
Abstract
CD19-targeted chimeric antigen receptor (CAR) engineered T/natural killer (NK)-cell therapies can result in durable clinical responses in B-cell malignancies. However, CAR-based immunotherapies have been much less successful in solid cancers, in part due to “on-target off-tumor” toxicity related to expression of target tumor antigens on normal tissue. Based on preliminary observations of safety and clinical activity in proof-of-concept clinical trials, tumor antigen-specific messenger RNA (mRNA) CAR transfection into selected, activated, and expanded T/NK cells may permit prospective control of “on-target off-tumor” toxicity. To develop a commercial product for solid tumors, mesothelin was selected as an antigen target based on its association with poor prognosis and overexpression in multiple solid cancers. It was hypothesized that selecting, activating, and expanding cells ex vivo prior to mRNA CAR transfection would not be necessary, thus simplifying the complexity and cost of manufacturing. Now, the development of anti-human mesothelin mRNA CAR transfected peripheral blood lymphocytes (CARMA-hMeso) is reported, demonstrating the manufacture and cryopreservation of multiple cell aliquots for repeat administrations from a single human leukapheresis. A rapid, automated, closed system for cGMP-compliant transfection of mRNA CAR in up to 20 × 109 peripheral blood lymphocytes was developed. Here we show that CARMA-hMeso cells recognize and lyse tumor cells in a mesothelin-specific manner. Expression of CAR was detectable over approximately 7 days in vitro, with a progressive decline of CAR expression that appears to correlate with in vitro cell expansion. In a murine ovarian cancer model, a single intraperitoneal injection of CARMA-hMeso resulted in the dose-dependent inhibition of tumor growth and improved survival of mice. Furthermore, repeat weekly intraperitoneal administrations of the optimal CARMA-hMeso dose further prolonged disease control and survival. No significant off-target toxicities were observed. These data support further investigation of CARMA-hMeso as a potential treatment for ovarian cancer and other mesothelin-expressing cancers.
Introduction
O
Multiple studies in epithelial ovarian cancers 4 –7 have shown that the presence of tumor infiltrating lymphocytes (TILs) correlates with increased patient survival, suggesting that immune-based therapies could impact outcomes in this disease. The major immunotherapy platforms evaluated for ovarian cancer to date include vaccines, immune checkpoint antagonists, and adoptive T-cell therapies. 8 –10 One type of adoptive T-cell therapy utilizes chimeric antigen receptor (CAR) T cells—T cells engineered to express CARs—that specifically target tumor associated antigens (TAA). 11 CAR T cells provide both the antigen specificity of a monoclonal antibody and the antitumor activity of cytotoxic T cells in one product.
Multiple clinical trials have demonstrated that the adoptive transfer of CAR T cells specific for the CD19 cell surface antigen can result in significant disease regressions in leukemia patients. 12 An essential feature of these products is the long-term expression of CAR by the viral transduction of highly purified, activated, and expanded T cells. This requires a complex, inefficient, and costly manufacturing process that includes approximately 1–2 weeks of ex vivo T-cell expansion. 13 Furthermore, the stable expression of CAR may result in significant “on-target off-tumor” toxicity, particularly in solid tumors that express the antigen target. In the case of CD19+ malignancies, the deletion of normal CD19+ B cells can be clinically managed as needed by long-term intravenous (i.v.) immunoglobulin therapy to restore immunoglobulins in treated patients. 14 Additional significant toxicities associated with CAR T-cell therapy include tumor lysis syndrome, cytokine release syndrome (CRS), and neurotoxicities related both to CRS and to the conditioning regimen used. 15,16 These difficulties, coupled with additional challenges posed by the inefficient homing and migration of CAR T cells to the solid tumor microenvironment and its immunosuppressive milieu, have largely restricted the development of CAR T-cell immunotherapies to CD19-expressing hematologic malignancies. 17,18
Expanded T cells transfected with messenger RNA (mRNA) encoding CAR (CAR-T) have the potential to mitigate “on-target off-tumor” toxicities. Initial preclinical 19 –21 and human proof-of-concept studies in metastatic mesothelioma and pancreatic cancer 22,23 support the safety and antitumor activity of limited dosing with mRNA CAR-transfected and expanded T cells. In contrast to transduced CD19-CAR T cells, which are “living drugs” with the potential to expand in vivo and establish a memory response, mRNA-transfected CAR T cells are transient and do not persist, and thus would require multiple infusions for antitumor activity. 22 The short half-life of mRNA-transfected T cells may provide a prospective way to control potential “on-target off-tumor” toxicity by simply withholding further infusions of the cell product.
The possible advantages of mRNA CAR-transfected immunotherapies led to the further development of a streamlined, cGMP-compliant strategy for manufacturing peripheral blood mononuclear cells (PBMCs) transfected with antigen-specific mRNA CAR without prior expansion or activation, CARMA, as a potential platform for treating multiple cancers. Mesothelin was selected as the first target tumor antigen 24 to develop for several reasons. First, it is widely overexpressed by multiple solid cancers. 25 –28 Second, mesothelin has shown promise as a target for antibody-directed therapies. 29,30 Third, mesothelin-specific CAR T cells have shown evidence of safety and clinical activity in mesothelioma and pancreatic cancer in an early clinical trial. 23 However, the product in this proof-of-principle trial contained mouse mesothelin sequences, which could result in neutralizing antibodies to the cellular product. Thus, a CARMA product was developed utilizing human mesothelin sequences to decrease the potential for immunogenicity and allow for repeated administration in humans.
This study characterizes anti-human-mesothelin CAR mRNA transfected directly into unselected, unexpanded PBMCs that have not been activated (CARMA-hMeso) first in vitro, and then in vivo using a preclinical murine model of ovarian cancer. CARMA-hMeso demonstrated high viability and CAR-expression in vitro, with the ability to recognize and kill mesothelin-expressing tumor cells at low effector-to-target (E:T) ratios. There were no detectable differences in the killing of tumor cells when anti-human-mesothelin CAR mRNA was transfected into PBMCs or adherence-depleted PBMCs (peripheral blood lymphocytes [PBLs]), whether fresh or cryopreserved, or whether activated or not. In nude mice challenged with murine ovarian cancer cells transfected with human mesothelin, it was demonstrated that a single intraperitoneal (i.p.) injection of human CARMA-hMeso cells resulted in a dose-dependent inhibition of tumor growth, with longer survival compared to untreated control and CARMA-CD19 (irrelevant CAR)-treated groups. Weekly administration of CARMA-hMeso for 3 or 6 weeks improved disease control, resulting in longer survival compared to a single administration of CARMA-hMeso. No overt toxicities were noted following a single i.p. administration of up to 1 × 108 CARMA-hMeso cells, a single i.v. administration of up to 4 × 107 CARMA-hMeso cells, or a total of six weekly i.p. administrations of 5 × 107 CARMA-hMeso cells. Together, these results support further evaluation of the safety and efficacy of CARMA-hMeso cells for the treatment of ovarian cancer and other mesothelin-expressing solid tumors in humans.
Materials and Methods
Gene constructs used for electroporation
The CAR is schematically shown (Fig. 1A) and described below. All CAR molecules were cloned into the pcDNA3.1+ plasmid backbone (Thermo Fisher Scientific) and contained a CD8α signal peptide, defined single chain variable fragment (scFv), CD8 hinge/transmembrane domain (TM), and 4-1BB and CD3ζ intracellular domains; H and M designators indicate human and murine scFv, respectively:

The structure of three different antigen-specific mRNA chimeric antigen receptors (CARMA): two specific for mesothelin with human single chain variable fragment (scFv; CARMA-αhMeso-H) or murine scFv (CARMA-αhMeso-M), and one specific for CD19 with murine scFv (CARMA-αCD19-M). Isolated peripheral blood mononuclear cells (PBMCs) that have not been further selected, expanded, or activated are electroporated with CARMA using the cGMP-compliant MaxCyte GT® System™, and then either cryopreserved or used fresh in experiments.
The sequences of the scFv components used in the construction of the CAR cassettes were obtained from published sources. The Meso-H scFv sequence was obtained from the 2007 Cancer Letters publication, 31 the Meso-M scFv sequence was obtained from its U.S. Patent, and the αCD19-scFv sequence was obtained from FMC63-28z NCBI. The sequence of the final construct was confirmed by sequencing (GenScript).
mRNA synthesis
mRNAs encoding different CARs were produced by in vitro transcription using the XbaI-linearized DNA template. mRNA was synthesized in accordance with the manufacturer's protocol provided with the mMachine mMessage T7 Ultra Kit. mRNA was confirmed by 1% agarose gel, as recommended by the kit. The mRNA was isolated from the gel, formulated in double-distilled water with 0.1 mM of EDTA, aliquoted, and cryopreserved at −80°C.
Cells and cell lines
Healthy volunteer donor leukapaks obtained from 10–15 L blood volume processing were purchased from Key Biologics, and shipped overnight on wet ice packs. Density gradient centrifugation using Ficoll Paque Plus (cat# 17-1440-03; Thermo Fisher Scientific) was performed to isolate the PBMCs. PBLs were obtained by collecting cells in suspension after 2 h culture of isolated PBMCs in five-tier cell factory (cat# 05-539-098; Thermo Fisher Scientific).
The K562-Meso cell line was developed by transfecting wild-type K562 cells with a plasmid encoding human mesothelin and the G418 resistance gene (cat# RC202532; Origene). K562 cells expressing human mesothelin were then selected by treatment with G418. Single cell clones were further selected by limiting dilution assay. The selected clones were expanded and assayed for expression of human mesothelin using purified rat monoclonal IgG2A α-human mesothelin as primary (cat# MAB32625; BD Biosciences) and α-rat IgG-PE as secondary antibodies (cat# F0105B; R&D Systems, Inc.). One clonal line was selected and designated as K562-Meso.
OP-1 cells were a gift from Dr. Dario Campana. 32 The cells were used to measure the target cell killing efficiency of αCD19CAR-M-transfected PBLs for the nonspecific control used in the animal studies. Wild-type and human mesothelin-engineered murine ovarian cancer cell line Defb29 Vegf-luc/Hmeso cells, all with GFP integration, were developed, as previously described. 33
Transfection of mRNA CAR
PBLs or PBMCs were washed in MaxCyte's Electroporation (EP) Buffer (cat# B201-100) at >20 × volume relative to the volume of the cell pellet. Washed cells were resuspended in MaxCyte EP buffer at 5 × 108 cells/mL and kept on ice. mRNA was added to this cell suspension to a final concentration of 200 μg/mL. The resulting cell and mRNA mixture was immediately transfected using the MaxCyte GT® System™, a cGMP-compliant electroporation system. Post transfection, the cells were allowed to recover for 20 min at 37°C and were subsequently incubated in RPMI-1640 culture medium supplemented with 10% fetal bovine serum (FBS) and 2 mM of L-glutamine for an additional 30 min. The resulting cells were then either cryopreserved or used immediately for functional studies.
Cryopreservation and thawing of cells
Cells were cryopreserved in CryoStor® freeze media (Biolife Solutions) at a concentration of 1–4 ×108 cells/mL, stored at −80°C overnight, and transferred to long-term storage in the vapor phase of a liquid nitrogen freezer. Cryopreserved cells were quickly thawed in a 37°C water bath. Immediately post thaw, 1 × Dulbecco's phosphate-buffered saline (PBS) supplemented with 0.1% human serum albumin was added to the cells: (1) initially dropwise for the half volume of thawed cells, (2) subsequently dropwise for 1 × volume of the thawed cells, and (3) then to a total of 10 × volume of the thawed cells. Resulting cells were centrifuged and re-suspended in either PBS for direct injection in the animal studies or in culture medium of RPMI-1640 supplemented with 10% FBS and antibiotics if used for in vitro studies.
Detection of CAR expression
The detection of CAR expression was performed using biotinylated rabbit anti-human IgG (H + L) Fab (code# 309-065-082; Jackson ImmunoResearch Laboratories, Inc.) as the primary antibody for αMesoCAR-H and biotinylated rabbit anti-murine IgG (H + L) Fab (code# 115-605-072; Jackson ImmunoResearch Laboratories, Inc.) as the primary antibody for αMesoCAR-M or αCD19CAR-M. After the cells were washed, they were stained with a secondary streptavidin antibody conjugated to PerCP (BD Pharmingen), and fluorescence-activated cell sorting (FACS) analysis was performed to detect CAR expression. In some studies, CD3-FITC and CD8-PE (BD Pharmingen) were used in addition to the secondary antibody for detecting the CAR expression.
In vitro cell killing assay
The target cell killing assay has been previously described. 34 Briefly, to evaluate killing of the K562-Meso target cells, the target cells were first loaded with calcein-AM (cat# C1430; Molecular Probe). After washing three times, 100 μL target cells at 105/mL (104 cells per well) were mixed with 100 μL human effector PBL at concentrations to reach the desired E:T ratios in duplicate culture wells of a U-bottom 96-well plate. After culturing the E:T cell mixtures overnight at 37°C in 5% CO2, cells were assayed for cytotoxicity using 20 s of time-controlled FACS acquisition and analysis. To evaluate killing of the target Defb29 Vegf-luc/Hmeso human ovarian cancer cells, the cells were plated in a flat-bottom 96-well plate 1 day prior the addition of human PBL effector cells. Cells from three wells were trypsinized for counting to determine the cell number per well just before the addition of effector cells. Effector cells (control or hMeso-transfected Defb29 Vegf-luc) at the desired E:T ratio were placed in duplicate wells. Following overnight culture, the mixture was rinsed with PBS and detached from the wells by addition of 100 μL trypsin to dissociate the viable target cells from the wells. Following detachment, 100 μL culture medium was added to neutralize the trypsin, and cells were assayed for cytotoxicity using 20 s of time-controlled FACS acquisition and analysis. The ratio of decrease in viable number of target cells in effector cultures versus control cultures was used to calculate the cytotoxicity using the equation: %Lysis = (N NonEffector – N Mix) × 100/N NonEffector, where N NonEffector is the viable target cell number without effector and N Mix is the viable target cell number with effector.
Biodistribution study
The biodistribution of CARMA-hMeso cells was evaluated following i.p. administration in NSG mice. NSG mice were purchased from Jackson Laboratories. On day 0 (D0), NSG mice (N = 5) received a single i.p. injection of 5 × 107 cryopreserved control PBLs or CARMA-hMeso cells. The biodistribution of control and CARMA-hMeso cells in the peripheral blood was evaluated by PE anti-human CD3 (BD Pharmingen) and FITC anti-human CD45 (BD Pharmingen) by FACS analysis on D1, D2, D4, and D7. On D8, the mice were euthanized, and the biodistributions of control and CARMA-hMeso cells in peritoneal washings and the spleen were evaluated by FACS analysis.
Animal tumor model studies
A series of four animal studies was performed, using transfected and cryopreserved CARMA-hMeso cells from different donors.
Tumor cell line
The Defb29 Vegf-luc/Hmeso ovarian cancer cell line was generated by transducing human mesothelin and luciferase into Defb29 Vegf-luc mouse ovarian tumor cells and was maintained in RPMI 1640 supplemented with 10% fetal calf serum and 100 IU/mL of penicillin, as previously described. 33 The expression of mesothelin on Defb29 Vegf-luc/Hmeso cells (ID8 H-Meso) and Defb29 wild-type control cells (ID8-WT) was confirmed by using primary rat anti-human mesothelin antibody (cat# MAB32625R&D Systems) with secondary anti-rat IgG–PE (cat# F0105B; R&D Systems). FACS (FACSCalibur; BD Biosciences) analysis was performed to detect the mesothelin expression.
Adoptive cell therapy experiments
Nude mice were challenged with 3 × 105 or 1 × 104 Defb29 Vegf-luc/Hmeso cells via i.p. injection on D0. On D3, the baseline luminescence intensity from the mouse abdomen was imaged using a Xenogen IVIS 200 (In Vivo Imaging System). The CARMA-hMeso cells were thawed and given as a single injection of a range of cell doses on D4 by i.p. or retro-orbital (r.o.) administration. Mice were then imaged weekly. In some experiments, mice were treated with CARMA-hMeso cells i.p. or r.o. weekly for 3 or 6 weeks. The survival data were recorded. The dose of tumors, the schedule of imaging, and the day of the treatment were based on previous publications. 33,35
All animal experiments were approved by the Johns Hopkins University Animal Care and Use Committee. Athymic nude mice were purchased from the Jackson Laboratory. Data are presented as mean ± standard deviation.
Results
Comparison of CARMA-mMeso- and CARMA-hMeso-mediated tumor cell lysis activity in vitro
CARMA-mMeso selected, activated, and expanded T cells have been reported to have antitumor activity.
36
CARMA-hMeso is a cellular immunotherapy product that utilizes CAR mRNA encoding a human origin scFv instead of a mouse origin scFv (known as CARMA-mMeso) as part of the CAR, which may have the advantage of reduced immunogenicity. The manufacturing process for CARMA-hMeso was fine-tuned to reduce time-consuming ex vivo manipulation. To confirm that CARMA-hMeso and CARMA-mMeso have similar antitumor activity against mesothelin-expressing target cells, their ability to kill human K562 cells expressing human mesothelin was evaluated in an in vitro cytotoxicity assay. Human mesothelin expression on K562-hMeso target cells was confirmed by flow cytometry (data not shown). The CAR T-cell viability was similar following transfection with either mouse mesothelin CAR mRNA or human mesothelin CAR mRNA (Supplementary Fig. S1A; Supplementary Data are available online at
CARMA-hMeso cells induce target-specific killing of human mesothelin expressing mouse ovarian tumor cells in vitro
The ability of CARMA-hMeso cells to recognize and kill Defb29 Vegf-luc/Hmeso tumor cells was evaluated in vitro. The expression of human mesothelin on Defb29 Vegf-luc/Hmeso cells was confirmed via FACS analysis (data not shown). When Defb29 Vegf-luc/Hmeso cells were co-cultured in the absence of effector cells or in the presence of control PBL effector cells (PBLs that were not transfected with hMeso targeted CAR mRNA), the Defb29 Vegf-luc/Hmeso cells continued to grow in monolayers (Supplementary Fig. S2A). However, when cultured in the presence of CARMA-hMeso cells, the Defb29 Vegf-luc/Hmeso cells were lysed, and the only remaining cells in the wells were CARMA-hMeso cells with small cell aggregates consistent with an activated T-cell morphology. The antigen-specific killing of Defb29 Vegf-luc/Hmeso cells, but not Defb29Vegf-luc cells, by CARMA-hMeso was incrementally increased across a range of E:T ratios (Supplementary Fig. S2B). No difference was found in the extent of cell lysis with directly transfected PBL or T cells that were expanded and activated by CD3/CD28 beads and then transfected (Supplementary Fig. S3).
Product characterization of transfected resting PBLs
For the clinical product, one manufacturing run using a single patient leukapheresis generates multiple cryopreserved aliquots of cellular product to enable repeat administrations. Therefore, the effect of cryopreservation was tested on CARMA-hMeso products by comparing the same CARMA product, one fresh (without cryopreservation) and the other cryopreserved. No significant differences in viability, transfection efficiency, or the phenotype of cell populations of the product were observed (Supplementary Fig. S4A). Furthermore, fresh and cryopreserved CARMA-hMeso cells showed comparable ability to kill mesothelin-expressing tumor cells in vitro (Supplementary Fig. S4B).
Biodistribution of CARMA-hMeso cells following i.p. injection in NSG mice
The biodistribution of CARMA-hMeso cells was evaluated following i.p. administration in NSG mice (Fig. 2). NSG mice were utilized for the biodistribution study because nude mice retain an active innate immune system that could result in increased clearance of CARMA-hMeso cells. Following a single i.p. injection, CARMA-hMeso cells were observed in the peripheral blood beginning on D2 (<1%) and increased through D7 (approximately 3%), the last time point evaluated. On D8, approximately 80% of the CARMA-hMeso cells were detectable in peritoneal washings, and approximately 4% were detectable in the spleen. In vitro data show that CAR-hMeso expression is not detected by D7. So, the additional persistence of the autologous PBLs that have lost the CAR-hMeso expression is not likely to serve any killing function. The biodistribution for control PBL and CARMA-hMeso cells was similar, confirming the MaxCyte transfection process does not negatively impact cell trafficking in NSG mice.

Biodistribution of CARMA-hMeso cells following intraperitoneal (i.p.) injection in NSG mice. Cryopreserved control peripheral blood lymphocytes (PBLs) or CARMA-hMeso cells at 5 × 107 were injected i.p. into NSG mice on day 0 (D0).
Intraperitoneal injection of CARMA-hMeso cells delays ovarian tumor growth in vivo
To evaluate the activity of CARMA-hMeso cells in vivo, nude mice were challenged i.p. with 3 × 105 Defb29 Vegf-luc/Hmeso cells. On D3 before treatment with CARMA-hMeso cells, there was no difference in tumor burden as reflected by the luminescence intensity among groups (Fig. 3). In mice treated with nonspecific CAR T cells (CD19 CAR) at doses of 1 × 107 and 1 × 108 cells, there was a very strong luciferase signal in the abdomen that was indistinguishable from the luciferase signal observed in mice treated with PBS alone. Mice injected with 1 × 107 CARMA-hMeso cells showed similar tumor signals in the abdomen, which indicated the treatment had no effect at this low cell dose. In contrast, mice injected with 1 × 108 CARMA-hMeso cells showed significantly lower tumor signals in the abdomen at all time points. To determine if it is necessary to incubate the CARMA-hMeso cells in vitro to allow time for hMeso CAR expression prior to injection, CARMA-hMeso cells were placed in a 37°C incubator with 5% CO2 for 3 h prior to injection. There was no difference in efficacy between CARMA cells recovered in the incubator and CARMA cells injected immediately after thawing. Thus, all remaining animal studies were performed using CARMA-hMeso cells immediately post thaw. Notably, it was observed that Defb29 Vegf-luc/Hmeso produces very aggressive tumors, and mice challenged with 3 × 105 tumor cells were dead after just 1 month. Due to the high lethality of Defb29 Vegf-luc/Hmeso cells, mice were challenged with a lower dose of tumor cells (1 × 104) for the remaining experiments.
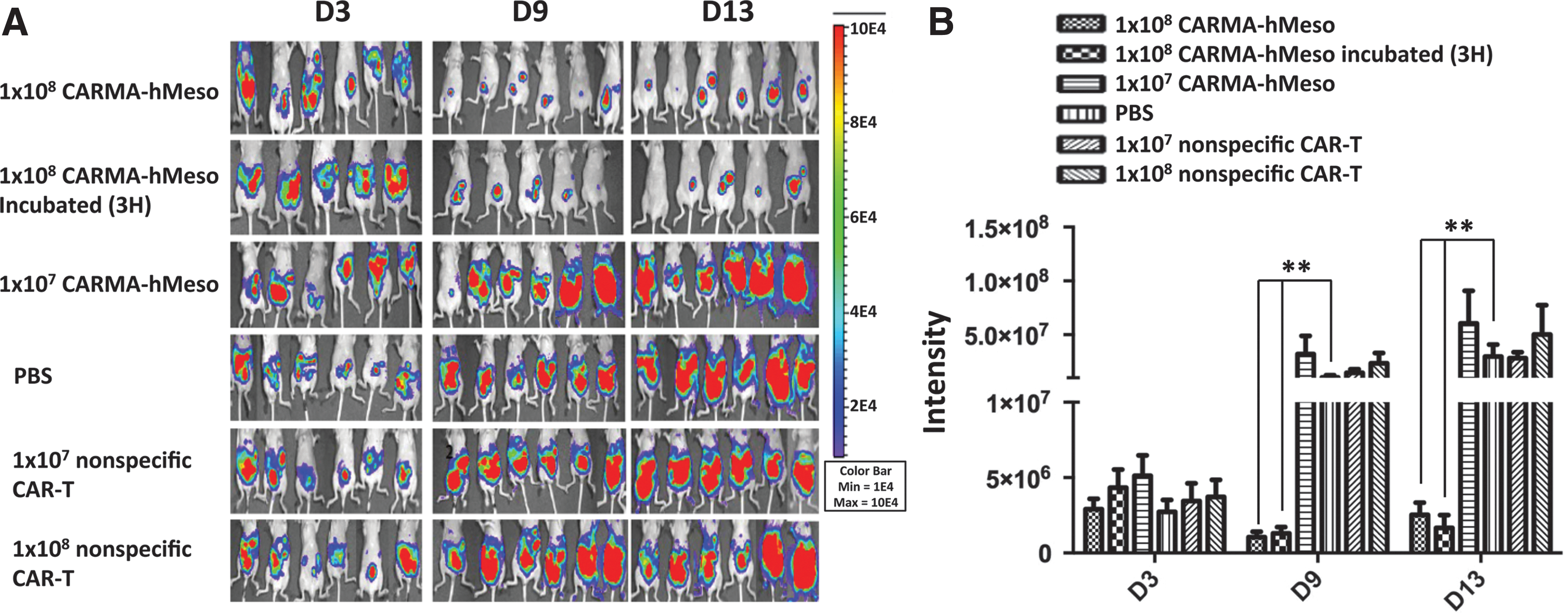
Injection i.p. of CARMA-hMeso cells controlled tumor growth in a mouse hMeso-expressing ovarian tumor model in nude mice. Mouse ovarian tumors expressing hMeso were initiated by i.p. injection of 3 × 105 Defb29 Vegf-luc/Hmeso into nude mice on D0. On D3, the baseline luminescence intensity from the mouse abdomen was imaged using IVIS. The CARMA-hMeso cells were thawed and injected i.p. on D4, and the mice were imaged once a week.
CARMA-hMeso cells were also prepared from PBLs following removal of adherent cells (e.g., monocytes and macrophages) by culturing PBMC in a five-tier cell factory for 2 h. To determine if antitumor effects could also be observed when adherent cells are present, PBMCs were transfected with the CARMA-hMeso-PBMCs and evaluated for antitumor activity in mice. Mice were challenged i.p. with 1 × 104 Defb29 Vegf-luc/Hmeso ovarian tumor cells on D0. On D4, PBS or 5 × 107 CARMA-hMeso-PBMCs were injected i.p. Mice treated with CARMA-hMeso-PBMCs showed stronger antitumor effects than mice treated with PBS control (Supplementary Fig. S5).
CARMA-hMeso cells have antitumor activity when given i.p. but not i.v.
The antitumor activity of CARMA-hMeso cells was evaluated following i.p. or i.v. (via r.o.) administration (Fig. 4). When compared to the control group, mice treated with both 1 × 107 and 1 × 108 CARMA-hMeso cells by i.p. injection had significant antitumor effects at all time points, and injection of 1 × 108 cells was more effective than the injection of 1 × 107 cells. Treatment i.v. was evaluated by injecting 2 × 107 or 4 × 107 CARMA-hMeso cells r.o. The tumor burden of mice treated with either dose r.o. was similar to control untreated mice, suggesting that the administration of meso-CAR T cells i.v. was ineffective. To evaluate this further, 2x107 CAR T cells r.o. were injected for three consecutive days. Repeat i.v. administration did not result in either a lower tumor burden or a survival benefit in treated mice.
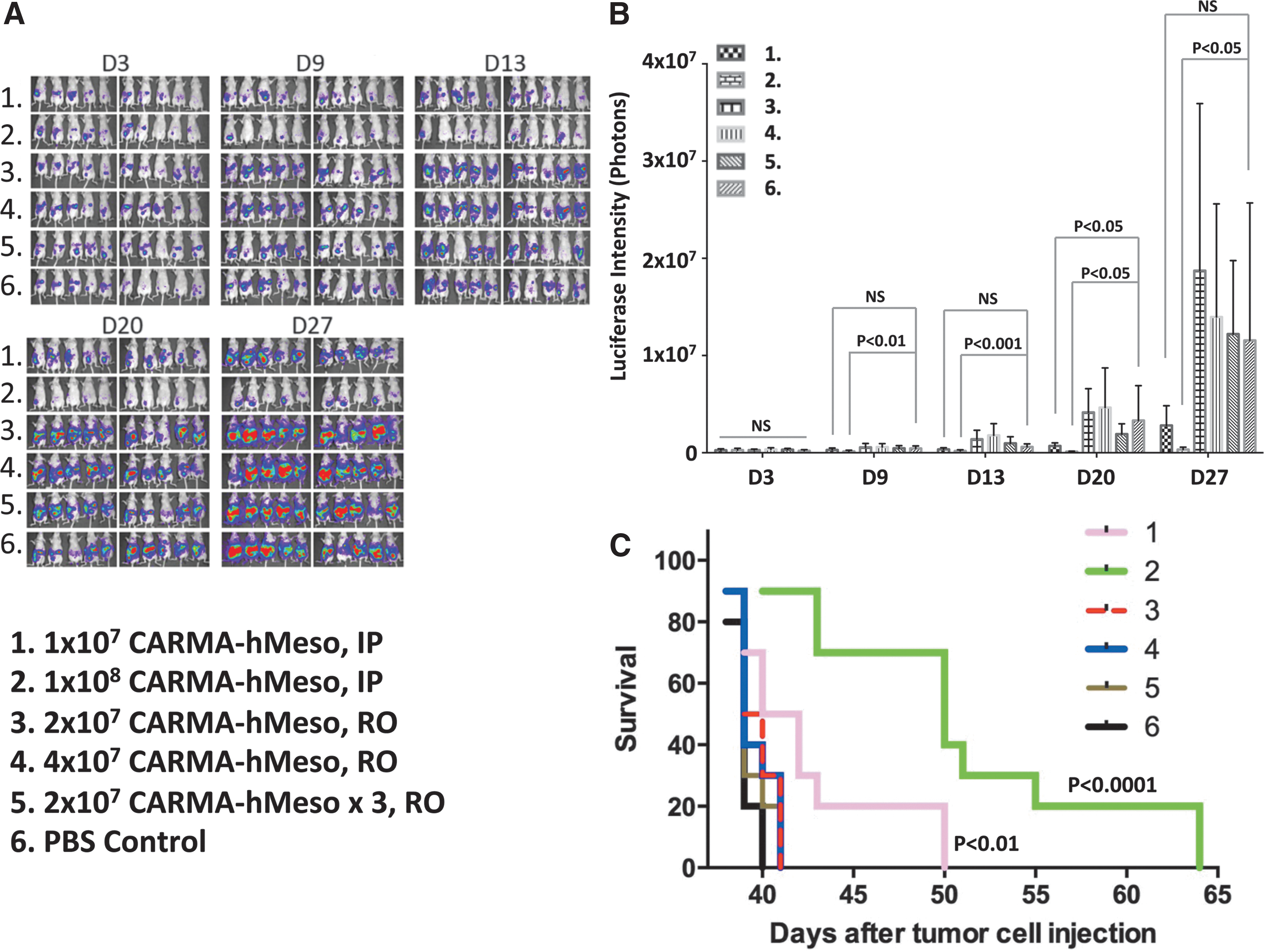
Intravenous injection of CARMA-hMeso cells did not have treatment effects on ovarian cancer in vivo. Defb29 Vegf-luc/Hmeso cells (1 × 104) were injected i.p. into nude mice on D0. On D3, the baseline luminescence intensity from the mouse abdomen was imaged using IVIS. The CARMA-hMeso cells were thawed and injected on D4 by i.p. or retro-orbital (r.o.) injection with different cell numbers, and treated mice were imaged once a week. Two days after the first injection, the continuous injection was administered daily for a total of three injections. The non-treatment group was the control group.
Weekly i.p. administration of CARMA-hMeso cells was more effective than a single i.p. administration
A single i.p. injection of CARMA-hMeso cells effectively controlled ovarian tumor growth in vivo, but this treatment effect was short-lived. To overcome the limitation of transient expression of the CAR on T cells, the study tested whether repeated i.p. injections of CARMA-hMeso cells would result in more effective ovarian tumor growth control. Nude mice were challenged i.p. with 1 × 104 Defb29 Vegf-luc/Hmeso ovarian tumor cells on D0. Tumor-bearing mice were then given 1 × 107 or 5 × 107 CARMA-hMeso cells i.p. on D4, D11, and D18. A weekly administration schedule was selected based on in vitro data demonstrating target cell lysis activity through approximately 7 days of in vitro culture (data not shown). Mice treated with a single injection of 1 × 107 or 5 × 107 CARMA-hMeso cells were injected on D4. Weekly administration of 5 × 107 CARMA-hMeso cells resulted in better tumor control than a single injection of 1 × 107 or 5 × 107 CARMA-hMeso cells (Fig. 5A). Furthermore, six weekly i.p. injections of 5 × 107 CARMA-hMeso cells was more effective in controlling tumor growth than three weekly i.p. injections of 5 × 107 CARMA-hMeso cells (Fig. 5B). These results indicate that serial weekly i.p. injections of CARMA-hMeso cells more effectively decreased tumor burden and improved survival than a single administration of these cells, and that higher doses of CARMA-hMeso cells were more effective at controlling ovarian tumor growth in vivo than lower doses of CARMA-hMeso cells.
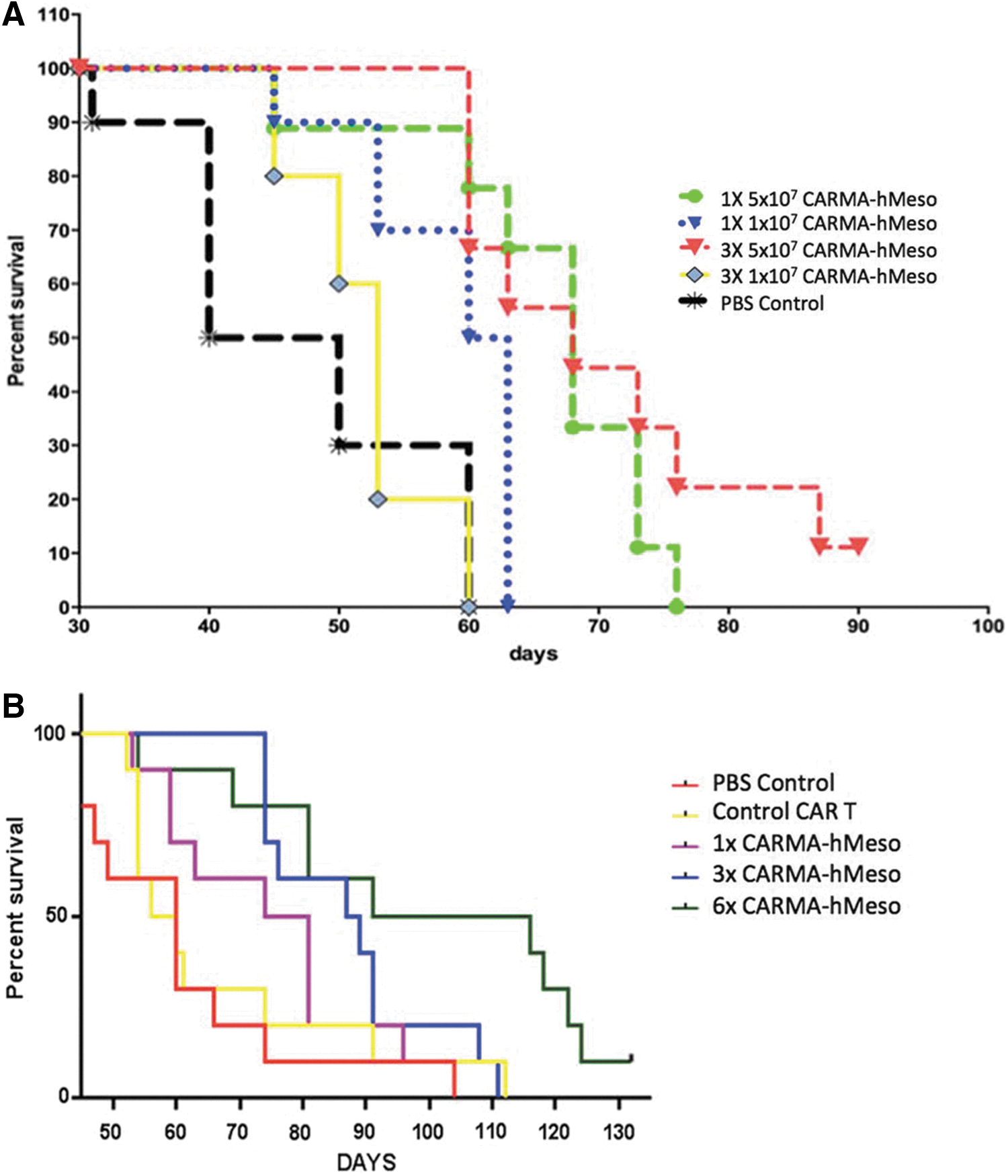
Repeated i.p. injection of CARMA-hMeso cells showed better activity in a mouse hMeso-expressing ovarian tumor model compared to a single injection. Mouse ovarian tumors expressing hMeso were initiated by i.p. injection of 1 × 104 Defb29 Vegf-luc/Hmeso cells into nude mice (N = 10/group) on D0.
Discussion
Here, the development of a cellular immunotherapy product, CARMA-hMeso, is described. CARMA-hMeso is produced by transiently transfecting PBLs with CAR mRNA produced in vitro from a plasmid containing the following nucleotide sequences: CD8α signal peptide, human origin scFv targeting human mesothelin, CD8 hinge/transmembrane domain, and the 4-1BB and CD3ζ intracellular domains. CARMA-hMeso T cells in the PBLs bind to mesothelin-expressing cells, activating T-cell dependent antitumor activity through signaling via CD3ζ and the costimulatory molecule 4-1BB. The study demonstrates that this cellular product can lyse ovarian tumor cells expressing human mesothelin in vitro, and control the growth of murine ovarian tumor cells expressing human mesothelin in vivo in a mouse model. Six key findings critical for product development and clinical translation emerged from these studies. First, the fully human CAR specific for human mesothelin (CARMA-hMeso) has similar lytic activity as the mouse CAR specific for human mesothelin (CARMA-mMeso), with the advantage that fully human CARMA-hMeso should be less immunogenic. Second, expression of the CAR is transient in vitro, potentially providing a means of mitigating potential “on-target off-tumor” toxicity in vivo in humans. It was not possible to evaluate toxicity in the mouse models, since CARMA-hMeso does not recognize murine mesothelin. Third, there were no differences in the composition and potency of PBLs transfected fresh and cryopreserved, or PBLs transfected fresh and evaluated without cryopreservation. Fourth, there was no apparent impact of adherent cells on the ability of CAR-transfected PBLs to control tumor growth in vivo. Fifth, weekly i.p. administrations of CARMA-hMeso control tumor growth more effectively than a single i.p. administration. Finally, i.p. administration of CARMA-hMeso effectively controlled tumor growth, but i.v. administration did not.
The use of transiently transfected CAR mRNA has the potential advantages of reduced toxicity to normal tissue triggered by retroviral transfections, and reduced target-specific off-tumor toxicity due to the transient pattern of CAR expression. 37 The platform developed here also confers the ability to target solid tumors based on antigen expression, and also provides a cost-effective, automated, cGMP-compliant clinical/commercial manufacturing process. There have been studies of mRNA CAR-transfected cells specific for a range of targets other than mesothelin, including CD20, 38 CD33, 39 NKG2D, 40 folate receptor, 41 and GD2. 42 Modified cells specific for these targets are known to induce hematopoietic stem cell toxicity or other organ-specific toxicities. These and other challenges associated with optimizing mRNA CAR approaches to permit meaningful clinical responses, as demonstrated in a study comparing mRNA CAR versus viral vector CAR delivery targeting the GD2 antigen, 42 suggest that systematic scientific studies are warranted.
Here, some of these challenges were specifically addressed by deploying a nonviral approach applicable to multiple solid cancers using mRNA CAR transfection to permit prospective control of “on-target off-tumor” toxicity. Collaborating with investigators at the University of Pennsylvania and the National University of Singapore, translation of mRNA CAR transfection has been enabled into ex vivo selected, activated, and expanded T cells/natural killer cells into preclinical models and human clinical trials targeting multiple tumor-associated antigens for both hematological malignancies and for solid cancers. 40,43 –47 Preliminary results from these studies indicate that mRNA CAR is a safe, nonviral strategy for engineering tumor-targeted immunotherapy. This approach has resulted in measurable antitumor activity in animals and preliminary evidence of clinical activity in man. 22,23
Human proof-of-concept trials of mRNA CAR transfected into selected, activated, and expanded T cells (mRNA CARTs) have provided supportive evidence for the use of CARMA. In these studies, a dose of 1 × 109 cells (>95% CAR+) was well-tolerated and did not result in any detectable “on-target off-tumor” toxicity. CAR protein expression was detectable in vivo but decreased incrementally over time due the dilutional impact of cell division. In some patients, a measurable reduction in tumor burden was observed about 2 months after cell administration. 22,23 Preclinical studies have shown that that repeat dosing of mRNA CARTs results in improved antitumor activity, and may promote long-term survival. 20,45,46
The initial human clinical trials used a CAR molecule that included mouse scFV anti-human mesothelin sequences (mMeso), which share homology and cross-react with human-mesothelin (hMeso). Initial evaluation of repeat dosing of mRNA-CART-mMeso in a patient who experienced a reduction in tumor burden after one dose of cells resulted in anaphylaxis, 48 likely due to human anti-mouse antibody that developed as a result of the initial treatment. Multiple cycles of treatment could thus not be evaluated in this mRNA-CART-mMeso trial. To mitigate the risk of developing anti-drug antibodies and facilitate repeat dosing, a fully human scFv that targets human mesothelin for CARMA-hMeso product manufacture was selected and tested. Notably, even though CARMA-hMeso uses a human mesothelin construct, no significant differences in K562-Meso tumor target cell lysis were observed when CARMA-mMeso or CARMA-hMeso effector cells were evaluated in an in vitro tumor killing assay.
The CARMA product platform capitalizes on the ability to transfect mRNA encoding CAR molecules rapidly into PBMCs and the deployment of a cGMP and regulatory compliant automated closed system for cell processing. 49 With the leukapheresis product in hand, the entire manufacturing process can be completed in a few hours, generating multiple cryopreserved cell aliquots suitable for repeat dosing. The process developed does not require use of viral vectors or extensive ex vivo manipulations and expansion over multiple days. The product characterization studies here showed that there were no statistically significant differences in viability, transfection efficiency, or ability to cryopreserve transfected CARMA-hMeso cells. In addition, fresh and cryopreserved CARMA-hMeso cells showed comparable cytotoxicity for tumor cells in vitro.
Most of the in vivo studies presented here used CARMA-hMeso cells prepared from PBLs following removal of adherent cells with a 2 h panning step. However, one study evaluated PBMCs transfected directly with the CARMA-hMeso-PBMCs. In this study, CARMA-hMeso-PBMCs also demonstrated in vivo antitumor activity. These findings suggest that it may be possible to decrease the overall cell processing time for future CARMA products further, but additional head-to-head comparisons between CARMA-hMeso-PBLs and CARMA-hMeso-PBMCs from the same donor preparation are needed to confirm these findings.
The CARMA strategy was used here for the treatment of human mesothelin-expressing mouse ovarian tumor cells in nude mice. Mesothelin was selected as the target antigen for CARMA-hMeso based on its high expression in human ovarian cancers and low expression in normal human tissues. Although data for other immunotherapies targeting mesothelin suggest that this target is safe, 50 “on-target off-tumor” toxicity could not be evaluated, since CARMA-hMeso does not recognize mouse mesothelin. CARMA-hMeso therapy resulted in decreased tumor burdens and increased survival in treated mice, with larger doses and repeat administrations resulting in better tumor growth control and improved survival. The need for repeat dosing to improve efficacy likely reflects the transient pharmacodynamics of CARMA-hMeso.
The mechanism of action of CARMA is more similar to the “hit and run” mechanism of ADCC-enhanced monoclonal antibodies or bi-specific antibodies such as BiTEs 51 than to the long-lived mechanism of conventional viral-vector transduced CD19 CARTs, 15 which relies on creating a niche for engraftment and expansion of CARTs. 16 Accordingly, CARTs frequently have associated toxicities related to “on-target-off-tumor” effects, the induction of CRS, neurotoxicity, and/or other changes that may require lymphodepleting regimens for patient conditioning. 14 CARMA-hMeso may also require patient conditioning or lymphodepleting regimens, 46 but it is possible that less aggressive regimens could be effective, since the modified cells intrinsically have a limited life-span. This requires further study.
Although no benefit was observed following i.v. administration of CARMA-hMeso in the mouse tumor model, the tumors were confined to the abdomen, which may not fully reflect metastatic ovarian cancer in humans where tumor cells may be observed outside the peritoneum. Thus, both i.p. and i.v. administration may be important to achieve optimal responses in humans. 22,23 Furthermore, the use of nude mice instead of NSG mice may have contributed to the lack of effects because nude mice have an active innate immune system that may have helped clear the CARMA-hMeso cells delivered i.v. The biodistribution study in NSG mice did show distribution of CARMA-hMeso cells into the blood, spleen, and distant lymph nodes at 4 days after a single i.p. administration.
CARMA represents a unique platform for the rapid manufacture and delivery of tumor-targeted immunotherapies using fully human CAR mRNA transfected into PBLs, potentially allowing for repeat cycles of treatment. With the CARMA platform, multiple aliquots of CARMA can be rapidly manufactured within hours from a single leukapheresis using a cGMP and regulatory compliant, automated, closed system, resulting in a consistent, high-quality product in a practical time frame and at reasonable cost. These data suggest that the manufacturing of CARMA-hMeso cells is feasible, and can produce multiple doses of tumor antigen-specific CARMA for repeated administration. Together, these in vitro and animal studies support the evaluation of i.p. CARMA-hMeso cells as a treatment for patients with platinum-resistant ovarian cancer.
Footnotes
Acknowledgments
We thank Dario Campana for providing the OP-1 cells. We also thank Jeremy Foote and Andrew Wang for assistance with the figures. Writing assistance was supported by MaxCyte, Inc., and provided by Biologics Consulting. Financial support for these studies was provided by MaxCyte, Inc., and NCI P30 CA006973. This work was presented as a poster at the 2017 annual meeting of the American Association for Cancer Research.
Author Disclosure
These studies were funded by MaxCyte, Inc. C.F.H. and L.A.E. received financial and material support from MaxCyte, Inc. L.L., A.V., C.A., P.N., R.S., and M.V.P. are employees of and have personal financial interests in MaxCyte, Inc.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.