
Abstract
The treatment of leukemia/lymphoma by chimeric antigen receptor (CAR) redirected T cells with specificity for CD19 induced complete remissions in the majority of patients, with a realistic hope for cure. However, recent follow-up data revealed a substantial risk of relapse through leukemic cells that lack the CAR targeted antigen. In this situation, a bispecific CAR with binding domains for CD19 and CD20 is aimed at recognizing leukemic cells with only one cognate antigen. The anti-CD20-CD19 bispecific CAR induced a full T-cell response upon engagement of CD19 or CD20 on target cells showing a true “OR” gate recognition in redirecting T-cell activation. T cells with the anti-CD20-CD19 CAR efficiently killed patients' chronic lymphocytic leukemia cells in vitro. The bispecific CAR T cells cleared pediatric acute lymphocytic leukemia with a mixed CD19+CD20+/CD20− phenotype from the blood and bone marrow of transplanted mice, while anti-CD20 CAR T cells left CD20− leukemic cells behind without curing the disease. Data indicate the superior anti-leukemic activity in the control of leukemia, implying that the anti-CD20-CD19 bispecific CAR T cells may reduce the risk of relapse through antigen-loss leukemic cells in the long term.
Introduction
T
Basically, there are at least three options to redirect T cells by CARs in order to target those malignant B cells that lack CD19 or CD20: (1) the administration of two mixed T-cell products 11 —T cells with anti-CD19 CAR and T cells with anti-CD20 CAR; (2) T cells that are engineered to co-express the anti-CD19 CAR and the anti-CD20 CAR, each CAR competent to drive full T-cell activation 12 ; (3) T cells that are engineered with one bispecific CAR with binding domains for both CD19 and CD20, respectively. The feasibility of targeting two antigens by a bispecific CAR was previously shown by targeting Her2/neu (ErbB2) and CD19. 13 Although both antigens are usually not co-expressed by the same cell, the so-called tandem CAR (TanCAR) showed proof-of-principle of the “OR”-gated computation of signals, that is, the CAR mediated redirected T-cell activation upon engagement of either Her2 or CD19. Binding to both antigens was basically not required for T-cell activation. However, the bispecific anti-Her2-CD19 CAR T cells were less strongly activated against Her2+ cells than against the Her2 and CD19 double-positive cells. In another experimental model, the frequency of tumor relapse was reduced in the B16ova/Pmel tumor model by bispecific CAR T cells in comparison to the respective monospecific CAR T cells. 14,15 T cells with a bispecific CAR targeting both CEA and TAG-72 or PSMA and PSCA killed the cancer cells with either antigen. 16,17
The present study demonstrates that the anti-CD20-CD19 bispecific CAR shows a clear Boolian “OR”-gate signal computation in order to activate the engineered T cells upon engagement of either cognate target. Such CD20-CD19 bispecific CAR T cells are superior in the treatment of transplanted pediatric ALL with a heterogeneous CD20+/– phenotype over CD20 monospecific CAR T cells, which were not capable of controlling the disease in a mouse model. The data imply that there is a high probability of relapse in the long term when using CD20-CD19 CAR T cells to treat B-cell malignancies.
Materials and Methods
Patient samples, cell lines, and reagents
All studies involving human blood cells were approved by the University Hospital Cologne Institutional Review Board (IRB; reference no. 01-090). T cells were isolated from the peripheral blood of healthy donors by density centrifugation and were stimulated by the agonistic anti-CD3 antibody OKT3 (200 ng/mL), the agonistic anti-CD28 antibody 15E8 (50 ng/mL), and interleukin-2 (IL-2; 500 IU/mL) for 48 h. Written informed consent and IRB approval of the University Hospital Tübingen (#27/2008B01, 213/2014BO2) and Cologne (#11-319) were obtained for the use of pediatric B-cell precursor (BCP) ALL bone marrow and CLL blood samples for this study. ALL and CLL cells were isolated by Ficoll Paque Plus (GE Healthcare, München, Germany) centrifugation after negative selection (RosetteSep human B-cell enrichment cocktail; StemCell Technologies, Köln, Germany). The Raji lymphoma cell line (ATCC CCL 86) was obtained from the American Type Culture Collection (ATCC). Primary human T cells, CLL cells, ALL cells, and Raji cells were cultured in RPMI 1640 medium (Invitrogen Life Technologies, Karlsruhe, Germany) supplemented with 100 IU/mL of penicillin, 100 mg/mL of streptomycin, 2 mM of L-glutamine, and 10 % (v/v) fetal calf serum (FCS; Invitrogen Life Technologies). HEK293 cells (ATCC CRL-1573) were modified by DNA transfection to express CD19, CD20, or CD19 and CD20, respectively. HEK293T cells (ATCC CRL-11268) express the SV40 large T antigen. HEK293 cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen Life Technologies) supplemented with 100 IU/mL of penicillin, 100 mg/mL of streptomycin, 2 mM of L-glutamine, and 10 % (v/v) FCS. The anti-CD19 monoclonal antibody (mAb; clone FMC63) was purchased from Merck Millipore (Darmstadt, Germany), and the anti-CD20 mAb (clone 2H7) and anti-CD3 mAb from Biolegend (San Diego, CA). The fluorescein isothiocyanate (FITC)-conjugated anti-CD3 mAb, the phycoerythrin (PE)-conjugated anti-CD19 mAb, and the allophycocyanin (APC)-conjugated and VioBlue-conjugated anti-CD20 mAb were purchased from Miltenyi Biotec (Bergisch Gladbach, Germany). The goat anti-immunoglobulin G (IgG) antibody and its biotin- and (PE)-conjugated F(ab′)2 fragment were purchased from Southern Biotechnology (Birmingham, AL). The anti-interferon (IFN)-γ antibody NIB42 and the biotinylated anti-IFN-γ antibody 4S.B3 were purchased from BD Bioscience (San Jose, CA). Recombinant IL-2 was purchased from Novartis Pharma (Nürnberg, Germany). The APC Annexin V Apoptosis Detection Kit was purchased from BD Biosciences Pharmingen (Heidelberg, Germany). Carboxyfluorescein succinimidyl ester (CFSE) was purchased from Invitrogen, and the red fluorescent dye PKH26 was purchased from Sigma–Aldrich (Taufkirchen, Germany). The FITC-conjugated anti-mouse CD45 mAb (clone 30-F11), PE Cy7-conjugated anti-human CD19 mAb (clone HIB19), APC-conjugated anti-human CD20 mAb (clone 2H7), and Brilliant Violet 711 (BV711)-conjugated anti-human CD3 mAb (clone SK7) were purchased from Biolegend. The PE CF594-conjugated anti-human CD10 mAb (clone HI10A), the APC H7-conjugated anti-human CD45 mAb (clone 2D1), and the FITC-conjugated anti-human CD45RO mAb were purchased from BD Biosciences. The PE-conjugated anti-human CD3 mAb was purchased from Miltenyi Biotec. The APC-conjugated anti-human CD62L mAb was purchased from ImmunoTools (Friesoythe, Germany).
CARs and scFv-Fc proteins
The anti-CD19 and anti-CD20 CARs have been described in detail previously. 18 –20 The retroviral expression cassettes for the scFv-Fc-CD28-CD3ζ CARs have been described earlier. 21 The anti-CD19 scFv, derived from the mAb FMC639, and the anti-CD20 scFv, derived from the mAb 2H7, were used to engineer the monospecific CARs anti-CD19scFv-Fc-CD28-CD3ζ and anti-CD20scFv-Fc-CD28-CD3ζ, respectively. The bispecific anti-CD20-CD19 CAR was obtained by linking the anti-CD19 and anti-CD20 scFvs by a (glycin4serin)4 linker.
Flow cytometry
Data were recorded using a FACSCanto II or a FACS LSR II cytofluorometer equipped with FACS-DIVA software v6.1.3 (BD Biosciences).
T-cell modification
Human peripheral blood T cells were retrovirally transduced to express the CARs, as recently described in detail. 21 Briefly, T cells were isolated by density gradient centrifugation, stimulated by incubating with anti-CD3 OKT3 and anti-CD28 15E8 agonistic antibodies, and transduced on day 2–3 by γ-retrovirus containing supernatants or by co-culture with virus producing 293T cells. Retroviruses were produced by 293T cells upon transient transfection with the DNA of the GALV encoding helper plasmid, the gag/pol encoding plasmid, and the plasmid encoding the respective CAR. CAR expression by the T cells was monitored by flow cytometry using a PE-conjugated F(ab′)2 anti-IgG1 antibody, which binds to the CAR extracellular Fc spacer, and a FITC-conjugated anti-CD3 antibody to identify the T cells.
Binding assays
Serial dilutions of cell culture supernatants containing the recombinant proteins anti-CD19 scFv-Fc, anti-CD20 scFv-Fc, or anti-CD19-CD20 scFv-Fc or a scFv-Fc of irrelevant specificity were incubated for 1.5 h at 4°C with HEK293 cells (105 cells). Bound scFv-Fc proteins were detected by a PE-conjugated anti-IgG (Fc) antibody and recorded by flow cytometry.
Cell conjugate formation assay
HEK293 CD19+ cells were labeled with PKH26 and mixed with CFSE labeled HEK293 CD20+ cells (each 105 cells) in the presence of the scFv-Fc proteins (1 μg/mL) anti-CD19scFv-Fc, anti-CD20scFv-Fc, anti-CD20-CD19scFv-Fc or anti-CD20-TNPscFv-Fc, or anti-TNP-CD19scFv-Fc, sedimented for 15 s at 1,000 g, and incubated for 30 min on ice. The cells were gently re-suspended and analyzed by flow cytometry. PKH26/CSFE double fluorescent particles represent conjugated HEK293 CD19+ and HEK293 CD20+ cells. For blocking, cells were incubated for 30 min with the anti-CD19, anti-CD20, or an anti-CD3 antibody (10 μg/mL) prior to co-incubation with the respective scFv-Fc proteins.
CAR-mediated T-cell activation
CAR T cells were cultivated in RPMI 1640 medium, 10% (v/v) FCS, without stimuli for 24 h, washed, and co-incubated in increasing numbers with tumor cells (1.5 × 104 cells/well) for 48 h in 96-well round-bottomed plates. Specific cytotoxicity of CAR T cells was monitored by a 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide salt (XTT)-based colorimetric assay (Cell Proliferation Kit II, Roche Diagnostics, Mannheim, Germany). Reduction of XTT to formazan by the viable tumor cells was colorimetrically monitored. Maximal reduction of XTT was determined as the mean of 12 wells containing tumor cells only, and the background as the mean of 12 wells containing RPMI 1640 medium, 10% (v/v) FCS. Non-specific formation of formazan due to the presence of T cells was determined from triplicate wells containing T cells in the same number as in the corresponding experimental wells. Cytotoxicity (%) = [1 – OD(experimental wells – corresponding number of T cells)/OD (tumor cells without T cells – medium)] × 100. Alternatively, specific elimination of target cells was recorded by flow cytometry. CFSE labeled HEK293, HEK293 CD19+, CD20+, and HEK293 CD19+ CD20+ cells were mixed (each 2.5 × 104 cells) and co-incubated with 2 × 105 CAR T cells. After 48 h, cells were stained with the PE-conjugated anti-CD19 mAb and APC-conjugated anti-CD20 mAb and recorded by flow cytometry. CFSE+ cells were gated, and specific cytotoxicity (%) was calculated: [1 – (number of HEK293 cells(with CAR T cells)/(number of HEK293 cells(with T cells))] × 100. Cell culture supernatants were analyzed for IFN-γ by enzyme-linked immunosorbent assay using the matched pair IFN-γ specific antibodies NIB42 and B133.5. Briefly, IFN-γ was bound to a solid-phase mAb NIB42 and detected by the biotinylated mAb B133. The reaction product was visualized by a peroxidase-streptavidin conjugate and ABTS as substrate (both from Roche Diagnostics).
Patient-derived leukemia in the xenograft mouse
NOD.Cg-PrkdcscidIL2rgtmWjl/SzJ (NSG) mice (The Jackson Laboratory, Bar Harbor, ME) were maintained under specific pathogen-free conditions in the research animal facility of the University of Tübingen, Germany, according to German federal and state regulations (Regierungspräsidium Tübingen, K1/14). Patient-derived leukemia was established in grafted mice, as described. 22 Briefly, 2 × 106 blasts were injected intravenously (i.v.) into 8- to 12-week-old NSG female mice randomized to the treatment groups. Ten days after leukemia cell injection, mice were injected i.v. with either 2 × 107 CAR T cells (each group n = 5) or were untreated as control (n = 4). Mice were euthanized 7 weeks after the start of treatment.
Histology
Mouse tibiae were fixed for 24 h in 4% (w/v) paraformaldehyde and decalcified for 48 h with OSTEOSOFT (Merck Millipore) before embedding in paraffin. Tissue slide sections were stained with hematoxylin and eosin. For immunostaining, heat-induced epitope retrieval was performed using antigen unmasking solution (Vector Laboratories, Burlingame, CA). Slides were blocked using Fc Receptor Blocker solution and Background Buster (Innovex, Aachen, Germany) and stained with the mouse anti-human CD10 mAb (clone 56C6; Dako, Hamburg, Germany) followed by incubation with the horseradish peroxidase-coupled anti-mouse IgG antibody (Sigma–Aldrich) and were visualized by 3,3′-Diaminobenzidine (DAB; Vector Laboratories). Slides were analyzed by light microscopy using a BX51 microscope (Olympus, Hamburg, Germany) equipped with the cell soft imaging system.
Results
Bispecific anti-CD20-CD19 CAR binding domain recognizes CD19 and CD20
The binding domain of the bispecific CD20-CD19 CAR consisted of the anti-CD20 scFv (VH-linker-VL), derived from the Leu16 mAb, linked by a 20 amino acid (Gly4Ser)4 linker to the anti-CD19 FMC63 mAb (VH-linker-VL). The corresponding monospecific CARs contained solely the respective scFv for binding (Fig. 1A). CD19 and CD20 scFvs were alternatively linked by a 40 amino acid linker. To demonstrate the specificity in binding, the extracellular CAR domains were produced in a soluble, non-membrane anchored form, and these bispecific scFv-scFv-Fc domains were incubated with engineered HEK293 cells, which stably expressed CD19 or CD20 or both. The bispecific anti-CD20-CD19 scFv-Fc domain bound to cells with CD19 or CD20 or both, but not to cells lacking both antigens (Fig. 1B), clearly indicating that the bispecific binding domain required only one cognate antigen for binding. The bispecific CAR binding domain with the 40 amino acid linker showed basically the same binding (data not shown). In contrast, the monospecific anti-CD19 scFv-Fc bound to CD19+ cells, not to CD19− cells, and the anti-CD20 scFv-Fc bound to CD20+ cells, not to CD20− cells.
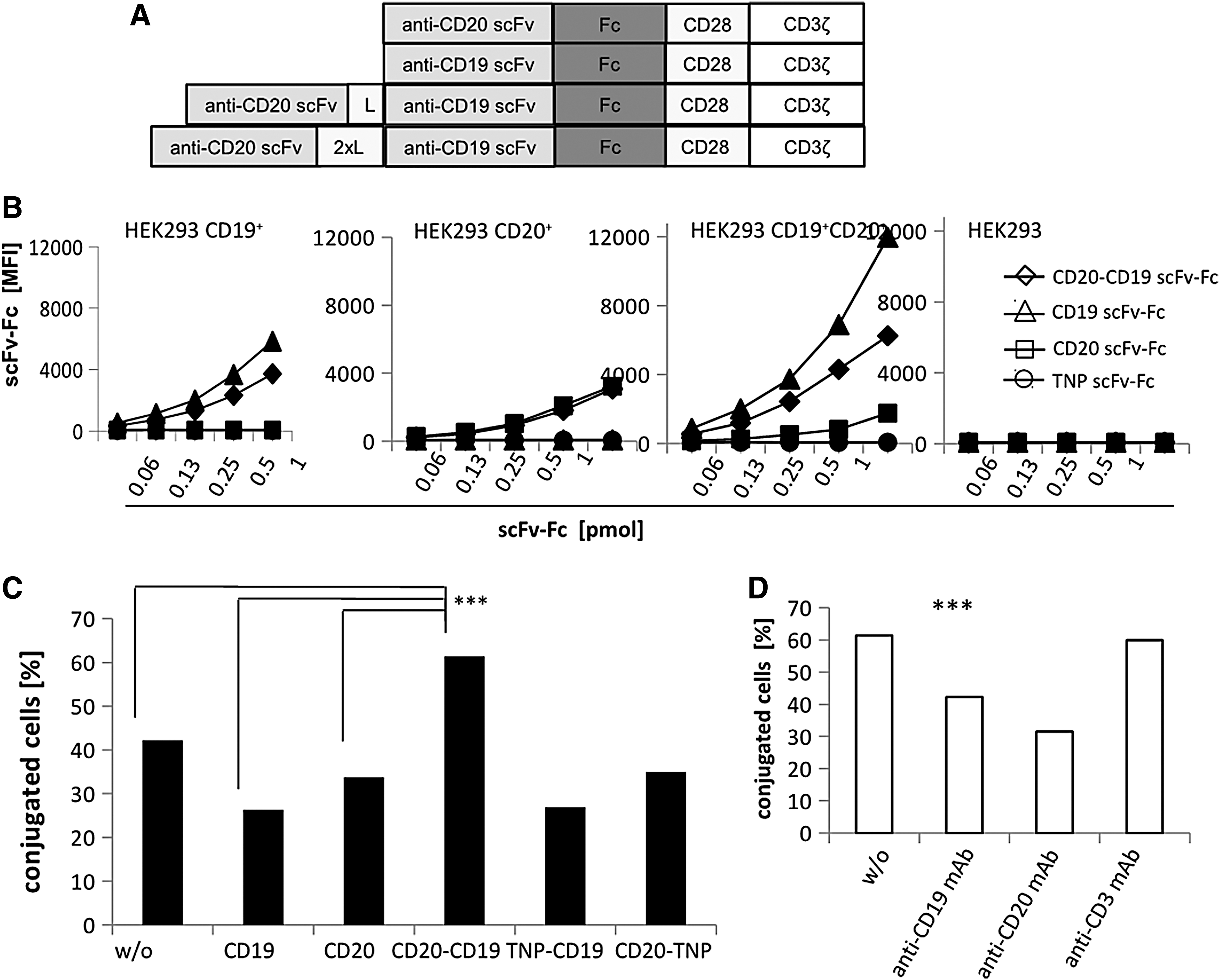
The bispecific anti-CD20-CD19 scFv domain bound to CD20 and CD19.
To address whether binding simultaneously to CD20 and CD19 can occur, the bispecific anti-CD20-CD19 scFv-Fc was incubated with CD19+ CD20− and CD19− CD20+ HEK293 cells labeled with PKH26 and CSFE, respectively. Simultaneous binding of the bispecific protein increased the number of cell conjugates between the PKH26-labeled CD19+ cells and the CFSE-labeled CD20+ cells, as revealed by flow cytometry (Fig. 1C). For comparison, the monospecific anti-CD19 scFv-Fc and anti-CD20 scFv-Fc binding domains did not increase the number of cell hetero-dimers. Moreover, the data indicated that binding to CD19 is not impaired by the CD20 binding domain in the bispecific antibody and vice versa. The bispecific format itself was not responsible for the increase in conjugate formation, since the bispecific anti-CD20-TNP scFv-Fc and the anti-TNP-CD19 scFv-Fc, both harboring the anti-TNP scFv of irrelevant specificity in the respective position, did not increase the number of cell hetero-dimers (Fig. 1C). The hetero-dimer formation of target cells by the anti-CD20-CD19 scFv-Fc is specific, since adding anti-CD19 mAb and anti-CD20 mAb, which competed in binding to the respective epitope, diminished the number of cell hetero-dimers, whereas adding anti-CD3 mAb did not (Fig. 1D).
“OR”-gate antigen recognition by the bispecific CAR: redirected T-cell activation upon encounter of CD19 or CD20 or both
The bispecific CD20-CD19 CAR in the tandem scFv-scFv format with a 20 amino acid Gly-Ser linker and the order anti-CD20scFv-anti-CD19scFv was used in the entire analysis. The bispecific CAR and the respective monospecific CARs were expressed by blood T cells upon γ-retroviral transduction (Fig. 2A). All CARs were equally potent to induce T-cell activation, since antigen-independent cross-linking by an anti-IgG antibody, which binds to the common spacer in the extracellular CAR moiety, induced IFN-γ and IL-2 release to nearly the same levels (Fig. 2A). Engagement of either CD19 or CD20 on engineered target cells initiated T-cell activation by the bispecific CAR indicated by the dose-dependent release of IFN-γ (Fig. 2B). Engaging both antigens did not increase IFN-γ release by the bispecific CAR T cells compared to engagement of either antigen. T cells with the bispecific CAR also mediated cytolysis of the target cells with CD19 or CD20 or both, whereas cells lacking both targets were not eliminated (Fig. 2B).
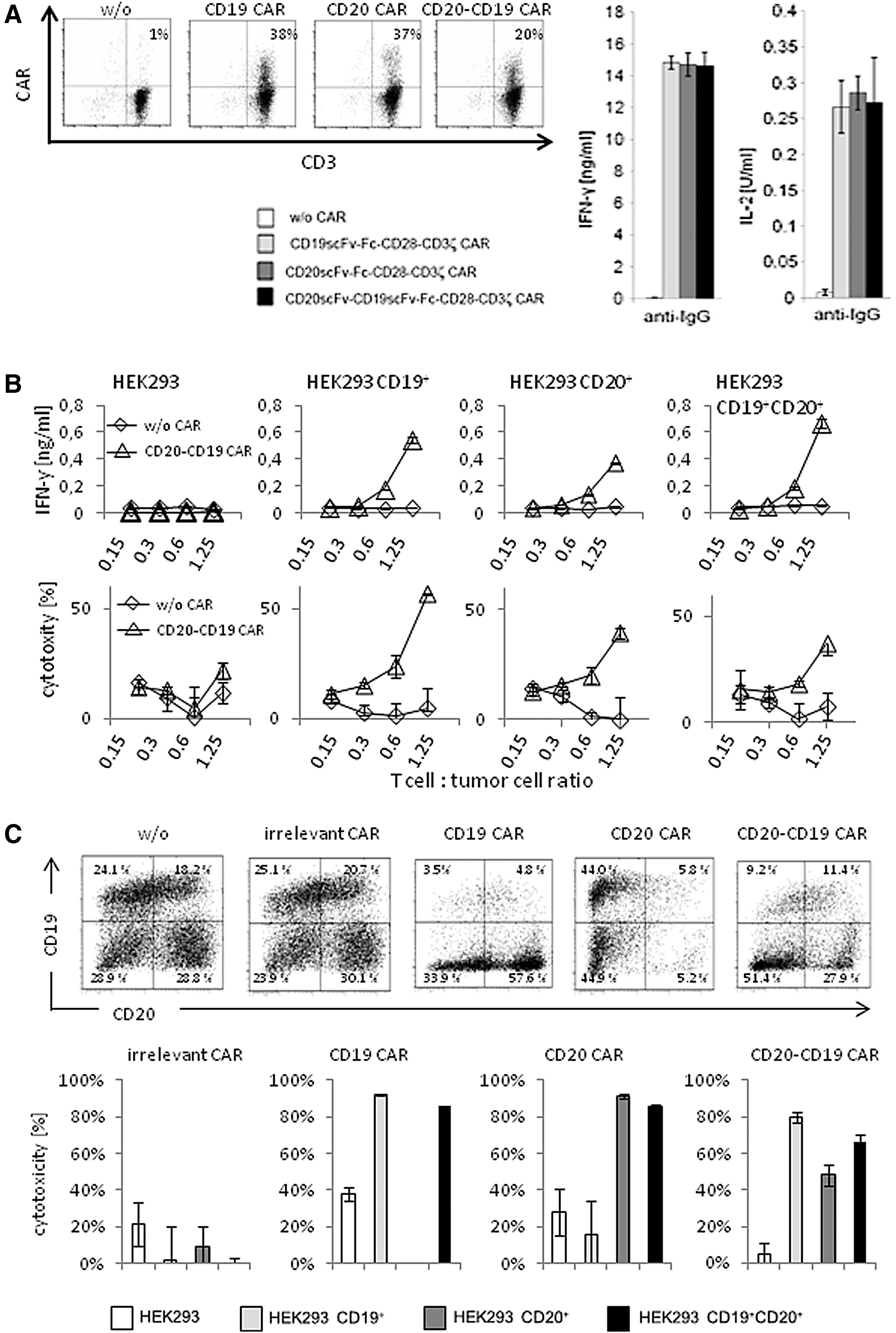
The anti-CD20-CD19 CAR redirected T-cell activation toward both CD19+ and CD20+ target cells.
Bispecific CAR T cells were capable of eliminating target cells with either antigen in a mixed target cell pool; the anti-CD20-CD19 CAR T cells eliminated cells with CD19 or CD20 or both (Fig. 2C). There was no substantial preference for either target cell subset. In particular, there was no preferred killing of target cells with both antigens, which is in accordance with a previous report. 23 As controls, the monospecific anti-CD19 and anti-CD20 CAR T cells selectively eliminated CD19+ or CD20+ tumor cells, respectively. T cells with a CAR of irrelevant specificity did not kill any cells in the assay. The assays demonstrated that the bispecific anti-CD20-CD19 CAR mediated T-cell activation upon engaging CD19 or CD20 or both, indicating a clear “OR”-gate integration of signals.
Bispecific CAR mediated T-cell activation against patients' leukemic cells in vitro
T cells with the bispecific CAR also exhibited cytolytic activity toward leukemic cells from patients. As summarized in Fig. 3A, T cells with the bispecific CAR efficiently induced cell death of CD19+ CD20+ CLL cells in vitro, indicated by Annexin V staining. Basically the same was observed with T cells redirected by a monospecific anti-CD19 or anti-CD20 CAR. For comparison, the bispecific CD20-CD19 CAR T cells eliminated cells of the established Raji lymphoma line as did anti-CD19 CAR and anti-CD20 CAR T cells.
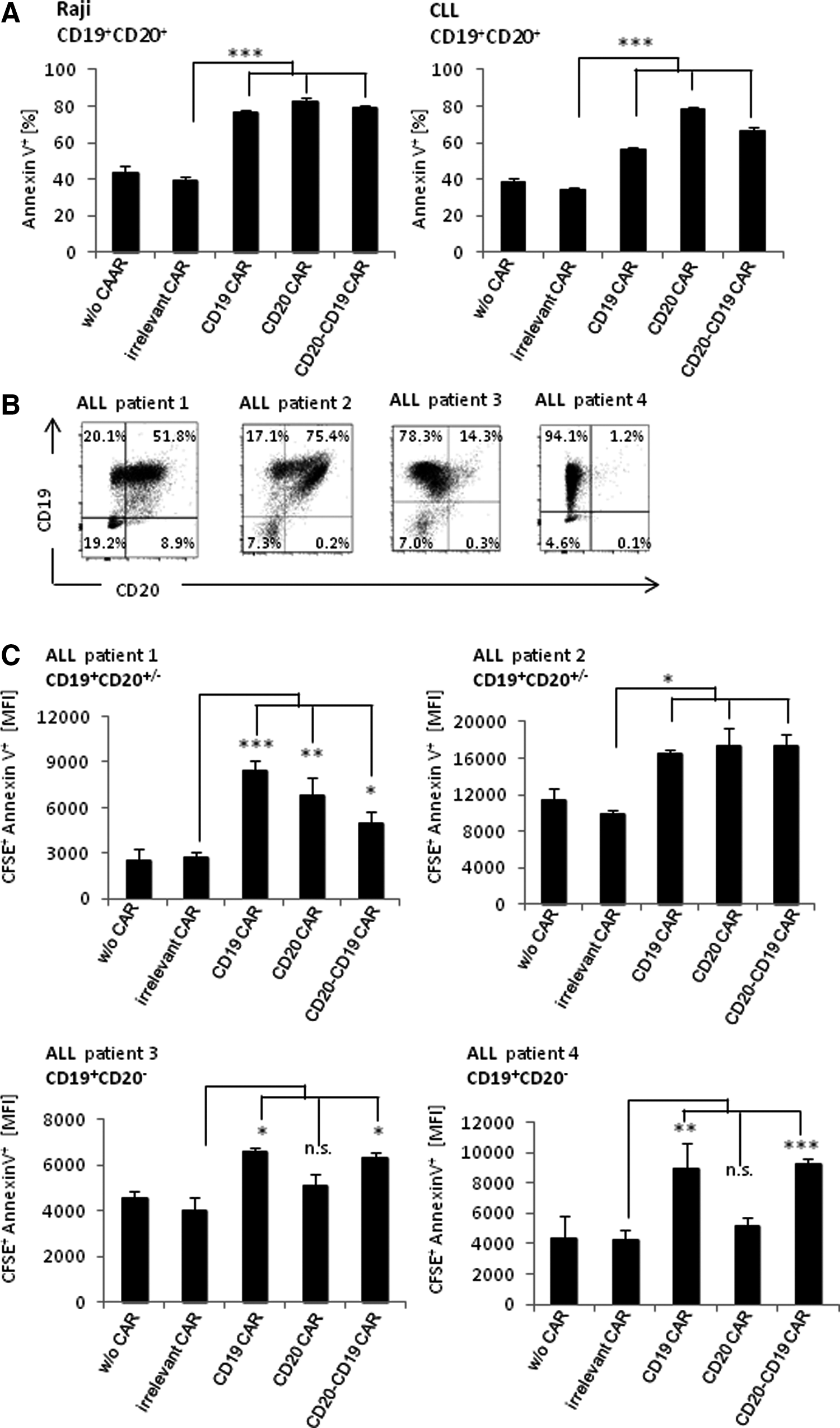
The anti-CD20-CD19 CAR mediated T-cell cytotoxicity toward primary leukemia cells that lack CD20.
The pediatric ALL cells are frequently heterogeneous with regard to CD20. Such heterogeneity can substantially limit the therapeutic efficacy of the cell therapy with anti-CD20 monospecific CAR T cells. ALL cells from four pediatric patients were recorded for CD19 and CD20 expression, revealing substantial heterogeneity, in particular with respect to CD20 levels (Fig. 3B). The elimination of patients' ALL cells with a heterogeneous CD20 expression was therefore monitored in vitro by mono- versus bispecific CAR T cells. While ALL cells with both antigens were equally eliminated by the monospecific and bispecific CAR T cells, CD19+ CD20− ALL cells were eliminated by the bispecific anti-CD20-CD19 CAR T cells as by T cells with the anti-CD19 CAR, but not by T cells with the anti-CD20 CAR (Fig. 3C). The data underline the benefit of the bispecific CAR T-cell targeting toward both CD19 and CD20 in eliminating heterogeneous leukemic cell populations.
Targeting by bispecific CAR T cells eradicated transplanted pediatric ALL, while T cells with the monospecific CD20 CAR did not control the disease
The power of the bispecific CAR T cells in controlling leukemia with heterogeneous cell populations with respect to CD20 expression was explored. Pediatric patients' ALL cells with a CD19+ CD20+/– phenotype were efficiently eliminated by T cells with the bispecific CAR in vitro but less by T cells with the monospecific CAR (Fig. 4A). To test for the anti-leukemia activity, the ALL cells were transplanted into immune-deficient mice, where the leukemia established within 1–2 weeks with at least 0.1% circulating human CD19+ CD10+ leukemic cells in the peripheral blood and 0.3% in the bone marrow. At day 10, T cells with the bispecific CAR T cells were adoptively transferred to the mice by i.v. injection; the corresponding monospecific CAR T cells were applied for comparison. T cells with the bispecific CAR eliminated the CD19+ CD20+/– leukemic cells from peripheral blood until week 7; no signs of leukemia were detected in treated mice (Fig. 4B). The same was observed in mice treated with CD19 specific CAR T cells as expected. However, T cells with the anti-CD20 CAR did not eradicate the disease and did not fully eliminate the CD20+/– leukemic cells; peripheral blood and bone marrow still harbored human ALL cells in nearly unchanged numbers. Bone marrow infiltration by leukemic cells was confirmed by immune histology recording human CD10 expression (Fig. 4C). For comparison, T cells with CAR of irrelevant specificity had no substantial effect. Taken together, the data demonstrate the superiority of the anti-CD20-CD19 bispecific CAR T cells in clearing established leukemia with a heterogeneous CD20+/– pattern in a xenograft model.
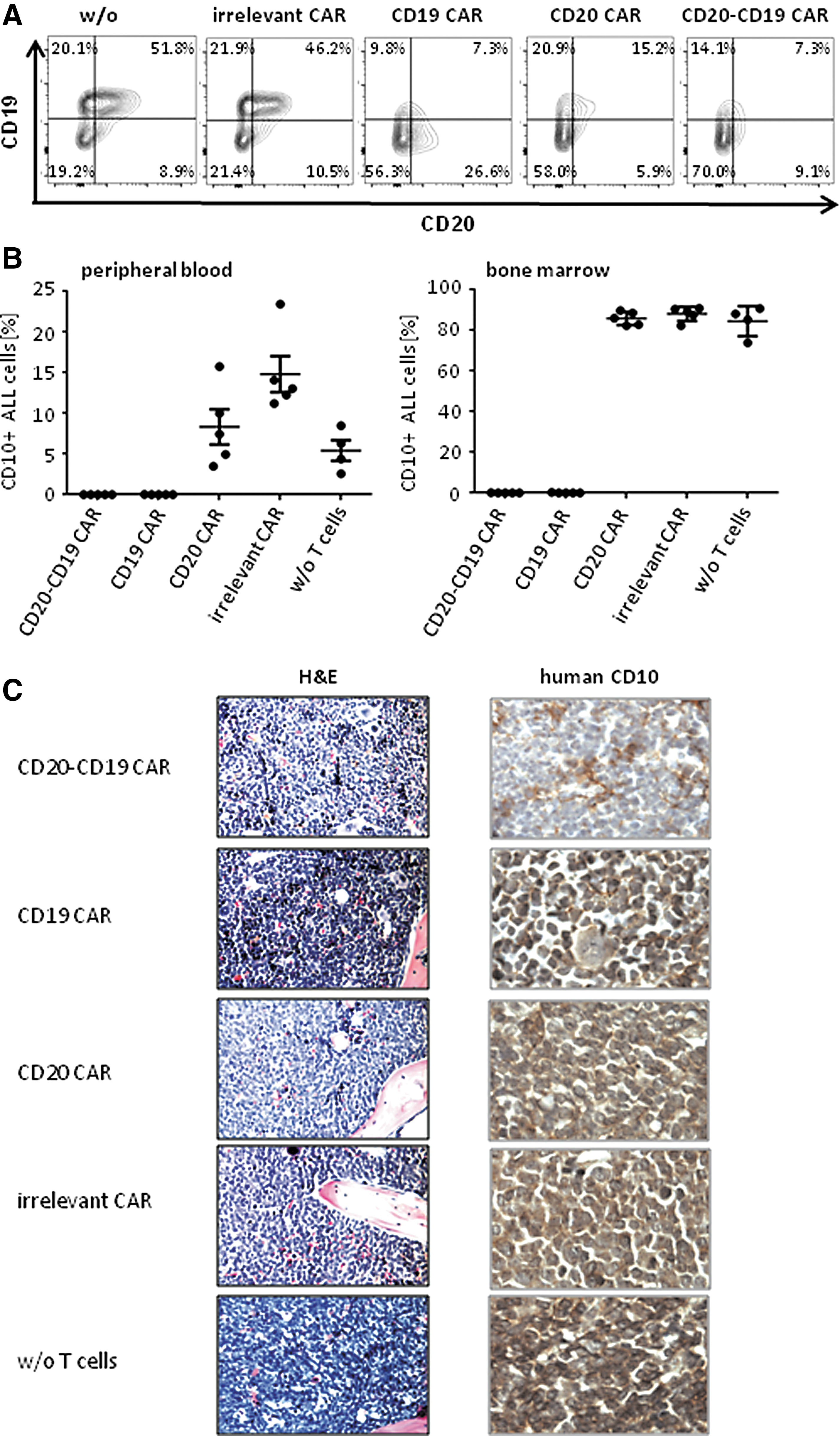
The anti-CD20-CD19 CAR T cells eliminated CD19+CD20+/– leukemia more efficiently than anti-CD20 CAR T cells in an ALL xenograft.
Discussion
The clinical situation of leukemia relapse through residual leukemic cells after CAR T-cell therapy demands bispecific CAR T cells, which are also efficiently activated when engaging only one target antigen. T cells engineered with the anti-CD20-CD19 CAR fulfill the criteria. Binding to CD19 or CD20 on target cells induced nearly the same level of T-cell activation with respect to cytokine release and redirected cytotoxicity as binding to target cells with both CD19 and CD20, showing a true Boolian “OR”-gate function. Although the overall structural requirements of monospecific CARs are known, the design of an optimized bispecific CAR still requires empiric exploration. Whereas the bispecific CAR harbored the two scFv domains in the order #1 VH-VL and #2 VH-VL, Zah et al. 24 used the #1 VL-VH and #2 VH-VL orientation in order to reduce the risk of mispairing of the respective scFv domains. The present study explored a flexible 20 and 40 amino acid Gly-Ser linker between the two scFvs. The CAR with the longer scFv linker performed equally as the CAR with the short scFv linker. It is reasonable that in order to make contact with the cognate antigen on the target cell, the position of the respective scFv within the CAR has to be carefully adjusted. CD19 is a single-pass transmembrane, immunoglobulin-like molecule with a long extracellular moiety, whereas CD20 is a multi-pass transmembrane molecule with a membrane near epitope. Given these structural differences, CD20 targeting requires a long extracellular CAR domain and CD19 a short domain. Therefore, the anti-CD20 scFv was set at the membrane distal position and the CD19 scFv at the membrane proximal position of the CAR in accordance to other reports. 24,25
Targeting CD19 or CD20 in the treatment of B-cell leukemia/lymphoma has the advantage that the antigens are nearly exclusively expressed by cells of the B-cell lineage; the risk of on-target off-tumor toxicity is low. Since CD19 and CD20 are involved in promoting B-cell survival, 26,27 the probability for the simultaneous loss of both antigens is also low, making dual targeting a potent safeguard against the survival of malignant B cells that lost one of the antigens. Accordingly, the anti-CD20-CD19 bispecific CAR T cells proved superior in controlling CD19+CD20+/– leukemia compared to CD20 CAR T cells in a xenotransplant model (cf. Fig. 4). CD19 CAR T cells were likewise effective, as expected.
Engineering T cells with the bispecific CAR in comparison to two CARs also has some technical advantages. The genetic construct is more compact, which facilitates vector packaging and improves transduction efficiencies. Bispecific CAR T cells represent one cell product in contrast to T-cell pools with different CARs, making the clinical grade production much easier. Upon activation in vivo, two CAR T-cell products may amplify at different degrees, with the risk of losing one CAR T-cell product, which is not the case for bispecific CAR T cells.
Moreover, bispecific anti-CD20-CD19 CAR T cells have the benefit of improved avidity to the double antigen-positive target cells compared to monospecific CAR T cells, in particular at low antigen densities. However, in the case of targeting leukemic B cells, the levels of the targeted CD19 and CD20 are far beyond threshold. In targeting other antigens at lower levels, the increase in avidity may more substantially impact T-cell activation.
In the near future, early-phase trials will explore the power of bispecific CAR T cells in controlling B-cell leukemia/lymphoma with heterogeneous CD20 and CD19 expression. Alternative antigen combinations will be envisioned as potential targets. Particular attention needs to be drawn to screening for subsets of malignant cells, which may give rise to relapse upon loss of the targeted antigen. Targeting more than two potential antigens associated with the malignancy may thereby strengthen the power of the CAR T-cell therapy in the long term.
Footnotes
Acknowledgments
We thank G.H. Fey, University Erlangen-Nuremberg, for discussion during the initial phase of this project. This work was supported by the Deutsche José Carreras Leukämie-Stiftung, München, the Deutsche Forschungsgemeinschaft, Bonn, the Else Kröner-Fresenius Stiftung, Bad Homburg v.d.H., the Wilhelm Sander-Stiftung, München, and the Medical Faculty of the University of Cologne.
Author Disclosure
The authors have no conflict of interest to declare in the submission of this work.