
Abstract
Oncolytic viruses (OVs) are quickly moving toward the forefront of modern medicines. The reward for the decades of research invested into developing viral platforms that selectively replicate in and lyse tumor cells while sparking anticancer adaptive immunity is presenting in the form of durable therapeutic responses. While this has certainly been a concerted global effort, in this review for the 25th anniversary of the European Society of Gene and Cell Therapy, we focus on the contributions made by European researchers. Research centers across Europe have held central roles in advancing OVs, from the earliest reports of coincidental viral infections leading to antitumor efficacy, to advanced mechanistic studies, and now through Phase I–III trials to imminent regulatory approvals. While challenges still remain, with limitations in preclinical animal models, antiviral immune clearance, and manufacture restrictions enforced by poor viral yields in certain cases, the field has come a very long way in recent years. Thoughtful mechanistic integration of OVs with standard of care strategies and other newly approved therapies should provide potent novel approaches. Combination with immunotherapeutic regimes holds significant promise, and the ability to arm the viral platform with therapeutic proteins for localized expression at the tumor site provides an opportunity for creating highly effective synergistic treatments and brings a new age of targeted cancer therapeutics.
Introduction
T
Europe has played a dominant role in the history of OV research. From its infancy, reports came from different parts of the continent of anticancer responses following virus infection. One of the first records was from an Italian group led by De Pace in 1912, demonstrating tumor regression of cervical uterine carcinoma following inoculation with an attenuated rabies vaccine. He was probably the first person worldwide to attribute tumor regression to cytolytic virus activity: “un'azione citolitica sulle cellule neoplastiche.” This work sparked much interest and led to the initiation of animal studies, resulting in the earliest demonstration of viral oncolysis, as assessed in mouse models. 2 In the 1950–1960s, a flurry of cases were published. Patients with leukemia were reported to enter remission following treatment with Epstein–Barr virus (administration of glandular fever patient serum), 3 and later positive responses with Langat and Kyasanur Forest disease virus were observed, 4 to name but a few. In Latvia, a wild-type nonpathogenic enteric cytopathic human orphan type 7 (ECHO-7), known as Rigvir, was the first OV worldwide to be granted national regulatory approval. While not sufficient to meet the regulation standards across Europe or in the United States, Rigvir has been used in Latvia since 2004 to treat melanoma. 5,6 Whether naturally oncotropic, genetically attenuated for normal tissue, or engineered to target and replicate specifically in tumor cells, the European research community has always been at the forefront of OV development. The adenovirus Ad5dl1520 was the first genetically modified OV to be trialed in patients and was found to have an excellent safety profile, with significant anticancer effects in injected tumor nodules. This agent, commonly known as the “Onyx virus,” was pioneered by Onyx Pharmaceuticals in Richmond, California. It was later sold to Shanghai Sunway Biotech and gained regulatory approval for clinical use in China in 2005. However, the very first clinical trials were actually performed in Glasgow, United Kingdom, under the stewardship of Ian Ganly and Stan Kaye. 7
As the field advances, more complex treatment regimens are being explored. Due to their distinct mechanisms of action and generally excellent safety profiles, OVs are undergoing extensive investigation in combination with both traditional chemotherapies and novel immunotherapies. OVs hijack tumor cell protein production machinery for amplification of new viral particles, and this mechanism can often be harnessed for in situ high-level expression of an additional therapeutic protein encoded within the virus genome. This ability to genetically “arm” the virus platform with another biologic enables its targeted expression within (and secretion from) infected tumor cells, thereby avoiding delivery-related systemic toxicities. This approach affords many possibilities for combining therapeutic agents with mechanistic synergies. In addition, some viruses can act as sensitizing agents to DNA damage-inducing cytotoxic drugs, 8 or as immune stimulators to enhance the efficacy of immune checkpoint blockers 9,10 or bispecific T-cell engagers. 11 In parallel to recent breakthroughs in the cancer immunotherapy field have come major strides in the efficiency of genome editing techniques such as clustered regularly interspaced short palindromic repeat (CRISPR) technologies. CRISPR-mediated engineering of virtually any virus strain with relative ease to generate attenuated, cancer selective, targeted OVs is now becoming achievable. 12 A London-based group has successfully explored CRISPR/Cas9 approaches to simplify the engineering of vaccinia virus (∼190 kb) recombinants, enabled the efficient incorporation of transgenes, 13,14 and designed a comprehensive set of CRISPR guide RNAs targeted to each of the viral genes to allow rapid gene knockdown and the study of individual gene functions. 15 Simplifying the manipulation of such large virus constructs and increasing the speed at which new recombinants can be engineered should dramatically accelerate research progress.
A continuous theme in the OV field is the close partnership between university and industry, and the ability to build quickly on one another's findings. These close links between European companies and academia may go some way to explaining how Europe has shown such strength in this field. This review will report on the major classes of OVs under investigation and, within each section, focus on the contribution of the European community to their advancement into clinical practice (Fig. 1, Table 1).
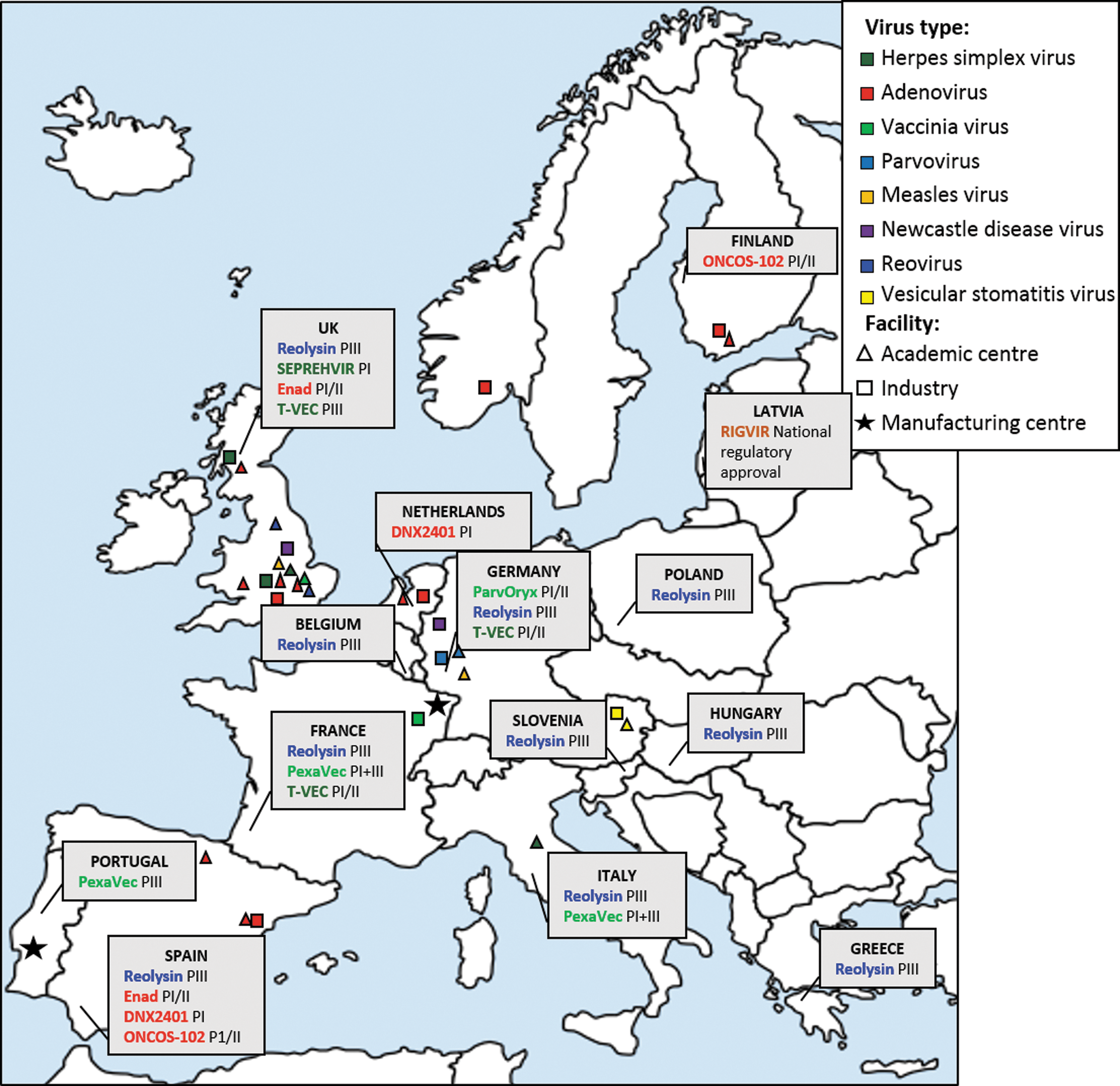
Major centers for oncolytic virotherapy across Europe. The map highlights the countries partaking in clinical trials for oncolytic viruses. Data obtained from
Summary of a selection of recently completed or ongoing clinical trials in Europe
n/a, non-applicable; HSV-1, herpes simplex virus type 1; GM-CSF, granulocyte macrophage colony-stimulating factor; Ad, adenoviruses; VV, vaccinia viruses; PV, parvovirus.
Herpes Simplex Virus
Herpes simplex virus (HSV) type 1 is an enveloped double-stranded DNA virus and the basis of the first widely approved OV in the Western world, Amgen's Imlygic. Although HSV research has been a global effort, several of the key steps along the way, not least the development of Imlygic, took place in Europe. 1,16 –20
The oncolytic potential of HSV was first explored in the early 1990s with the study of mutant strains that could only replicate in dividing cells and were inactive in most normal and nondividing cells. One of the earliest strains, “1716”, was identified in Glasgow. This agent lacked neurovirulence in mice but could be grown in tissue culture. 18 It had a functional deletion in ICP34.5, a gene essential for supporting virus replication in post-mitotic cells by allowing protein synthesis that would otherwise be shut off by the host antiviral response mediated through protein kinase R (PKR). 21,22 In animal models, intracranial administration of HSV1716 into mice harboring brain tumors was well tolerated and prolonged survival. 23,24 Subsequently, HSV1716 was approved by the British Gene Therapy Advisory Committee for safety studies in humans with recurrent malignant glioma. 19 Across several trials, clinical use of HSV1716 (later called Seprehvir® under stewardship of Virttu Biologics) was shown to be both feasible and safe.
One limitation of ICP34.5 deletion mutants is a tendency to attenuate virus activity to some extent in cancer cells compared to the wild-type virus. To compensate, it is possible to make other changes in the HSV backbone to enhance cancer cell lysis. HSV US11 is a tegument protein gene product usually expressed late on during infection and incorporated into the capsid. It is capable of intercepting PKR, and early expression can compensate for some of the attenuating elements of ICP34.5 deletion. Mutations leading to early expression of US11 were found to improve oncolytic activity without compromising safety. 25 One way to achieve early US11 expression is to delete ICP47. This brings US11 under control of an early promoter. In addition, deletion of ICP47 is potentially a beneficial mutation for oncolytic immunotherapy because the protein otherwise blocks presentation of antigens on MHC1. Allowing improved antigen presentation (of both cancer antigens and virus antigens) should increase the immunogenicity of infected tumor cells, encouraging an effective immune response while further attenuating virus persistence in normal tissues.
These elements were brought together in the clinical isolate HSV-1 strain “JS1” to create JS1/34.5-/47-, 26 which was to become the backbone for ONCOvex, later called Imlygic (talimogene laherparepvec [T-VEC]). The choice of a fresh clinical isolate as the basis for an OV should not be overlooked. Laboratory-passaged viruses tend to become attenuated over time, whereas circulating strains maintain full virulence. Introducing selective mutations in the most virulent strains has the potential to provide the greatest therapeutic index. JS1/34.5-/47- was further augmented by the expression of granulocyte macrophage colony-stimulating factor (GM-CSF) to enhance the immune response in the vicinity of lysed tumor cells. In animal studies, expression of GM-CSF facilitated immunologic clearance of non-injected tumors, whereas the control virus (lacking GM-CSF expression) was only active in tumor nodules directly injected. 26
In 2002, the UK's Genetic Therapy Advisory Committee approved ONCovex for clinical evaluation, although the first trial results were not published until 2006. 27 The initial dose escalation trial was designed to assess safety and feasibility of intra-tumoral administration of doses ranging from 1 × 106 to 1 × 108 plaque forming units (pfu)/mL. The most common side effect (and encouragingly so) was local erythema and inflammation. This local inflammation was more pronounced in seronegative patients, consistent with the hypothesis that the events were virus driven. The dose escalation was followed by a patient cohort that settled on a preferred regime of a 1 × 106 as the first dose, followed 3 weeks later by repeating doses of 1 × 108 pfu/mL. The idea behind this regime was to minimize side effects, particularly in seronegative individuals. Biological responses measured included the presence of virus, expression of GM-CSF, and an observed flattening of some of the injected lesions. There were no complete or partial responses by conventional criteria, but altogether this was an encouraging start for an initial safety assessment. The antibody status of the patients did not appear to impact on any of the biological measures of activity, as may be expected following direct intra-tumoral administration.
Using the dose regime determined in the first study, a Phase II trial was initiated in patients with stage III and IV melanoma who were not amenable to surgery. 28 Of the 50 patients enrolled, eight patients had a complete response and a further five had a partial response. Most of these responses were durable, lasting more than 7 months and up to 31 months. In some patients, both injected and uninjected lesions responded, indicative of an immunologic response. Consistent with other immunologic interventions, some patients experienced apparent tumor progression prior to an eventual response.
The following Phase III trial, which led to Food and Drug Administration approval of T-VEC, enrolled 436 patients with unresectable stage IIIB, IIIC, and IV melanoma. This open-label study randomized patients at a ratio of 2:1 to receive intra-tumoral T-VEC or subcutaneous GM-CSF. In this trial, the durable responses were reported to be 16.3% for T-VEC and 2.1% for GM-CSF. The median time to treatment failure was delayed in the T-VEC arm at 8.2 months relative to GM-CSF arm with 2.9 months. The median survival did not reach a significance threshold (hazard ratio = 0.79; p = 0.51) with the numbers enrolled, although many commentators conclude it was very close.
The European dimension to this work is very strong, but it has been a concerted global effort in a scientific endeavor that made this a clinical reality. The initial trial took place in the United Kingdom, bringing together components and understanding of HSV-1 biology from all over the world. The technology was initially developed by a British Biotech company (BioVex), through the most challenging initial stages, before finally resting with Amgen to deliver the large Phase III trial and product approval.
Not resting on this success, the founder of BioVex, Robert Coffin, has established another company to develop the oncolytic approach further. It is interesting to note that with this new company, Replimune, the technology development side at least is based in Europe.
Adenovirus
Adenoviruses (Ad), a large family of non-enveloped linear double-stranded DNA viruses, have remained one of the most frequently employed OV types through the years. Their popularity relies on their ease of genetic manipulation and manufacture, detailed knowledge of their structure and infection profile, as well as their potent oncolytic effects. Much of the research has centered around the prototype Ad type 5 (Ad5), often attenuating this strain with E1A 29 –32 or E1B deletions (e.g., Ad5dl15207) to endow cancer specificity. A range of high-profile genetically modified Ad OVs have been developed in European centers, and this review describes some of the most clinically advanced Ad platforms. Some of the agents have been engineered for tumor-enhanced activity using tumor-associated promoters to regulate key virus genes, while others have been attenuated to make them dependent on the tumor phenotype for full replicative activity.
An example of an Ad regulated by a tumor-associated promoter is VCN-01, a product of the Institut Català d'Oncologia Barcelona spinout company VCN Biosciences. The virus was engineered to replicate selectively in tumors with a defective retinoblastoma (Rb) pathway (E2F-1 promotor controlling E1A expression), and modified to incorporate an integrin-binding motif Arg-Gly-Asp-Lys (RGDK) in the fiber for enhanced tumor targeting. 33 The virus encodes secreted PH20 hyaluronidase, an enzyme to degrade hyaluronan, a high molecular weight structural component of the tumor extracellular matrix that acts as a dense barrier to drug penetration and has been implicated in cancer metastasis. 34 Preclinical work using cell lines, mice, and Syrian hamster models has demonstrated a favorable safety profile and strong efficacy against a range of cancer types, including pancreatic cancer, retinoblastoma, osteosarcoma, and glioblastoma. In an academic trial (now closed) ICOVIR5, an earlier version of VCN-01 (lacks hyaluronidase expression) was injected systemically in 12 metastatic melanoma patients in a dose escalation up to 1 × 1013 vp. Dose-limiting hepatic toxicity established the recommended Phase II dose at 3.3 × 1012 vp. Detection of virus in biopsies indicated that tumor targeting is feasible by the intravenous (i.v.) administration route. However, a lack of antitumor efficacy supports the use of more potent Ads. Using VCN-01, there are two Phase I dose escalation clinical trials ongoing in patients with advanced solid tumors. These are designed to investigate the safety and tolerability of three intra-tumoral injections of VCN-01 (ID NCT02045589) or a single i.v. injection of VCN-01 (doses ranging from 1 × 1011 to 1 × 1013 vp/patient; ID NCT02045602) alone or in combination with nab-paclitaxel or gemcitabine. Pre-existing antibodies to Ad5 were assessed prior to therapy, and only patients with a titer <1/320 were included for the i.v. delivery trial. Viral activity was evident at well-tolerated doses (minor side effects consisting of transient ’flu-like symptoms, and one episode of dose-limiting toxicities in 1/23 patients at the highest vp). Increased tumor infiltration of cytotoxic CD8+ T cells was also observed. 31 Recent preclinical strategies have highlighted the potential and flexibility of OV arming by encoding cytokines 35,36 and other immune modulatory genes to redirect viral cytotoxic T-cell responses toward tumor clearance. 11 ICOVIR-15K was engineered to express a bispecific T-cell engager (BiTE) targeted to the epidermal growth factor receptor (EGFR). 11 Once secreted from the infected cancer cells, the BiTE linked EGFR on the cancer cells directly to nearby CD3+ T cells, resulting in T-cell activation, proliferation, and EGFR-targeted killing in in vitro and in vivo models. Such an approach represents a promising therapeutic combination and may warrant further investigation in a trial setting.
Following a licensing agreement with VCN Biosciences, ORCA Therapeutics B.V. is developing its lead oncolytic candidate ORCA-010 for clinical trials in treatment-naïve prostate cancer patients. The Ad5 backbone has an E1A deletion, mutant E3/19K, and a RGD insertion in the fiber knob, and was demonstrated to be highly effective in a range of solid tumors (prostate, ovary, lung), as assessed by cell line, primary cell, and in vivo assays. 37 After recently securing funding for the GMP production of ORCA-010, Phase I/II testing is expected to commence in the near future. The company is also investigating expressing the tumor suppressor p53 from their OV platform to augment the oncolytic effects and enhance cancer cell death. 38
A Helsinki-based research group led by Prof. Hemminki is working toward the clinical development of another oncolytic Ad5 candidate, this time expressing interleukin (IL)-2 and tumor necrosis factor alpha (TNF-α; TILT-123), with the aim of enhancing adoptive T-cell therapy. Similar to above, the virus contains an E2F promoter and a deletion in the Ad E1A gene, enabling selective replication in cells defective in Rb signaling. In this case, the Ad5 fiber knob domain was swapped for that of Ad3 to improve tumor cell uptake. 32 Data suggests that this Ad5/Ad3 cytokine expressing virus altered the tumor microenvironment to prolong T-cell persistence and proliferation, and ultimately improved adoptive tumor infiltrating lymphocyte therapy in Syrian hamsters and T-cell receptor transfer in murine models of melanoma. 39,40 Phase I clinical trial data using a similar Ad5/Ad3-derived platform but expressing GM-CSF (ONCOS-102 undergoing development by the Norwegian company Targovax) in combination with chemotherapy suggests good tolerability following intra-tumoral injection, with indications of clinical efficacy and immune stimulation. 41 Building on this, recently, several Phase I/II trials have commenced testing ONCOS-102 against melanoma (NCT03003676), mesothelioma (NCT02879669), and advanced peritoneal malignancies (NCT02963831).
As an alternative to Ad5-based OVs, Enadenotucirev (EnAd, formerly ColoAd1), a chimera of group B Ad types 11 and 3, was developed by Seymour and Fisher et al. at the British spinout company Psioxus Therapeutics, in collaboration with the University of Oxford and Hermiston and Kuhn at Schering AG. EnAd was generated by directed bioselection, a non-prejudicial approach in which a pool of Ads were passaged on colon carcinoma cells under conditions encouraging viral recombination. In this manner, the biology of the cancer cells determined which virus variants showed greatest potency. 42 As several “hallmarks of cancer” and “hallmarks of virus infection” overlap, viruses were able to exploit disrupted cancer pathways, making some virus genes redundant in tumors and leading to a loss of genes that were complemented by the tumor phenotype. This enabled isolation of a potent and highly selective recombinant virus. 42,43 Extensive safety studies using primary human cells and tissues highlighted EnAd's specificity for tumor cells, finding at least 100-fold less viral genomes in normal cells and no release of infectious virions. 43 Importantly EnAd retains its oncolytic activity for carcinoma cells in the presence of whole human blood, likely due to the low levels of neutralizing antibodies against the Ad11-derived capsid, a quality that contrasts to its highly sero-prevalent Ad5 counterpart. 44 This was a major contributing factor in the decision to prioritize EnAd for clinical development.
An ever-growing body of evidence suggests OV efficacy is a combination of direct cell lysis and induction of host anticancer immunity. To contribute to the latter, preclinical data demonstrated EnAd kills cancer cells in an proinflammatory fashion, with upregulation of damage-associated molecular patterns (DAMPs) such as HMGB1 and HSP70, linked to an influx of calcium, increased membrane localized calreticulin, and release of ATP. 45 These data are encouraging, as high levels of such markers are associated with the induction of an adaptive immune response. Early Phase I trials were performed to examine the optimal dosing, administration schedule, and the route of delivery in metastatic carcinoma patients. 46,47 Trial data suggest a regime of three consecutive i.v. injections of EnAd (6 × 1012 vp/dose) on days 1, 3, and 5 achieved optimal circulation kinetics of viral particles and was well tolerated. Tumor biopsies resected post i.v. treatment provided evidence of viral replication and recruitment of CD8+ T cells, indicating successful systemic delivery. Arming strategies with immunomodulatory agents to exploit EnAd's potential further is an area of active investigation. Recently, Psioxus Therapeutic Ltd. has teamed up with Bristol–Myers Squibb to assess the utility of combining EnAd with the PD1 inhibitor nivolumab for the treatment of patients with metastatic or advanced epithelial tumors, and results from this trial are eagerly awaited.
Parvovirus H-1
Parvovirus H-1 (H-1PV) is a non-enveloped linear single-stranded rodent DNA virus (∼5 kb) capable of replicating in neoplastic but not normal cells. The oncolytic effects are predominately mediated by the viral nonstructural protein (NS1), which is involved in the dysregulation of cell cycle, stress response, and viral life cycle. 48,49 H-1PV is effective at low multiplicities of infection and has activity against a range of tumor types, including, but not limited to, glioblastoma, 50 pancreatic ductal adenocarcinoma, 51 and lymphoma. 52 One of the earliest trials using H-1PV in Europe took place in France in 1993 and consisted of dose escalation of repeated intra-lesional H-1PV injections (1 × 108-1 × 1010 pfu) in patients with a range of different solid cancers. This trial led by Le Cesne et al. reported a good safety profile and viral genomes in injected and distant tumor sites indicative of systemic stability and targeting. 53
Unlike many other OVs, H-1PV can permeate the blood–brain barrier, making glioblastoma a particularly attractive target for parvovirus development. Encouraging data in preclinical rat models have been produced, showing complete regression of both rat and human glioma xenografts without any inflammation or toxicity to normal brain tissue following intracerebral and i.v. delivery. 50 Specific targeting following delivery via a noninvasive i.v. route makes repeated administration and access to multiple tumor sites feasible, although higher doses may be required. Based on the work of Prof. Jean Rommelaere et al. in Germany, a Phase I/II clinical trial was initiated in 2011 to study the effects of H-1PV in patients with recurrent glioblastoma multiforme (GBM; ParvOryx, NCT01301430). The trial was designed with two treatment arms. In the first group, the virus was injected both intra-tumorally and at time of resection into the walls of the tumor cavity. In the second group, H-1PV was delivered i.v. followed by into the tumor cavity at resection on day 10. 54 Results from the trial found ParvOryx to be well tolerated, with no signs of central nervous system pathology or dose-limiting toxicities. 55 The virus penetrated the blood–brain barrier, and using immunohistochemistry and fluorescence in situ hybridization techniques, intra-tumoral viral spread and replication in tumor tissue were observed at doses of 1 × 106–5 × 109 pfu. 56 Importantly, both antiviral and antitumor immune responses were demonstrated. In a follow-up compassionate use study led by Dr. Karsten Geletneky with six of the patients from the initial trial in whom tumors had recurred, 5 × 108 pfu of ParvOryx was re-administered followed by no treatment (1/6), chemotherapy (1/6), or the anti-angiogenic drug bevacicumab (5/6). 57 The combination of virus with bevacicumab improved survival times in patients with recurrent GBM compared to previous treatments, implying a potential synergistic effect between the two therapeutic agents, and this combination is undergoing further investigation. Due to the immune stimulatory effects of ParvOryx, another subset of recurrent patients were treated with the virus i.v., or intra-tumorally and i.v., followed by bevacicumab and the PD-1 blocker nivolumab. 58 The three patients also received valproic acid, and two were re-irradiated prior to administration of ParvOryx. In this combinatory compassionate use trial, patients experienced transient flu-like symptoms following virus injection. At 4–8 weeks following therapy, patient tumors regressed, as assessed by magnetic resonance imaging, and there was an improvement in clinical responses. While patient numbers were small, the results using ParvOxyx for GBM are very promising, and hence it is also under clinical investigation for other targets, with Phase I/II trials (NCT02653313) for metastatic pancreatic cancer currently recruiting.
Vaccinia Virus
Vaccinia viruses (VV) comprise a family of enveloped double-stranded DNA viruses, of which several strains are under investigation as anticancer agents, including the smallpox vaccine Wyeth strain, Lister, and the more virulent Western Reserve strain. Unique among DNA viruses, VV replication occurs entirely in the cytoplasm. Due to its genome size (∼192 kb), it can accommodate the insertion of large transgenes for enhanced therapeutic efficacy and “armed” VV-based OVs are progressing steadily through Phase I–III trials for multiple indications in both Europe and the United States.
Developed by SillaJen (formerly Jennerex) in collaboration with the French biopharmaceutical company Transgene, Pexa-Vec (JX594/pexastimogene devacirepvec) is an engineered Wyeth strain of VV with thymidine kinase (TK) inactivation and “armed” with GM-CSF. The TK inactivation limits viral replication to cells with high endogenous levels of phosphorylated thymidine, typical of rapidly proliferating cancer cells. In addition to the cellular TK levels, selectivity of this OV for tumor cells is reported to be dependent on EGFR/Ras pathway signaling and type I interferon (IFN) resistance. Pexa-Vec is currently undergoing clinical trials for multiple cancer types, with the lead indication being hepatocellular carcinoma (HCC). Headed by Transgene's partner in the United States, SillaJen, Pexa-Vec completed a Phase I and II trial with encouraging results, demonstrating virus infection of tumors following both intra-tumoral and i.v. administration, viral replication, and production of GM-CSF. 59,60 Following three direct intra-tumoral injections (days 1, 15, and 29) of Pexa-Vec in patients with unresectable HCC, there was a significant improvement in overall survival between patients treated with a high dose (14.1 months with 1 × 109 pfu) compared to those given the low dose of virus (6.7 months with 1 × 108 pfu). 60 Due to these promising outcomes and an acceptable safety profile, the virus has recently entered a Phase III trial (PHOCUS/NCT02562755) for advanced HCC. Patients (recruiting 600) will be split into two treatment groups, one receiving the OV and the tyrosine kinase inhibitor sorafenib (standard of care for HCC), and the other sorafenib only, and the primary endpoint will be overall survival. The results of the PHOCUS trial are eagerly awaited by the VV community. Additional exploratory trials in Europe include the combination of Pexa-Vec with checkpoint blockade, Pexa-Vec combined with Nivolumab in advanced HCC, and Pexa-Vec combined with Ipilimumab in solid tumors. Another OV in the pipeline at Transgene is TG6002, a VV with TK and ribonucleotide reductase (RR) deletions and encoding the FCU1 gene. 61,62 The FCU1 enzyme 63,64 converts the non-cytotoxic prodrug fluorocytosine (5-FC) to 5-fluorouracil (5-FU), a potent chemotherapeutic agent. Thus, expression from TG6002 enables high levels of localized activity at the tumor site. Transgene aims to enter TG6002 into clinical trials for glioblastoma later this year.
Several preclinical strategies to modulate host immunity for maximal oncolytic anticancer efficacy are under investigation. 65 –67 Yaohe Wang et al. at Barts Cancer Institute, London, developed a Lister strain of TK-deleted vaccinia virus expressing IL-10 (VVLΔTK-IL-10), with the aim of achieving IL-10-mediated suppression of an early antiviral immune response and prolonging the period of effective virus replication. 67 In murine models of pancreatic cancer, VVLΔTK-IL-10 showed a reduction in tumor size compared to parental VVLΔTK, decreased the number of virus-specific CD8+ T cells, and induced antitumor immunity. This work again reports on the potential of engaging the virus and host immune system to achieve synergistic responses. However, this contrasts to the many aimed at enhancing the immune stimulatory effects of the virus, and points to the importance of timing such responses and avoiding early viral clearance.
Measles Virus
Some of the earliest reports of oncolytic viral activity came from opportunistic infections of wild-type measles virus (MV) in patients with acute lymphoblastic leukemia (ALL), 68 Hodgkin's lymphoma, 69 and Burkitt's lymphoma, 70 leading to spontaneous tumor regressions. MV is an enveloped negative-strand RNA virus that enters cells via the SLAM, CD46, and nectin-4 receptors. The attenuated Edmonston vaccine strain of MV preferentially binds to CD46, a regulator of the complement system overexpressed on cancer cells, thus helping to confer tumor selectivity. Successfully vaccinating millions of people over the past 50 years, this virus has a well-established safety profile, and clinical results in its oncolytic form are very encouraging.
In Heidelberg, Germany, Prof. Ungerechts et al. have performed preclinical studies combining immunomodulatory agents with the vaccine strain of MV for enhanced therapeutic efficacy. Several agents have been investigated, including checkpoint inhibitors, 9 GM-CSF, 71 and IL-12. 72,73 In one such study, they encoded antibodies against the T-cell inhibitory factors CTLA-4 and PD-L1 within MV, and the “armed” viruses increased levels of activated CD8+ T cells and IFNγ release in vivo. 9 There was a delay in tumor progression and improved survival in mice treated with MV-CTLA-4/PD-L1 compared to the parental control. The group is now planning a Phase I/II clinical trial using MV for the treatment of pancreatic cancer. Patients with metastatic pancreatic adenocarcinoma will be treated with ultrasound-guided intra-lesional MV injections and i.v. delivery of anti-PD1 antibody. 10 The trial coordinators aim to resect sequential biopsies post treatment to study tumor infiltrating lymphocytes, assess cytokine and chemokines profiles, and perform immune receptor and transcriptome sequencing to identify potential response biomarkers. 74 Design of clinical trials with a strong research focus such as this will no doubt be very informative to the immune virotherapy community and will help to classify those immune signatures indicative of better responders.
At University College London, understanding and improving MV anticancer efficacy in adult ALL is a key focus of Prof. Fielding's lab. To improve the delivery of MV to cancer sites and bypass preexisting neutralizing viral antibodies, they loaded bone marrow–derived mesenchymal stromal cells (BM-MSC) with MV in vitro, prior to administering to mice with disseminated ALL, and observed successful targeting and handoff of virus from BM-MSC to ALL cancer cells in the presence of anti-MV antibodies. 75 While adding to the complexity of a clinical trial regime, cell delivery strategies may have the potential to bypass humoral immunity, improve systemic delivery, and afford the possibility of repeated dosing to enhance overall efficacy.
Prof. Russell began his scientific career at the University of London, and now based in the Mayo Clinic in the United States, he continues to be a major player in the OV field. His work using the MV Edmonston strain encoding the human thyroidal sodium–iodide symporter (NIS) reporter in clinical trials for myeloma has demonstrated very encouraging results. Early reports from the Phase I trial (NCT00450814) showed virus tumor targeting following a single dose (1 × 1011 pfu) of MV-NIS as assessed by NIS-mediated radioiodine uptake studies. 76 Two patients with relapsing drug-refractory myeloma, lacking anti-MV antibodies, showed impressive clinical responses, with reduced levels of circulating paraprotein and a reduction in bone-marrow plasmacytomas. Overall, the trial recruited 32 refractory myeloma patients divided into two cohorts, the first receiving MV-NIS i.v. and the second administered the immune modulator cyclophosphamide 2 days prior to i.v. virus. Recently published findings showed adverse effects consisting of fever, chills, gastrointestinal symptoms, and cytopenia. 77 After treatment, all but two patients (MV seropositive at start of study) treated with 1 × 109 TCID50 or higher showed persistent viremia at 72 h, and post therapy the majority of patients were positive for MV neutralizing antibodies. There were transient reductions in paraproteins in several of the patients and viral RNA was detected in bloods and urine. Eight patients were positive for radioiodine (123I) uptake scans indicative of MV infection. This trial concluded that MV-NIS was capable of safely targeting and replicating at tumor sites prior to immune clearance.
Reovirus
Reovirus is a ubiquitous non-enveloped double-stranded RNA virus reported to preferentially replicate in cells with dysfunctional RAS signaling pathways. RAS overexpression inhibits the dsRNA-activated PKR response, making cells less equipped to sense viral genomes and inherently sensitive to viral infection. Reolysin (developed by Oncolytics Biotech, Canada) is the commercial name for the wild-type nonpathogenic type 3 Dearing reovirus undergoing extensive preclinical and clinical evaluation for several indications, and recently completed Phase III trials for head and neck cancer.
While the majority of the clinical work with reovirus has been performed outside Europe, Prof. Melcher et al. in the United Kingdom have published important findings about its pharmacodynamics following i.v. administration in patients with colorectal cancer metastases. By performing “window of opportunity” studies and systemically administering reovirus prior to planned surgical resection of tumors, they found that despite the presence of anti-reovirus neutralizing antibodies, successful tumor targeting could be achieved and infectious reovirus could be recovered from the patient peripheral blood mononuclear cells (PBMCs) and granulocyte and platelet fractions. 78 There were no grade 3 or higher adverse effects observed, and immunohistochemistry of the resected tumor sections showed reovirus staining localized to cancer cells, rather than stroma or normal tissue. The report demonstrated that reovirus could effectively target tumor deposits due to its ability to “hitch hike” on circulating blood cells and circumvent the antiviral immune response to access the tumor. While animal models initially suggested that cyclophosphamide could facilitate reovirus delivery to tumors and improve efficacy by reducing neutralizing anti-reovirus antibodies, 79 this did not translate in clinical trials. A Phase I trial in patients with advanced cancer, using cyclophosphamide to decrease neutralizing antibodies, found that while the immunomodulatory drug did not influence antibody titers against the virus, reovirus could be detected associated with patient PBMCs 2 weeks following administration, again indicating that neutralizing antibodies are not detrimental to viral persistence in the blood and highlighting the differences between human and murine models. 80
Newcastle Disease Virus
Newcastle disease virus (NDV), an avian paramyxovirus with a negative single-strand RNA genome, was named after the location of its first poultry outbreak, Newcastle-upon-Tyne, England, in 1926. 81 There are three major strains of NDV, classified by their pathogenesis in birds as lentogenic (avirulent, no clinical manifestations), mesogenic (intermediate, mild disease), or velogenic (virulent, high mortality). While none of the strains are associated with serious disease in humans, the mesogenic and velogenic types demonstrate a lytic replication cycle in human tumor cells, and all strains are associated with antitumor immune responses. Tumors are susceptible to NDV infection due to defects in cellular IFN signaling and apoptotic pathways, as well as sensitivity correlating with Rac1 and Ras expression levels.
Europe has been a central hub for NDV clinical trials through the decades. A Phase II trial was conducted by in Kirchner et al. in Germany in the early 1990s, involving 208 renal cell carcinoma patients administered autologous tumor cells infected with NDV (lytic strain T73) and irradiated, in a regime with IL-2 and IFN-α. 82 The oncolysate and cytokines were well tolerated, and encouraging responses compared to historical data were reported. At a similar time in Hungary (1993), Csatary et al. administered the NDV strain MTH-68/H via inhalation to 33 advanced colorectal cancer patients in a Phase II placebo controlled trial. Clinical responses were observed in 55% of patients in the treatment group versus 5% in the control cohort, and better survival rates after 2 years following NDV therapy were reported (seven survivors in the virus group compared to none in the control). 83 Since this trial, there are reports of significantly greater numbers of patients (>4,000) being treated with MTH-68/H in various combination therapies with positive outcomes. However, more thorough evaluation and rigorous controls are required to confirm these findings. 84
Schirrmacher et al. in Germany have been advancing the non-lytic Ulster strain of NDV through Phase II and III clinical trials as part of their autologous tumor cell vaccine (ATV) approach. In this case, tumor cells are isolated from the patient, infected with NDV, and 200 Gy irradiation is applied to the infected cells to prevent cell proliferation but not affect viral replication. The live cell vaccine (rather than lysate as used above) is then injected intra-dermally into the patient. NDV upregulates MHCI and adhesion molecules on the tumor cell surface, while the cell triggers danger signals in response to NDV infection, effects that subsequently help to break immune tolerance. At the site of injection, the autologous cells invoke a delayed type hypersensitivity skin response (DHR), an effect indicative of antigen-specific memory T cells, and this in combination with the newly awakened, virally induced proinflammatory environment leads to antitumor responses. In a Phase II study against glioblastoma, tumors were surgically removed, and patients received radiotherapy prior to ATV-NDV treatment. Ex vivo the isolated tumor cells were expanded and treated, and patients were given eight intradermal vaccinations of ATV-NDV (1 × 107 cells) to the upper thigh. In the treatment group, survival rates were enhanced, with 91% of patients surviving 1 year, 39% for 2 years, and 4% for longer, a significant improvement compared to the control group, with 45%, 11%, and 0% survival rates, respectively. In addition, increased DHR, levels of tumor memory T cells, and CD8+ tumor-infiltrating T cells were reported. 85 A prospectively randomized controlled Phase II/III trial, using a similar approach but for an alternative indication, investigated the effects of ATV-NDV on the survival of colorectal cancer patients with hepatic metastases. 86 Following complete resection of liver metastases, patients received six doses of the treatment over a 6-month period. In a long-term, 10-year follow-up, the ATV-NDV-treated colon-cancer subgroup showed significantly increased overall survival and improved metastases-free survival times. The Immunological and Oncological Center (IOZK) in Cologne (Germany) are now working on a derivation of the ATV-NDV approach, combining it with dendritic cells for increased tumor-associated antigen-specific T-cell generation, and have described some encouraging findings. 87
All of the aforementioned NDV studies use wild-type strains of the virus. There are serious environmental and economic concerns regarding the use of mesogenic or velogenic NDV due to the risk of epidemic disease in poultry should an outbreak occur. This has led to tight regulatory controls, and no clinical trials with these strains have been performed since 2008, despite encouraging earlier results. MedImmune based in Cambridge, United Kingdom, in collaboration with their U.S. partners, have employed reverse genetics techniques to generate recombinant versions of the mesogenic T73 strain of NDV. 88 They created an attenuated virus exhibiting reduced replication in avian cells, thereby vastly improving the environmental safety profile, without affecting its oncolytic ability in human tumor cells. This was achieved by modification of the fusion protein (F) to decrease cleavage, an essential step for fusion with the cell surface and viral spread, and through a sequence insertion into the HN-L intergenic region of the genome. The recombinant virus was also engineered to express a foreign transgene, GMCSF (recNDV-GMCSF), for improved anticancer efficacy, without compromising viral titers. 88,89 This virus is undergoing further investigation and is thought to be the subject of upcoming human trials.
Vesicular Stomatitis Virus
Vesicular stomatitis virus (VSV) belongs to the rhabdovirus family, a group of enveloped negative-strand RNA viruses (11 kb) with rapid replication kinetics. By mutating the VSV matrix (M) protein, viral defenses against host IFN responses are ablated, and it becomes extremely sensitive to antiviral signaling. Thus, this mutation prevents replication in healthy human cells but makes the virus permissive for tumor cells with dysregulated IFN pathways. 90 The wild-type virus has neurotropic properties, which have been linked to neurotoxity in animal models. 91 Thus, considerable efforts have been invested into developing restricted attenuated VSV variants with a significantly improved safety profile while retaining the potent oncolytic properties. While not as advanced as some other OV types, clinical trials with VSV have commenced in the United States (NCT01628640 and NCT02923466).
Led by ViraTherapeutics, a spin out of Dr. von Laers' lab at the University of Innsbruck, Austria, the attenuated vaccine vector VSV-GP shows preclinical efficacy against several cancer types, including glioblastoma, melanoma, prostate and ovarian cancer. 92,93 VSV-GP was generated by replacing the native neurotropic VSV glycoprotein with that of the lymphocytic choriomeningitis virus (LCMV) WE-HPI strain. This replication-competent pseudotyped virus is non-neurotoxic and has been shown to be very effective against brain tumors while sparing normal cells, as demonstrated by in vitro studies and rodent models. 92 As well as detargeting from neurons, the LCMV glycoprotein helps to hide the virus from the host immune system. In contrast to the parental virus, VSV-GP does not induce viral-specific humoral immunity and boosts antigen-specific immune responses upon repeated delivery. This lack of neutralizing antibodies makes multiple i.v. administrations of the vector a strong possibility. 94 Evidence of synergy with other anticancer therapeutics has been observed. In a recent study against ovarian cancer, VSV-GP was less effective in tumor cell lines capable of responding to IFN, but addition of the JAK1/2 inhibitor ruxolitinib helped to reverse this. 93 In collaboration with Boehringer Ingelheim in Germany, ViraTherapeutics is now moving forward to develop VSV-GP alone and in combination with other therapies for Phase I clinical trials.
Current Challenges and Future Prospects
With several potential OVs to choose from but without direct clinical comparisons, the most appropriate virus for a particular indication will require careful consideration. Decisions to “arm” the virus with a further therapeutic rather than delivering the individual agents separately will depend on the packaging capacity of the virus, optimal dosing schedules, and licensing. Currently, OV manufacture, development, and regulatory approvals are different for each virus undergoing study, making the design of a standardized preclinical package and uniform trial design challenging, as well as inferring poor economic sense in some cases. Establishing a common OV GMP manufacturing facility in Europe may go some way to streamlining the process and reducing costs. Commercial scale amplification and purification of certain OVs can be problematic due to poor viral yields and occasional risk of genome drift, leading to virus variants following multiple passages (e.g., viruses lacking an endogenous proofreading DNA polymerase). Within the supportive European regulatory framework, a clearer path to achieving clinical success is being carved out. The European Medicines Agency (EMA) has a defined set of scientific guidelines to help in gaining authorization for human medicine applications, which includes technical specifications for product characterization of replicating viruses such as tumor selectivity, molecular variants with an altered profile, adventitious agent testing, and in vivo studies where possible.
An added element of complexity for the preclinical OV development stage comes from the fact that many of the most popular virus types are not permissive for nonhuman cells, making in vivo investigations in rodent models of very limited use. While small-animal studies provide some information on biodistribution profiles and particle-related toxicities, they very often cannot be employed to study the effects of virus infection, oncolysis, or innate and adaptive immune responses. By swapping mouse models for human tumor explants in the laboratory, which are much more informative and clinically representative, a better understanding can be gained of the therapeutic activity in a highly relevant patient setting. Evidence of tumor targeting following systemic administration of certain viral types such as vaccinia 59 and adenovirus 47 has been obtained from trial patients. However, upon contact with the bloodstream, pre-existing neutralizing antibodies and antiviral immunity in humans affect the ability of several viruses to target disseminated tumors. With these factors in mind, it may be beneficial to have more defined preclinical package requirements before applying for EMA authorization, for instance extensive replication studies in human primary cells from both normal and tumor samples, and assessment of virus activity in the presence of whole human blood. Small-scale mechanistic studies in humans (P0 trials) to establish proof of concept and which are strongly research orientated would play a key role in dissecting the mode of action, predicting dosing schedules and improving clinical viability.
As OVs continue to become a clinical reality, the focus must be on integrating them with standard treatments regimes and newly approved therapies. Designing studies in which the OV is used in combination with standard-of-care agents will help in gaining trial consent and provides an opportunity for synergistic effects. Work both in Europe and across the world continues in an effort to resolve the current challenges, with steady advances on all aspects. With major leaps forward in the clinical utility of OVs, the outlook for the field is very promising.
Footnotes
Acknowledgments
The authors would like to gratefully acknowledge support from Cancer Research UK (Programme grant #C552/A17720) and the Kay Kendall Leukaemia Fund (grant #KKL1050).
Author Disclosure
L.W.S. and K.D.F. own equity or share options in PsiOxus Therapeutics Ltd., which is leading the clinical development of Enadenotucirev. M.R.D. has no competing financial interests.