgcagcag-3′; sXBP1as: 5′-ccccactgacagagaaagggagg-3′, product length: 247 bp). The amount of gene transcript was calculated as the n-fold difference relative to the control gene actin as an internal control (forward primer: 5′-cgcgtccacccgcgag-3′, reverse primer: 5′-cctggtgcctagggcg-3′, product length: 193 bp). Results were expressed according to the formula 2ΔCt, where ΔCt represents the difference in threshold cycle between the target and control genes. Vector content measurements in liver samples were performed in genomic DNA of harvested samples isolated using the QIAmp Blood&Tissue DNA Mini-kit (Qiagen, Hilden, Germany). Genomic DNA was subjected to qPCR using specific primers, as described above for transgene expression. For normalization of the amount of genomic DNA, specific primers for murine GAPDH sequence were used (fw, 5′-ccaaggtcatccatgacaac-3′; rv, 5′-tgtcataccaggaaatgagc-3′). The genomic DNA samples were quantified by means of qPCR, as described for transgene expression.
Neurophysiological studies
The therapeutic efficacy of the inducible construct was measured by the impact of recurrent acute attack on sciatic nerve function. Amplitude and latency of the compound motor-evoked potential of the sciatic nerve were measured in the two hind legs of each of the animals, as previously reported. 7 Latency and amplitude were measured before and 45 days after the administration of 1.5 × 1010 gc/kg of a vector carrying the inducible or the constitutive promoter in groups of three AIP male mice. Three phenobarbital challenges were administered biweekly in those male AIP mice, starting 2 weeks after vector injection.
Statistics
All analyses were done in duplicate. The results were plotted as means ± standard deviation. For statistical analysis, data were transformed using the formula log(1 + x) in order to normalize the variances. Comparisons between two groups were analyzed by Student's t-tests. In the case of comparisons between more than two groups, data were analyzed by analysis of variance, and pairwise comparisons were made using Bonferroni's multiple comparison tests. The null hypothesis was rejected when p < 0.05. Statistical analyses were done using GraphPad Prism® v5 (GraphPad Software, Inc., La Jolla, CA).
Results
The insertion of two minimal active sequences of the human ADRES elements in phase with the promoter strongly enhanced transgene expression throughout exposure to the porphyrinogenic stimuli
The constitutive androstane receptor (CAR) is a multi-sensor receptor that translocates from the cytoplasm to the nucleus in the presence of a variety of factors, some of which have been identified as factors precipitating acute attacks. 16,17 In the nucleus, CAR dimerizes with cognate nuclear receptors and binds upstream ADRES elements and different enhancers of hepatic cytochromes (Fig. 1A). In order to use this system to modulate the expression of a transgene, this study started by determining the optimal combination of the minimal active sequence of the ADRES to achieve high levels of hepatic transgene expression in the presence of a variety of porphyrinogenic factors. Different plasmids were developed carrying one, two, or three ADRES elements cloned in random orientation with the liver specific human α-1-antitrypsin promoter with regulatory sequences from the human albumin enhancer (EalpAAT 7 ; Fig. 1B). Following hydrodynamic delivery into male wild-type BALB/c mice, the expression of luciferase transgene was measured after 2 days of phenobarbital exposure (90 mg/kg/day). The results revealed that animals receiving the plasmid carrying two ADRES motives in phase with the promoter (2 × ADRES-EalpAAT-luciferase plasmid) displayed a 100-fold increase in luciferase expression when compared to animals that received control luciferase plasmid (Fig. 1C). The inclusion of a third copy in phase with the promoter (3 × ADRES) or inverted (2 × ADRES + InvADRES) also enhanced the transgene expression in response to phenobarbital but to a lesser extent than the increase observed with the plasmid carrying the 2 × ADRES motive. Finally, the incorporation of a single ADRES element (1 × ADRES) or two copies in reverse orientation (2 × InvADRES) only induced a slight increase in luciferase expression in response to phenobarbital (Fig. 1C). These results clearly established how the incorporation of different numbers of ADRES elements with different orientations upstream of the EalpAAT promoter affects transgene expression. The induction of the luciferase transgene was maintained throughout the presence of phenobarbital (Fig. 1D). The expression returned to baseline values in the absence of the drug, but it was re-induced after re-administration of the same stimulus, reaching levels of expression similar to those achieved with the first induction (Fig. 1D).
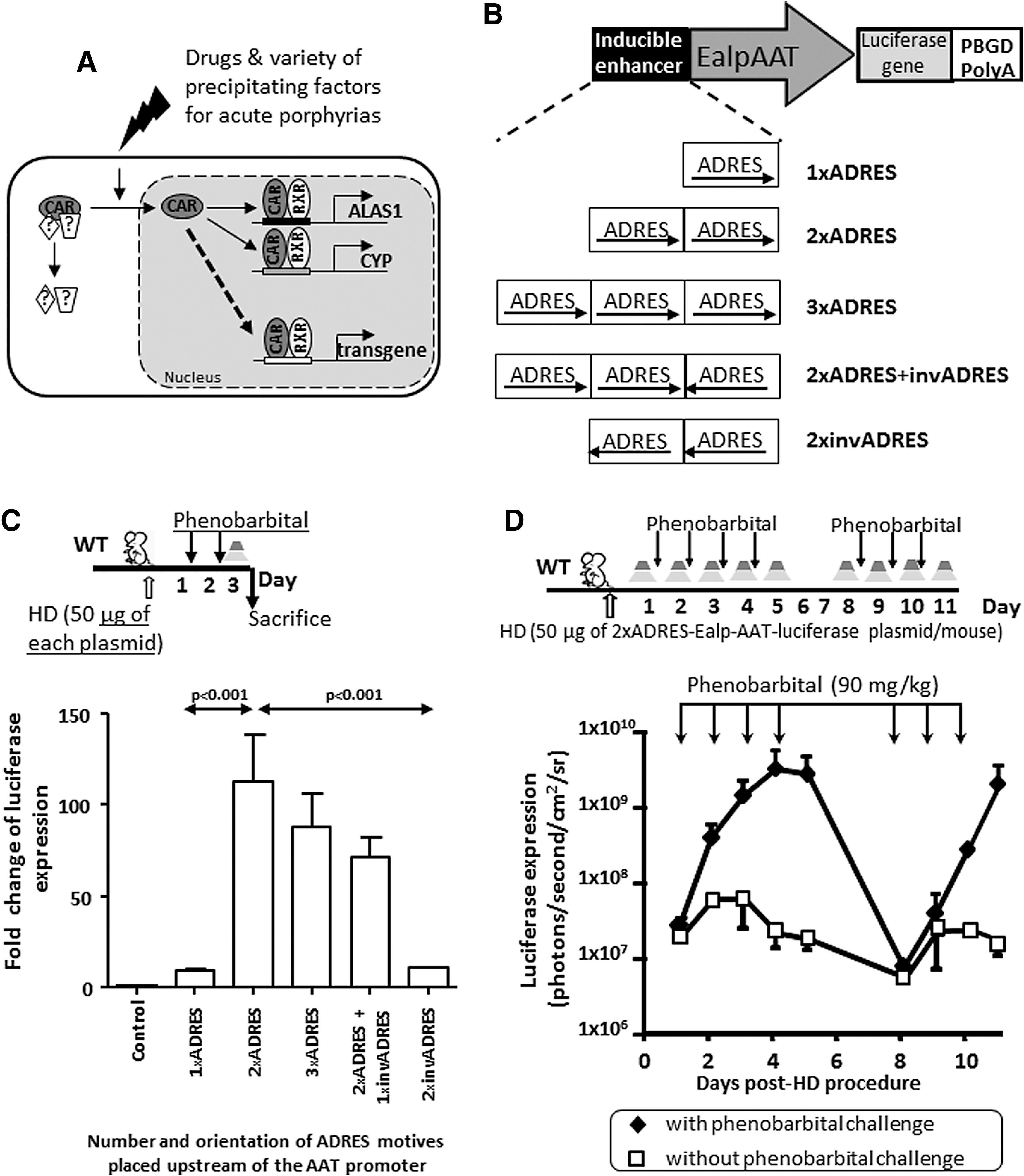
Effects of the incorporation of ADRES elements upstream of the promoter on the expression of luciferase throughout the phenobarbital exposure.
The inclusion of two ADRES elements in phase with the promoter also enhanced the expression of the transgene in presence of several porphyrinogenic stimuli, such as fasting (Fig. 2A and B), CT-1 (Fig. 2B and C), or estrogen (Fig. 2B, D, and E). An important point is that the induction of the transgene was maintained throughout the presence of estrogen stimulus (Fig. 2D and E) extended for 1 week. This is an important difference when compared to other inducer elements previously used in the context of viral vectors for gene therapy. 18 Thus, although the insertion of five tandem copies of an ERE in association with the promoter generated an initial enhancement of luciferase expression after the first exposure to estrogen (17α-ethinyl-estradiol), the induction effect progressively declines despite the presence of high amounts of estrogens (Fig. 2D and E).
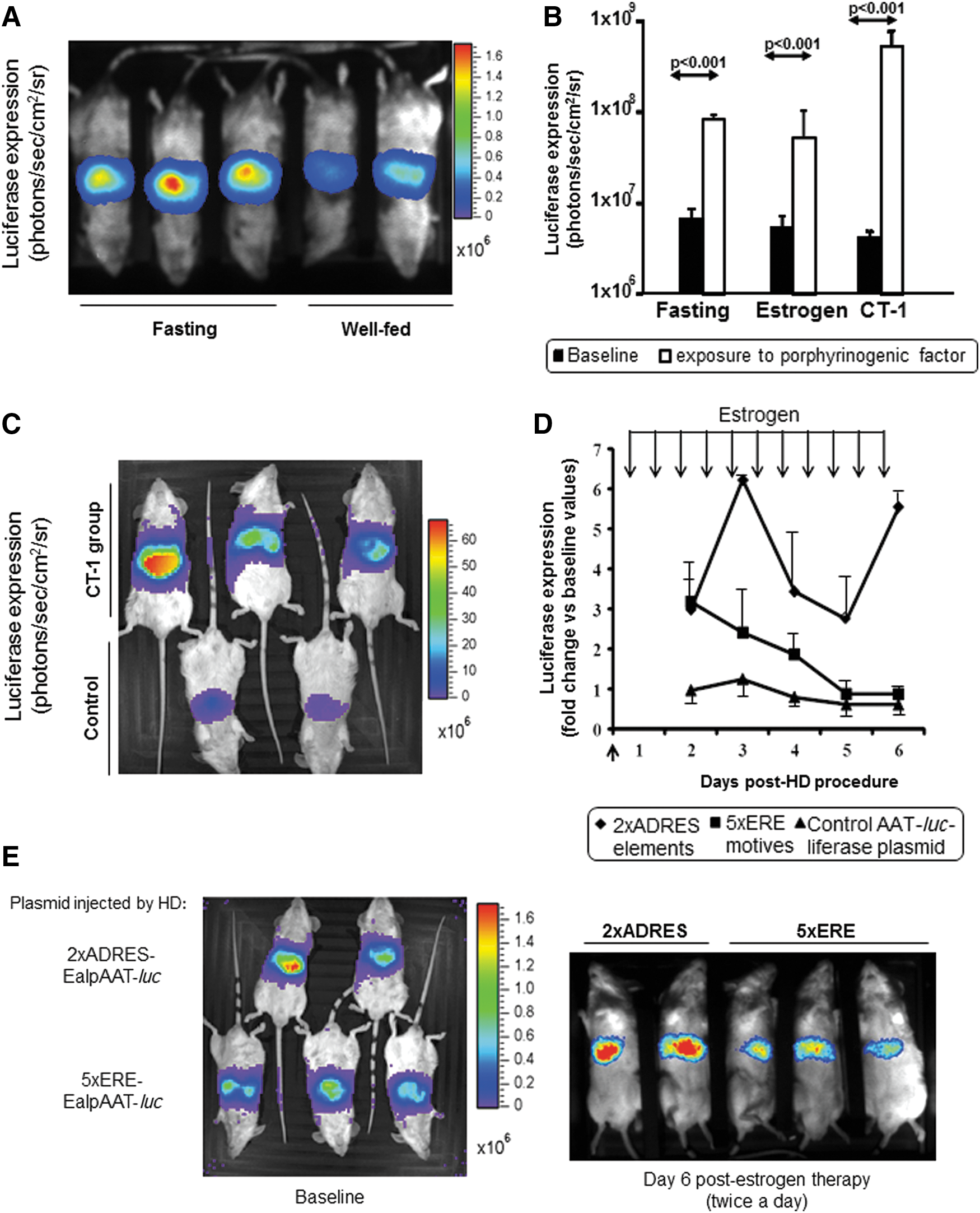
Effects of different porphobilinogenic stimuli on luciferase expression controlled by two ADRES elements upstream of the promoter.
Effect of the porphyrinogenic-responsive ADRES elements in an AAV backbone
The therapeutic effect of the incorporation of ADRES elements in a context of AAV vectors expressing PBGD was further assayed in female AIP mice. Groups of animals (n = 4) were intravenously injected with increasing doses of the control AAV vector (AAV2/8-EalpAAT-PBGD; Fig. 3, open columns) ranging from 7.1 × 109 to 3 × 1011 gc/kg. Another three groups of female AIP mice (n = 4) were intravenously administered with the ADRES vector (AAV2/8-2 × ADRES-EalpAAT-PBGD; Fig. 3, gray columns) at 7.7 × 109, 1.5 × 1010, and 2.3 × 1010 gc/kg. An extra group of untreated female AIP mice was also included (Fig. 3, black column). Thirty days after the vector administration, animals were placed in a metabolic cage to recover individual 24 h urine samples and to measure baseline porphyrin precursor excretions in AIP mice (upper limit of baseline excretion was represented as dotted line in Fig. 3A and B). In order to imitate a porphyria attack, animals were intraperitoneally injected with increasing doses of phenobarbital over four consecutive days. Porphyrin precursor excretions were measured following the last phenobarbital dose (Fig. 3A and B). As expected, the AIP mice that did not receive any AAV vector showed a high increase of both ALA (Fig. 3A) and PBG (Fig. 3B). Animals receiving the lowest doses were only partially protected against porphyrin precursor accumulation. Full protection, as evidenced by baseline excretion of both ALA and PBG levels, was obtained with a dose of 3 × 1011 gc/kg of the control constitutive AAV vector and a dose of 1.5 × 1010 gc/kg of the inducible vector carrying two ADRES sequences.
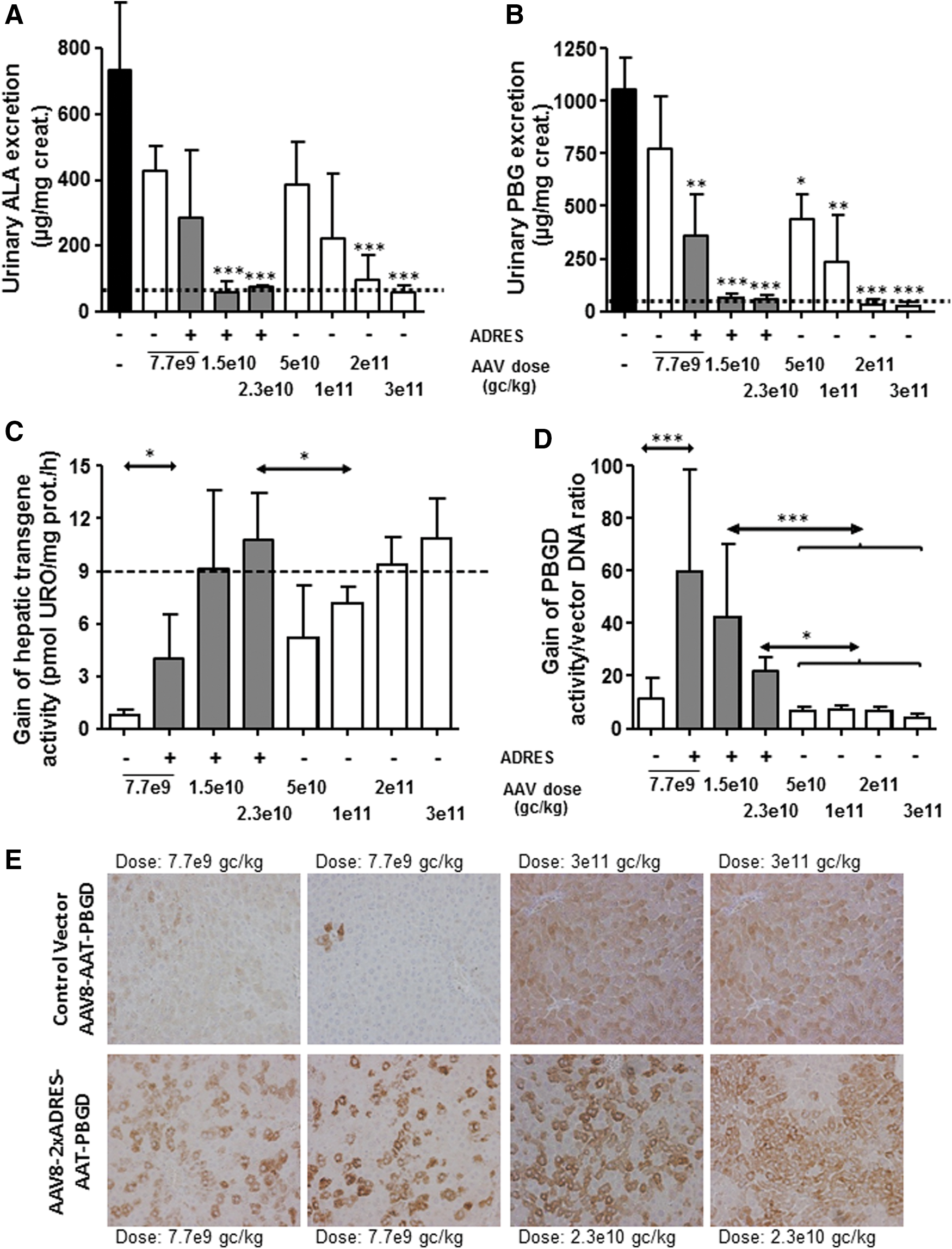
Dose effects of the incorporation of ADRES elements in an AAV vector carrying the human PBGD sequence in an experimental acute porphyria model. Therapeutic effect of AAV2/8-EalpAAT-PBGD (as control AAV vector) and AAV2/8-2 × ADRES-EalpAAT-PBGD viral vectors was comparatively tested in AIP mice 35 days after the administration of the indicated doses. Animals (n = 4 mice/group) were intraperitoneally treated with increasing doses of phenobarbital to imitate porphyria attacks. A group of AIP mice that were not injected and received no treatment with phenobarbital provided the baseline values. Urinary excretion of
The hepatic PBGD activity measured 24 h after the last phenobarbital dose showed that the gain of PBGD needed to normalize the hepatic PBGD in the mouse liver (Fig. 3C, dotted line) was obtained with a 10-fold lower dose of the vector carrying the two ADRES elements in tandem with the EalAAT promoter when compared to the control vector without ADRES elements (Fig. 3C). The gain of PBGD activity normalized by measurements of vector DNA content confirmed a dose-escalating abundance in the liver of transduced mice (Fig. 3D). Increased hepatic PBGD transgene expression in mice receiving the vector carrying two ADRES sequences was confirmed upon specific immunohistochemistry PBGD detection on formalin-fixed and paraffin-embedded liver sections from those mice (Fig. 3E). Thus, the effect of ADRES elements in an AAV backbone allowed the viral dose administered to be reduced while at the same time maintaining the protection against acute attack.
The inducible effect of two ADRES elements upon transgene expression was comparatively tested with the endogenous induction of hepatic PBGD mRNA in AIP mice exposed to a porphyrinogenic stimulus
The gain of exogenous PBGD mRNA expression induced after phenobarbital exposure was further assayed in female AIP mice. PBGD-deficient animals (n = 8/group) were intravenously injected with 2.3 × 1010 gc/kg of either AAV2/8 vectors carrying or not the two ADRES elements in tandem with the EalAAT promoter (Fig. 4A). Two extra groups of untreated female (AIP and wild type) mice were also included (n = 4). Thirty days after vector administration, four mice from each group were challenged with increasing doses of phenobarbital, and all animals were then sacrificed. Although all groups of mice showed similar DNA vector content in the liver (Fig. 4B), mice injected with the vector AAV-2 × ADRES-EalpAAT-PBGD showed a fivefold increase in exogenous PBGD mRNA levels when exposed to phenobarbital challenge (Fig. 4A).
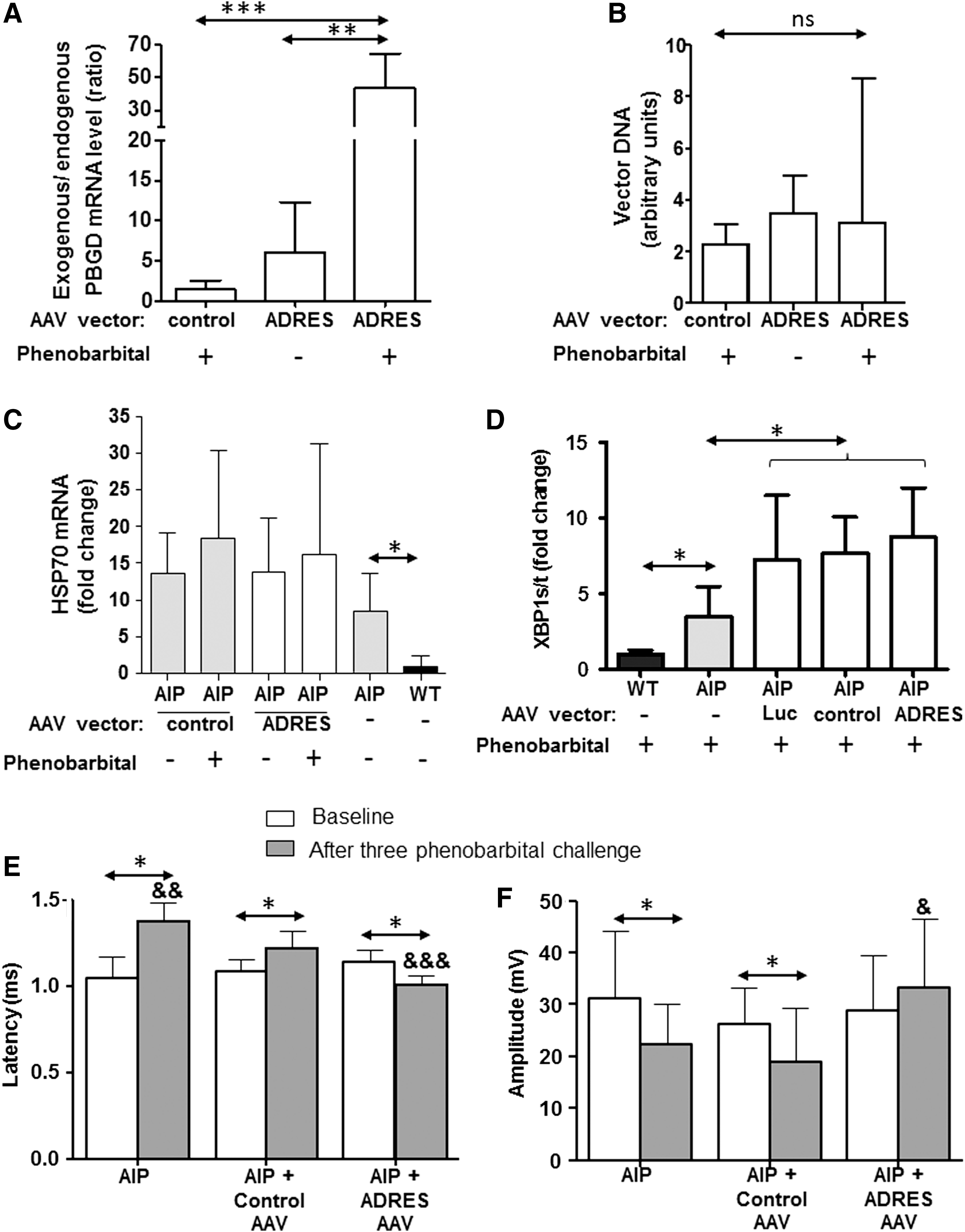
The insertion of two tandem ADRES elements into the promoter of an AAV vector led to enhanced PBGD transgene expression after the exposure to a porphyrinogenic stimulus. AIP mice treated with 2.3 × 1010 gc/kg of AAV2/8-2 × ADRES-EalpAAT-PBGD or AAV2/8-EalpAAT-PBGD were exposed to three doses of 90 mg/kg of phenobarbital (n = 4) and sacrificed 24 h later. A control group treated with the inducible vector but without phenobarbital exposure was also included. Liver samples were analyzed to measure
Since PBGD protein overexpression may compromise cytoplasmic protein folding homeostasis, HSP70 mRNA expression was analyzed as a measure of cytoplasmic stress in treated and untreated AIP mice (Fig. 4C). A higher expression of HSP70 mRNA implied disturbances in cytosolic folding machineries in AIP mice when compared to wild-type animals (Fig. 4C). As previously described, un-injected AIP mice showed a significant overexpression of HSP70 when compared to wild-type animals. However, the administration of AAV vectors with or without ADRES elements and the subsequent phenobarbital-induced overexpression of the PBGD did not alter HSP70 mRNA expression in AIP mice (Fig. 4C).
In the liver of AIP mice, deficiencies in protein folding in the lumen of the endoplasmatic reticulum probably cause the activation of an intracellular signaling pathway (the unfolded protein response) that initiates a transcriptional program to restore endoplasmatic reticulum folding capacity. 19,20 The unfolded protein response signaling, represented as the ratio of spliced XBP1 and total XBP1 mRNA expression, was increased in all groups of AIP mice injected with both constitutive and inducible vectors (Fig. 4D). As a control group, AIP mice were also injected with a control AAV vector expressing luciferase. This control group also increased the XBP1 ratio (Fig. 4D). To confirm that the overexpression of exogenous PBGD was not associated with the unfolded protein response, the correlation between XBP1 ratio and PBGD was analyzed in animals injected with increased doses of constitutive AAV expressing PBGD. No significant correlation was observed between those parameters in animals with a range of hepatic PBGD between 4 and 400 units (r = 0.015, not significant).
The therapeutic efficacy of the inducible construct was measured by the impact of recurrent acute attack on sciatic nerve function
Neurophysiological studies showed an increase in latency and a decrease in amplitude after repeated induction of acute porphyria attacks in this AIP mouse. 7 Latency (Fig. 4E) and amplitude (Fig. 4F) were measured before and 45 days after the administration of 1.5 × 1010 gc/kg of vectors carrying the inducible or the constitutive promoter. Three phenobarbital challenges were induced biweekly in those male AIP mice starting 2 weeks after vector injection. As shown in Fig. 3, a dose of 1.5 × 1010 gc/kg of the vector with the constitutive promoter did not protect against porphyrin precursor excretion. In line with this result, AIP mice injected with constitutive vector showed an increase in latency (Fig. 4E) and a decrease in amplitude values (Fig. 4F) compared to its baseline measurement. In contrast, the administration of the same dose of the new inducible vector protected the mice against the nerve dysfunction induced by consecutive attacks (Fig. 4E and F).
Discussion
PBGD gene therapy represents a promising therapeutic option for patients with AIP suffering recurrent attacks, despite the regular prophylactic administration of intravenous hemin. Recently, a first-in-human Phase I open-label liver-directed gene therapy clinical trial for AIP revealed that AAV-EalpAAT-PBGD administration was safe, but metabolic correction was not achieved at the doses tested (up to 1.8 × 1013 gc/kg). 9 Higher doses and/or more efficient vectors are needed to obtain therapeutic expression levels of the transgene. However, increasing the AAV doses could be a potential concern due to the immunogenicity associated to viral capsids. 21 Previous results in AIP mice strongly suggest that a high expression of PBGD was safe. 7 Clinical benefit is achieved after partial transduction because those hepatocytes overexpressing PBGD could metabolize the heme precursors produced by non-transduced neighboring cells. 15 Thus, the development of an inducible vector can significantly reduce the therapeutic dose for a given vector.
The combination of a transcriptional enhancer of the mammalian ALAS1 gene (ADRES elements) in the liver-specific EalAAT promoter of AAV vector supported a strong induction of transgene expression in the presence of the porphyrinogenic stimuli triggering the acute attack of porphyria. However, the introduction of the ADRES enhancer element may drive ectopic expression of the transgene. To minimize this ectopic expression, the AAV serotypes were carefully selected, choosing those with a narrow liver tropism. 22 An expression cassette was also designed with both enhancers and promoter sequences of hepatic genes. These hepatic elements assure liver specificity. 11 Finally, it is important to consider that the NR1I3 gene (also known as CAR, CAR1, MB67) shows a preferentially hepatocyte expression. 23
The ADRES element has been previously reported to contribute to the transcriptional activation of ALAS1 and certain P450 cytochrome genes in response to its interaction with nuclear receptors. 24 These nuclear receptors bind as homodimers or heterodimers to cognate response elements consisting of two hexameric half-sites. The sequence, orientation, and the distance between these half-sites account for the specificity of a given response element to a particular nuclear receptor dimer. This study cloned a 173 bp core ADRES element in the distal 5′-flanking region of the human ALAS1 gene (from −20918 to −20745 bp upstream the ALAS1 transcriptional start site). 16 This ADRES element included the predicted nuclear receptor DR4 and DR5 response elements, which bind the heterodimer nuclear receptor formed by CAR and pregnane X receptor (PXR) in the presence of 9-cis retinoic acid X receptor (RXR). The ability of the PXR and CAR to transactivate the 173 bp ADRES was previously demonstrated in HepG2 human hepatoma cells in response to a variety of drugs, such as phenobarbital (400 μM), propylisopropylacetamide (250 μM), rifampicin (10 μM), metyrapone (400 μM), or clotrimazole (10 μM). 16 In vivo studies in the AIP mice in this study confirm the role of ADRES elements in the strong transcriptional activation of the PBGD transgene when two tandem copies were operably linked to a liver-specific chimeric EalAAT promoter of a recombinant AAV gene therapy vector. Interestingly, the expression of the therapeutic transgene was induced in real time in response to a variety of endocrine and exogenous signals.
The main advantages of using endogenous ADRES sequences is that no chimeric factors are involved in the activation of the transgene and that activation occurs when it is most needed, that is, when porphyrinogenic factors increase ALAS1 transcription and precipitate acute porphyria attacks. Indeed, transgene overexpression was maintained throughout the whole period of exposure to porphyrinogenic factors (as reported with phenobarbital and estrogen exposure), as shown in mice injected with a plasmid carrying the luciferase marker gene. This is a significant difference when compared to other enhancer elements, such as EREs, where transgene induction disappeared, even though estrogen stimulus was maintained. Nevertheless, a drop in luciferase expression was observed in our system between days 3 and 5 (Fig. 2D). This behavior suggests a tachyphylaxis phenomenon after continuous estrogen stimulus. This sustained increase could be a pivotal point for women suffering recurrent acute attacks associated with hormonal factors.
The AAV vector carrying two ADRES tandem copies in phase with the promoter strongly induced transgene PBGD expression during the full period of exposure to porphyrinogenic factors. An intrahepatic overexpression of the PBGD protein, as obtained after PBGD gene therapy, is safe and does not cause disturbances in the folding machineries in continuous expressions >20-fold the endogenous PBGD 7 or higher levels sustained for >1 year in mice 19 and nonhuman primates. 25
The present results in AIP mice strongly suggest that the inclusion of ADRES elements into the AAV vector provides clinical benefit, even at a low dose, because the hepatocytes overexpressing high amounts of PBGD could metabolize the heme precursors produced by non-transduced neighboring cells. 7 In this way, full protection against phenobarbital-induced acute attack was obtained with a dose of 1.5 × 1010 gc/kg of the inducible AAV and with a dose of 2 × 1011 gc/kg of the control AAV (Fig. 3C). Thus, the new vector, including the ADRES enhancer sequences, could reduce the effective dose by 10 when compared to a control AAV vector carrying the same promoter and transgene. Accordingly, the exogenous PBGD mRNA level showed a 40-fold increase in mice injected with 2 × ADRES-EalpAAT-PBGD in response to phenobarbital exposure when compared with the control vector (Fig. 4A).
The clinical relevance of the inducible vector was measured by the impact of recurrent acute attack on sciatic nerve function. Neurophysiological studies regarding sciatic nerve conduction velocity in AIP mice demonstrated increased latency values associated to porphyrin precursor accumulation. 7 In this study, latency values were also longer for animals receiving a vector with constitutive promoter than the inducible one. The protective effect of the inducible AAV vector was more evident in the recorded amplitude values. Collectively, these results suggest that inducible vector protects against functional block of the compound muscle action potentials induced by porphyrin precursors generated in the liver from AIP mice.
Finally, the results confirmed that the human ADRES sequence is also recognized by murine nuclear receptors. Thus, the results obtained in mice may be translated to clinical trials.
In conclusion, these studies support the inclusion of two ADRES elements before the AAT promoter in gene therapy vectors for AIP as an enhancer element of therapeutic PBGD expression in the presence of the same factors that trigger the transcriptional activation of the endogenous ALAS1 gene and trigger the acute attack of porphyria. The insertion of two tandem ADRES elements in the promoter driving the expression of the PBGD transgene of an AAV vector allows the effective dose to be lowered by 10 while maintaining protection against an acute attack. Therefore, an AAV-PBGD vector carrying this regulatory element would be expected to increase therapeutic transgene expression markedly in clinical trials of AAV-mediated gene therapy for AIP or, analogously, for the other acute porphyrias.
Footnotes
Acknowledgments
We thank Sara Arcelus for her technical assistance. Drs. Unzu, Prieto and Ávila are acknowledged for their helpful discussion and scientific support. This work was supported in part by grants from the Spanish Fundación Mutua Madrileña, Spanish Fundación Eugenio Rodríguez Pascual, the organization Friends of the University of Navarra, the European VII Framework program—Project AIPGENE (grant FP7-Health-2010-261506), and Spanish Institute of Health Carlos III (FIS) co-financed by European FEDER funds (grant numbers PI09/02639, PI12/02785, and PI15/01951 and PI16/00668 funds). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Disclosure
No competing financial interests exist.