
Abstract
Hematopoietic stem cell–directed gene therapy (HSC-GT) provides an innovative treatment option for hematological disorders. Gene therapy promises to cure the disease “at the root” and is therefore exceptional in its potential, but also formidable in its challenges, as long-term side effects are hard to predict and clinical experience remains limited. Many excellent reviews on the topic by designated experts in the field of HSC-GT have come forth, elucidating the successes and pitfalls in the various clinical studies. This review attempts to discuss what we understand from those studies to represent current state of the art with respect to vectors, stem cell transduction, and pretransplant preparatory regimes, what limitations may remain, and which types of diseases may be more suited for HSC-GT than others (targets). We thus discuss the available vector platforms (tools) and preclinical/clinical and basic research (tricks) that contribute to our current understanding of HSC-GT, as well as some overarching principles we can conclude from these. The field has also learned from previous shortcomings, although some of the major concerns of the past, specifically insertional mutagenesis, may not be of relevance for future trials. This very positive development in HSC-GT, however, has to compete with the improvements in hematopoietic stem cell transplantation or enzyme-replacement therapy, leaving a narrow margin for gene therapy.
THE CONCEPT OF HEMATOPOIETIC STEM CELL–DIRECTED GENE THERAPY
T
The first successes in HSC-GT were achieved with gammaretroviral long terminal repeat (LTR) vectors, which were used for treating primary immunodeficiencies, namely X-linked severe combined immunodeficiency (X-SCID), adenosine-deaminase deficiency (ADA-SCID), X-linked chronic granulomatous disease (X-CGD) and Wiskott-Aldrich-Syndrome (WAS). 4 –8 However, the initial euphoria was deflated by the occurrence of severe adverse events in some of the patients. In consequence of the vector integration in the genome and due to enhancer mediated interactions between the viral vector enhancer/promoter and cellular genes, a considerable fraction of patients developed leukemia due to upregulated expression of proto-oncogenes. Six children enrolled in gene therapy studies to treat X-SCID developed T-cell-acute lymphoblastic leukemia, 9 –11 two young adults and two children treated for X-CGD presented with myeloid dysplasia 12,13 and seven of the nine treated children with Wiskott-Aldrich-Syndrome developed T-cell-acute lymphoblastic leukemia or acute myeloid leukemia, some of them even both. In contrast, all patients transplanted with HSC transduced with a gammaretroviral vector expressing adenosine deaminase for the treatment of ADA-SCID (i.e., a gene transfer tool no different from the above) were successfully treated without developing leukemia. 5,14,15 One of the retroviral vectors for gene therapy in ADA-SCID patients (Strimvelis) is now approved by the European Medicines Agency for the treatment of children with this disease who lack a suitable stem cell donor. 16
In more recent trials, with the aim to prevent potential genotoxicity and to improve gene transfer and expression, retroviral vectors in the self-inactivating (SIN) configurations have been explored, including lentiviral vectors. Using these tools, thus far no severe adverse events due to genotoxicity have been reported in these trials. Lentiviral vectors have been successfully applied for the correction of WAS, 17,18 X-SCID, 19 Sickle Cell disease 20 X-linked adrenoleukodystrophy, 21 and metachromatic leukodystrophy (MLD). 22,23 In addition, gene therapy for X-SCID with SIN gammaretroviral vectors proved to be efficacious without inducing leukemia. 24
The mechanisms by which retroviral vectors cause insertional mutagenesis is today well studied; however, as to why this happened more often in one disease than the other we can still only speculate. That said, further pondering is futile since improvements in vector technology have removed (most of) the genotoxic potential of the vectors. Genotoxicity removed from the equation, plenty of other hazards remain, their frequency and severity depending on the underlying disease, including stem cell extraction, stem cell quality (before and after gene transfer), intensity of the conditioning regime, and in vivo selection of gene modified cells after transplantation.
The Tools
Retroviral vectors for HSC transduction
Retroviral vectors are the ideal tools to achieve integrating gene transfer. They can be derived from different genera, and today several are well, albeit variably well, developed, namely the gammaretrovirus, lentivirus, alpharetrovirus, or foamyvirus vectors. 25 –29 Foamy viral vectors are of interest because they can encode larger expression cassettes; however, foamy- and alpharetroviral vectors have not been used for clinical HSC-GT yet. All platforms follow the same principle of separating the viral structural genes (gag, pol, and env, in the case of lentiviral vectors in addition the rev) from the gutted gene transfer vector to achieve a vector system that can transduce target cells but will not produce replicating particles. In place of the removed viral genes, transgenes can be encoded. In the classical gammaretroviral vector, transcription is initiated from the viral enhancer/promoter in the unique region (U3) of the 5′ LTR. The first generations of these vectors were optimized for high levels of gene expression in hematopoietic cells; 30 –32 however, the strong activity of the viral enhancer is the main cause of insertional transformation as it can inflict strong and long-range enhancer-mediated interactions with cellular promoters. Therefore, the relocation of the enhancer/promoter from the LTR to an internal position was a significant step to improve safety and efficacy of retroviral gene therapy. This vector configuration is called self inactivating (SIN). 33,34 During viral vector production a strong promoter in the 5′ LTR produces maximal amounts of viral vector genomes. The deletion in the U3 of the 3′LTR is copied to the 5′ LTR during reverse transcription, producing a viral DNA with two flanking LTR with deletions in both U3 regions. After completion of RT in the cytoplasm, the viral DNA forms the pre-integration complex (PIC) in which the viral integrase has hydrolyzed the cytosine-adenine bases at the ends of the DNA. The PIC enters the nucleus. While gammaretroviral vectors depend on cell mitosis to approach the genome during the phase the nuclear membrane is dissolved, 35 lentiviral vectors can passage the nuclear membrane via the nuclear pores. Alpharetroviruses are reported to have an intermediate behavior. 36,37 Lentiviral integrations are also more frequently found in chromatin in the nuclear periphery, probably due to the lentiviral nuclear entry mechanism. 38,39
The integration of the viral genome in the host genome occurs semirandomly, is influenced by additional factors, and the vectors follow the integration site preference of their parental virus. All retroviruses prefer to integrate into actively transcribed genes and regions where DNA is bent (around nucleosomes). Gammaretroviruses integrate preferentially into promoter regions upstream of the transcription start site of genes and CpG rich regions, 40 while lentiviruses tend to integrate into coding regions downstream of the transcription start site and gene dense regions. 41 Alpharetroviruses integrate close to randomly with a propensity of ∼50% in hitting gene regions. 42,43 Foamyviruses, on the other hand, avoid integration into gene bodies but still target promoter regions to some extent. 44
Before integration occurs, the PIC is tethered to the genome by tethering factors, in case of lentiviruses, lens epithelial-derived growth factor. Gammaretroviruses are guided by the bromodomain and extraterminal domain family of proteins BRD2–4. The bromodomains target acetylated lysines at histones H3 and H4, while the extraterminal domain binds the viral integrase. By deletion or mutations of the bromodomain and extraterminal domain protein binding domain of the integrase, the integration pattern of gammaretroviral vectors can be changed. 45 In this case the integrase is fused to proteins that allow chromatin targeting, gammaretroviral vector integration can be guided and may become safer. 46
Preclinical assays to assess genotoxicity
The integration site preference will to some extent affect the safety of the vector. However, in addition to the characterization of integration site patterns, it is important to evaluate the safety of the retroviral vector in functional assays. These assays have been developed in vivo based on serial bone marrow (BM) transplantations in wildtype mice, or using tumor prone mouse models. C57BL/6 mice transplanted with BM cells transduced with gammaretroviral LTR vectors containing viral enhancer/promoters develop leukemias at low frequency (∼1–2 in 100 transplanted mice). 47 The change of the vector architecture to SIN by maintaining the viral enhancer in the internal position alone does not prevent leukemia induction in mouse models. 48 In contrast, no leukemias have been reported when using SIN gammaretroviral vectors with cellular promoters, although the integration site preference remains the same. In agreement with this observation, in the clinical gene therapy trial treating X-SCID with SIN gammaretroviral vectors the integration site distribution shows the same pattern as the LTR-driven gammaretroviral vectors but reduced incidences of integrations near potential oncogenes. This observation may be interpreted different ways, one of which may indicate that although the same preference exists the weaker enhancer activity of the internal promoter (in this case, the elongation factor 1 short promoter) prevented selection of clones with a growth advantage due to insertional activation of cellular gene promoters.
Selection of dominant clones (clonal dominance) precedes leukemia, and therefore, the evaluation of the clonal composition after transplantation of retrovirally transduced cells can be used as surrogate to assess vector safety. 49 –51 In this sense, the presence of strong clones could be correlated with vector integration sites close to (proto-) oncogenes. Hematopoietic clonality in mice transplanted with cells transduced with retroviral vectors with and without viral enhancer/promoter is distinctly different. 52 –54
Gene modification with lentiviral vectors has not been associated with oncogene activation by enhancer-mediated activation in patients yet. This may be attributed to the use of cellular promoters in lentiviral vectors instead of the HIV viral promoter. Recent long-term follow-up studies in HIV patients under highly active antiretroviral therapy (HAART) have shown evidence for clonal selection of cells with integrated HIV copies. In some patients, clones with intronic integrations in BACH2 and MKL2 were enriched. 55,56 By the same token, investigations using the more sensitive in vivo readout system by transplantation of transduced BM cells of the tumor-prone Cdkn2a deficient mouse strain revealed that also integrated lentiviral vectors can accelerate tumor onset 57 and that this effect is mediated by the strength of the promoter used in the vector. 58 Using the in vitro immortalization (IVIM) assay, which reads out insertional mutants with a replating phenotype, 59 the immortalization potential of lentiviral vectors with viral promoters in the internal position was also clearly demonstrated; however, the incidence is reduced by half compared to gammaretroviral vectors. 60 This can be explained by the different integration site preference of lentiviral vectors compared to gammaretroviral vectors that will less often hit the relevant position in Mecom, which is the most often activated and selected gene in the IVIM assay. However, some low replating frequency can be observed when using the locus control region in lentiviral globin vectors, 61 which is in line with the potential of the locus control region to activate gene expression in hematopoietic cells. 62 A new molecular surrogate assay for genotoxicity assessment that was developed based on the IVIM assay provides a more sensitive platform to evaluate vector safety 63 and will be very useful in the future to define differences in activation potential of vectors with weak promoters.
In addition, the presence of splice sites in vectors can interfere with cellular splicing. In a patient treated to cure beta thalassemia by lentiviral globin transfer, a dominant clone evolved with the vector integration in the third intron of the high mobility group AT-hook 2 gene (HMGA2). This integration led to the production of a truncated transcript that lacked the regulatory let-7 miR-target site in the 3′UTR causing a substantial increase of HMGA2 mRNA. 64 In a preclinical in vitro assay in BaF3 cells, splice interference from retroviral vectors can be assessed because vector integration upstream of the first coding exon of the growth hormone receptor can activate the gene by using splice sites in the vector and transform the interleukin-3 (IL-3)–dependent BaF3 cell line to growth with bovine growth hormone. 65 This very sensitive assay, however, can only test vectors that transcribe from a promoter upstream of the splice site (e.g., the promoter contained in the LTR upstream of the viral leader) thus is not suited for the current SIN vector design. 66
Although the introduction of splice sites in vectors should be avoided, so far all leukemias in HSC-GT were induced by enhancer-mediated (proto)-oncogene activation. In light of this, the incorporation of insulators with enhancer-blocking function in the vector may be of advantage, although formal proof that these insulators reduce the risk on insertional transformation in vivo is limited. 53,58 Additionally, insulators in the viral vector LTRs may shield the vector from the surrounding genome and may prevent silencing of the promoter in the vector. The loss of transgene expression turned out to be one of the major problems in the clinical trial treating patients with X-CGD. 13 However, in this study, a gammaretroviral vector containing complete LTRs was used. The viral LTR sequences and primer binding site can be recognized by proteins that in turn will trigger the shutdown of expression from the integrated provirus by methylation of CpG islands in the enhancer/promoters and histone modifications. 67,68 This mechanism is well described in embryonic stem cells but is also found during establishment of HIV latency. 69,70 Current-generation vectors may be less prone for recognition, as they contain only limited stretches of viral sequences. To prevent silencing in gene therapy vectors, researchers have investigated the use of chromatin opening elements, of which the one derived from the human HNRPA2B1/CBX3 genomic locus is most often used. 71,72 Through a series of optimization steps, this element was depleted of splice sites and reduced in size. 73 Introduction of this element can support transgene expression from retroviral vectors in HSC. 74,75
Finally, the preclinical evaluation of genotoxicity in human rather than mouse cells is frequently demanded but has so far not provided applicable information. This is probably due to the fact that human CD34+ cells cannot easily be transformed by insertional mutagenesis in vitro, and also not after transplantation in NOD/SCID/IL2rg−/− (NSG) mice. Possibly the incidence of such events is too low to be monitored because the number of transduced and transplanted CD34+ cells is usually low (as low as 5 × 104 cells/mouse). The interpretation of the potential safety of retroviral vectors because no leukemia was observed in the NSG transplantation model should, therefore, be considered with caution, as relevant positive controls (inducing leukemia in the mice by the respective mechanisms) are missing.
To summarize the above, it appears that the enhancer strength of the promoter has the major impact in activating cellular genes, that using the SIN configuration of the vector allows the use of weaker cellular promoters, and that the incorporation of splice sites should be avoided. This will reduce the likelihood to deregulate cellular gene expression and thus lower the risk for cell transformation. The integration site preference, however, can only be influenced by the choice of retroviral vector platform. In light of this, gene editing by introducing DNA breaks at defined positions in the genome followed by homology-directed repair will become very interesting in the future (reviewed elsewhere 76,77 ). However, this procedure relies on the transfer of multiple components into HSC at the same time which currently remains challenging.
The Tricks
HSC maintenance: what is best?
Ex vivo cultivation of HSC is a prerequisite for their efficient genetic modification; however, this implies removing stem cells from their natural habitat, depriving them of their supportive microenvironment. Moreover, gammaretroviral transduction is dependent on cell division. The same is true for homology-directed double-strand break repair during gene editing, which is most efficient during S/G2 phase of the cell cycle. Therefore, HSC need to actively cycle in vitro at least once, which is usually accompanied by loss of repopulation capacity. 78,79 Additionally, it was recently demonstrated that prolonged ex vivo cultivation (from 36 hours on) of human CD34+ cord blood (CB) cells reduced their repopulation capacity. 80 Clearly, defined ex vivo culture conditions are important for the maintenance of efficiently modified HSC for gene therapy.
HSC are cultured in cytokine-replete medium and the impact of cytokines/cytokine cocktails to maintain or expand HSC has been thoroughly investigated (see Table 1). Evaluation of cytokines was based on their potential to promote proliferation of human long-term culture-initiating cells and the preservation of repopulation capacity. Of the cytokines tested, only the ligand for fms-like tyrosine kinase 3 (FLT3) and thrombopoietin (THPO) were able to expand long-term culture-initiating cells as single factors, whereby the most efficient combination was FLT3-L, stem cell factor (SCF), and IL-3. 81 Another study came up with the combination of THPO, FLT3-L, and SCF as most potent in increasing cobblestone area–forming cells and preserving marrow repopulation capacity comparable to freshly isolated CD34+Thy1+Lin− cells. 82 Specifically in the context of gene therapy it was also suggested that FLT3-L provides survival signals to CD34+ cells during retroviral transduction. 83 Murine BM cells could efficiently be expanded with a combination of SCF, IL-3, and THPO, whereby THPO shortened the time to first cell division. 84 THPO was further proven as a potent factor for HSC expansion and maintenance of repopulation capacity in long-term whole mouse BM cultures followed by transplantation. 85 Recently published work concludes that the combination of FLT3-L, THPO, SCF, and IL-3 is the best-established prestimulation condition in HSC gene therapy trials. 86 However, IL-3 has been implicated in reduced repopulation potential of HSC and the predominant support of progenitors in short- and long-term cultures prior xenotransplantation, questioning its undeterred use. 87,88
Overview of cytokines and small molecules used to expand hematopoietic stem cells in vitro
Angptl, angiopoietin-like; BM, bone marrow; CAFC, cobblestone area-forming cell; CB, cord blood; FLT3, fms-like tyrosine kinase 3; HSC, hematopoietic stem cell; IGF, insulin-like growth factor; IL, interleukin; LDA, limiting dilution assay; LTC-IC, long-term culture-initiating cells; LT-HSC, long-term HSC; SCF, stem cell factor; SRC, severe combined immunodeficiency repopulating cells; THPO, thrombopoietin; VCN, vector copy number; n.d., not determined.
In a screen for HSC-supportive factors, Zhang and colleagues furthermore identified angiopoietin-like 2 (Angptl2) and Angptl3 as potent inducers of ex vivo long-term (LT)-HSC expansion. 89 The same group later additionally reported Angptl5 and insulin-like growth factor (IGF)-binding protein 2 as similarly potent expansion factors based on the measurement of SCID repopulating cells. 90 However, these factors have not been used in HSC-GT trials. A chemical compound screen in the zebrafish embryo identified prostaglandin E2 as capable of expanding HSC. 91 Cultivation of mouse whole BM cells with the stable derivate 16,16-dimethyl-PGE2 (dmPGE2) alone resulted in a 2.3-fold expansion of LT repopulating cells. In a further study dmPGE2 incubation of whole BM cells could confirm HSC expansion and coupled it to mechanisms of increased survival and homing capacity with higher expression of CXCR4 and survivin and reduced caspase-3 activity. 92,93 A phase 1 trial evaluated the reproducibility of these data in patients, by stimulating cord blood (CB) stem cells with dmPGE2, but remained inconclusive. 94 The impact of dmPGE2 on retroviral transduction was tested within a xenotransplantation model. Here, dmPGE2 treatment of CB CD34+ cells did not improve the overall human engraftment in NSG mice but did significantly increase the proportion of engrafted transduced cells. 80 Anyhow, the overall high vector copy number reported (2–4 no PGE2; 4–6 PGE treated) are not comparable to vector copy number reported from gene therapy trials using the same multiplicity of infection (MOI); however, this may be due to the use of cord blood–derived CD34+ cells. Nevertheless, they could link improved transduction to an endocytosis-dependent early step in viral transduction.
In a large screen for molecules capable of promoting HSC expansion, Boitano and colleagues identified StemReginin-1 (SR1), a purine derivative and inhibitor of the aryl hydrocarbon receptor, as a potent inhibitor of HSC differentiation during cytokine induced proliferation. 95 They calculated a 17-fold increase in SCID repopulating cell numbers compared to the cytokine only controls. In our own studies, SR1 expanded phenotypical human CD34+ CB cells in vitro but did not increase human engraftment in NSG mice after retroviral transduction. 96,97 The use of SR1 expanded CB units was further evaluated in a phase 1/2 trial, reporting a 330-fold increase in CD34+ cells. Nevertheless, only 6 of 20 transplanted patients showed a dominant chimerism of the SR1 expanded CB unit and further 5 a mixed chimerism with the simultaneously transplanted unmanipulated CB unit, while 6 patients repopulated dominantly with the unmanipulated graft (and 3 not at all). 98 Therefore, CD34+ cell expansion in vitro by SR1 does not guarantee for engraftment. Based on their reported anti-apoptotic and differentiation inhibiting effects, SR1 and dmPGE2 were evaluated in zinc-finger nucleases (ZFN)-mediated gene editing. Interestingly, while dmPGE2 increased the fraction of transduced cells, its combination with SR1 seems to reduce this effect, reflected in a lower frequency of transduced cells in the transplanted mice of the SR1/dmPGE2 group compared to dmPGE2 only group, 99 which leads to the suggestion that dmPGE2 is sufficient to improve gene modification and engraftment.
More recently, Fares and colleagues proposed the pyrimidoindole derivative UM729 and its 10–20 times more efficient analog UM171 as a new small molecule for ex vivo HSC expansion, apparently exerting its effect independently of aryl hydrocarbon receptor inhibition. 100 Cultivation with UM171 was reported to suffice to preserve the most primitive human HSCs (CD34+CD90+) over 7–14 days of culture. UM171-expanded and lentivirally transduced mobilized peripheral blood (PB) HSC did not show significantly improved engraftment potential compared to a minimally cultured and transduced control. 80 Similarly, a preclinical test of UM729 addition to cytokine cocktails during lentiviral-based HSC gene transduction did not suggest a clear effect. 101
To summarize, current HSC in vitro transductions are conducted in cytokine supported media, which preserve the HSC phenotype and engraftment potential. The most reliable combination is SCF, THPO, FLT3L, and IL-3. The addition of small molecules, like SR1 or UM171, seemed to boost the cytokine induced effect, but this was not necessarily accompanied with improved engraftment. Further, with the exception of above cytokine cocktail, none of the compounds reaching a clinical trial prevailed, despite promising data from pre-clinical studies and thus questioning the predictability of these as models for human hematopoietic stem cell transplantation (HSCT). The available and most often used models (NOD/SCID-IL2rg−/−) IL2rg-deletion (NSG mice) or truncation (NOG mice) have some critical drawbacks, which are as follows: (a) human HSC quiescence is poorly promoted by the murine stem cell niche, (b) differences of the murine and human HSC phenotype, anticipating varying demands on the microenvironment and cytokines supplied for efficient engraftment, (c) the inability of human megakaryopoiesis and erythropoiesis to evolve in the humanized mouse, and thus (d) second engraftment is not well supported. Attempts of solving these problems have been made with different human cytokine knock-in mice or even the transplantation of human fetal tissue, 102 –105 albeit with limited clinical relevance thus far.
Although new factors and small molecules have been identified in the past to expand HSC, the large screens did not come up with an exceptional molecule. This might be the reason that we still rely on the classical culture conditions for HSC-GT. Therefore, one could think of other more HSC-related mechanisms that have been reported for in vivo HSC expansion, like antagonizing CXCR4. Thus, pharmacological CXCR4 blockade was associated with expansion of the most primitive HSC pools in BM, apparently due to cell cycle recruitment. 106 This alternative approach might also help to overcome the difficulties associated with gene editing protocols, relocating the expansion of modified cells to the in vivo niche which better supports their maintenance. Did we reach a limit of in vitro expansion because of an intrinsic limit of HSC self-renewal before exhaustion? In this respect, Bernitz and colleagues used a label-retaining transgenic mouse model and counted a maximum of five divisions before LT-HSC lost their full capacity of reconstitution. 107 Whether this is predictive for the human system remains to be shown. However, in principle this suggests that with further increased HSC expansion, we would inevitably lose LT-HSCs. Lower numbers of HSCs would amount to fewer clones contributing to hematopoiesis after transplantation, which brings up another important concern in the field of gene therapy: clonal diversity after HSCT and the risk for clonal dominance.
Clonality—the model matters?
In HSC-GT, due to the occurrence of leukemias induced by vector integration and following clonal dominance, the clonality of hematopoiesis is of special interest and is routinely monitored during the follow up of gene therapy patients by analysis of the vector integration sites. To evaluate the information gleaned from these studies, it is of foremost importance to understand the clonal dynamics of normal hematopoiesis and hematopoiesis after transplantation.
Our knowledge about clonal diversity during human steady state hematopoiesis is limited, but animal models can contribute to our understanding. In humans, clonal dominance is observed during aging and is associated with a higher risk of leukemia development or myelodysplastic syndromes, although the causality is not clear. In the context of gene therapy, the observation of dominant clones could be either the result of a low number of transplanted engrafting HSC, or the result of insertional mutagenesis.
It can be assumed that clonality depends on the number of transplanted cells. For transplantation of unmodified autologous HSC from BM or mobilized PB HSC, a minimum of 2 × 106 CD34+ cells/kg body weight is recommended for successful engraftment, and higher doses may be associated with faster myeloid engraftment. 86,108,109 The fact that, on a per-kilogram basis, mice typically receive at least one log-scale higher “stem cell” doses may impact on the comparability of clonality studies between mouse and man.
In attempt to track HSC in native hematopoiesis, a doxycycline-inducible expression of a hyperactive Sleeping Beauty (HSB) transposase was used in the mouse (M2/HSB/Tn mouse model). A defined DNA transposon will mobilize and integrate at a new unique genomic position upon HSB transposase activation, generating trackable genetic marks. This transposon-based marking and analysis in mice suggested that native hematopoiesis is mostly driven by long-lived committed or multipotent progenitors (MPP), since <5% of LT-HSC tags were found in mature cell types. Instead, transposon tags were more often shared between LT-HSC and mature progeny following transplantation. However, only 30% of the LT-HSC pool was labeled, leaving open how the majority of this pool contributed to steady-state hematopoiesis. 110 Another study employed Cre-recombinase-mediated fluorescent labeling of HSC in a tamoxifen-inducible and Tie2 locus restricted fashion. This system is tightly controlled and resulted on average in 1% labeled LT-HSC. Such fluorescently labeled hematopoiesis also suggested that steady-state hematopoiesis is driven by short-term (ST) self-renewing HSC and progenitors more so than by LT-HSCs, similarly to what was reported with the transposon tags. 111 The authors calculated about 5,000 active HSC clones in the mouse representing 30% of the total HSC pool. They also described a strong lineage bias of HSC and an increased participation of LT-HSC after transplantation. Similar observations are reported from lentiviral vector mediated barcoded HSC. Transplanted HSC showed a strong lineage bias and low correlation was found between LT-HSC and progeny detected barcodes. 112 Recently, also human HSCs were reported to acquire early lineage determination, which is inherited to their progeny. 113
As mentioned before, xenotransplantation models for the investigation of human HSC behavior suffer from several shortcomings. Thus, non-human primate studies are desirable for better comparison to human, regarding not only life span, but also comparable phenotypic characteristics. A short-term (1–9 months; 4 animals) and a long-term (up to 12 years; another 4 animals) clonal tracking study in rhesus macaques reported that reconstitution early after transplantation (around 3 months) is characterized by the fluctuation of clones, of which many are unilineage. Persisting ST- and LT-tags started to contribute to hematopoiesis from 2–4 months and 7–13 months after transplantation, respectively. HSC clones with balanced myeloid and lymphoid output were observed in the long run of the study and replaced those with biased output. Clonal contribution was fixed approximately 3 months after transplantation, of which the largest clones also represented low differentiation bias. 114 –116
Monitoring of lentiviral integration sites in WAS patients after gene therapy revealed a low output of CD34+ cells towards the different lineages during the first 3 months after transplantation. HSC- and MPP-derived integration sites were detected from 3–6 months onwards and identical clones reached a stable level around 12 months with stabilized multilineage output. Back to steady-state hematopoiesis, clonal diversity increased and showed stable polyclonality, approximately 1,200 clones within the CD34+ pool supporting hematopoiesis. 117 Patients from a gammaretroviral vector-based gene therapy for ADA-SCID also were successively monitored for clonal diversity. In the individual patients a poly- or oligoclonal situation persisted over time, but the authors remarked that only patients with high complexity showed sufficient B-cell reconstitution. This could not be correlated with transplanted cell dose or transduction efficiency, but with the age of the patient (and his stem cells) at the time of transplantation. The estimated total number of integration sites in the patients varied from 50 to 25,000. 15,118 Similar pool sizes of hundreds to thousands of clones were detected in another lentiviral based gene therapy trial for WAS. None of the clones accounted for more than 10% of the population, revealing a polyclonal distribution. 17 Based on the reported clone numbers in the different trials one would eyeball a content of 103 to 104 LT-HSC clones within a normal graft. Are these numbers realistic? Considering that approximately 0.01% of CD34+ cells are LT-HSC, and assuming a 100% transduction efficiency you could expect around 3,500 LT-HSC clones based on the transplantation of 7 × 106 CD34+ per kilogram and a 5 kg child. Either this example significantly underestimates the number of HSC, or the reported clonality estimate includes progeny of long-lived precursor and progenitor cells in addition to HSC.
As summarized in Fig. 1 the kinetics of progenitors early after transplantation and LT-HSC taking over hematopoiesis in the long run are most similar between human and the reported non-human primate studies. Based on the reported clinical data the early phase after transplantation would be reflected by a clonal succession model and end up with a combination of a “clonal stability” and “dynamic repetition” model of hematopoiesis. 119 The murine model in general seems to favor the overall reconstitution by MPPs; LT-HSC contribution is found quite early after transplantation but to a low amount, ending up with a more oligoclonal situation with stronger differentiation bias. As life expectancy in mice is highly different to human, the exact comparison of the reconstitution kinetic is difficult to judge.
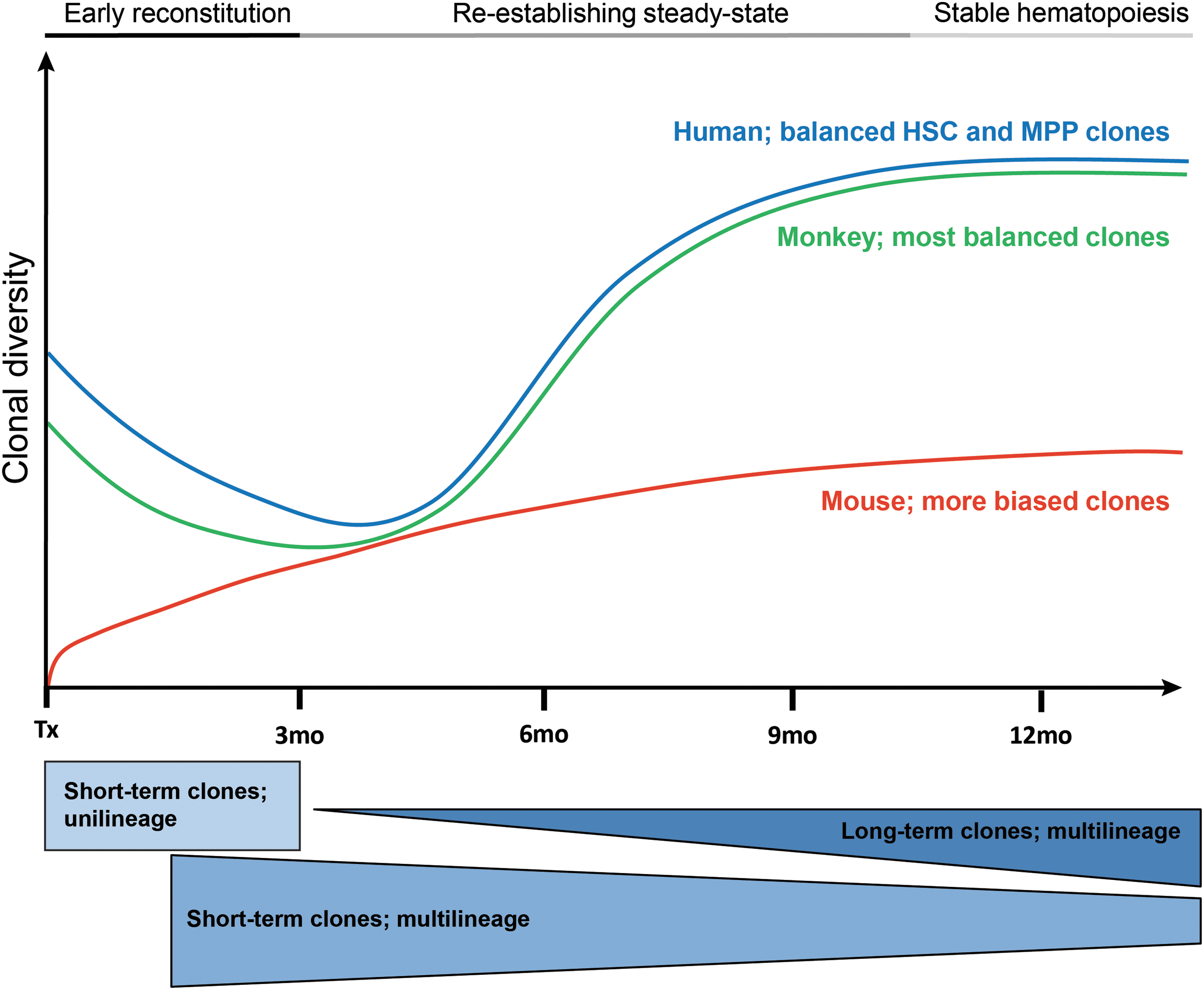
Comparison of hematopoietic reconstitution in different species after gene marking or even gene therapy. In monitored Wiskott-Aldrich Syndrome patients and non-human primate studies clonal succession and increasing clonal diversity over time characterize immature and mature hematopoiesis early after transplantation. Detected short- and long-term clones were similar in their kinetics and shifted from unilineage to multilineage output at steady-state as a result of hematopoietic stem cell-directed (HSC) and multipotent progenitors (MPP) contribution. 114 –117 Why murine hematopoiesis remains oligoclonal (characterized by the fluctuation of clones and less clonal stability) for extended periods of time remains unclear but elicits questions regarding its value as a model. 54,112,146 –149
As vector safety has massively improved over the past years, the question arises: What other factors would contribute to HSC fitness and the clonal outcome after transplantation? In a comparison of culture conditions for lentiviral transduction of murine HSC clonal diversity was highest when short prestimulation and prolonged postexpansion conditions were applied. 120 Further, transplantation of insufficient numbers of HSC may contribute to leukemogenesis. This was recently demonstrated in the murine model of SCID-X1. Rag2−/− γc−/− mice transplanted with low numbers of wildtype hematopoietic stem and progenitor cells developed leukemia, while this was not the case when higher numbers were transplanted. 121 Similarly, T-cell acute lymphoblastic leukemia was reported in the absence of a donor T-cell progenitor supply in mice. 122 Furthermore, in some patients, HSC may be impaired due to persisting infections. This was demonstrated for HSC of CGD patients. 123 In CGD patients as in mice, constantly elevated levels of inflammatory cytokines activate HSC into a cycling state, impairing HSC function. The impact of inflammatory cytokines on HSC maintenance was recently demonstrated in murine models. 124 Finally, the transduction by retro/lentiviral vectors may cause innate immune responses in HSC, on the one hand interfering with transduction efficiency, and on the other hand triggering p53-dependent apoptosis of transduced HSC. 125 –127
Concluding from the reported data, clonal dominance seems not to play a major role within the latest gene therapy trials. However, it is not clear how culture conditions, massive expansion, and genetic modification would skew clonal outcome in the long run. If considering genomic instability causal for age-related leukemia, the risk would be much higher for HSC-GT patients to develop those. Although gene editing methods are supposed to be less genotoxic in terms of irregular gene expression, how off-target activity of the nuclease would affect genomic stability of HSCs has not been demonstrated. The observation that gene therapy for young patients is more efficient than for older ones moreover suggests that the disease pheno/genotype plays a so far under-considered role for the outcome of hematopoietic reconstitution. Thus, a sufficient number of (ideally, young) HSCs and controlled vector integration would mainly determine the balance of clonal dominance and diversity.
The Targets
HSC-GT first and foremost can target genetic disorders of the hematopoietic system; lineage-specific diseases can be addressed by using lineage-specific promoters, diseases affecting all hematopoietic cells by ubiquitous or hematopoietic-specific promoters. In theory, any genetic disorder amenable to HSCT should be a suitable target for HSC-GT, and in fact, the ever-increasing safety of allogeneic HSCT has been and continues to be one of the greatest competitors to more widespread application of HSC-GT approaches, as patients with suitable donors have for the most part been excluded from HSC-GT protocols. Given the very high level of safety now provided by improved stem cell harvesting, vectors, transduction and conditioning protocols, the current preference of HSCT over HSC-GT, when possible, may need to be revisited.
In selecting targets for HSC-GT, we posit that inability to rescue the phenotype with allogeneic HSCT (and the clinical necessity to resort to such drastic therapeutic approaches) should preclude, for ethical and economic reasons alike, HSC-GT approaches for that diseases—after all, allogeneic HSCT provides 100% competent cells compared with a couple percent for the genetically engineered autologous HSC. So what is left is a rather narrow corridor of eligibility of a patient for HSC-GT which will constantly have to be redefined as progression on all fronts is being made. Three areas, primary immunodeficiencies, inborn errors of metabolism, and hematological diseases have seen most activity. In the interest of brevity, only the at least partially effective disease entities are briefly discussed, although some information is also to glean from the unsuccessful ones.
Primary immunodeficiencies
Primary immunodeficiencies have been both obvious targets for HSC-GT and comparatively easy in achieving engraftment in many of these diseases. Adenosine deaminase deficiency (ADA-SCID) was the first disease treated with blood-cell directed gene therapy. Although strictly speaking, an inborn error of purine metabolism, characterized by accumulation of (deoxy)adenosine in all cells, which inhibits ribonucleotide reductase and thus prevents DNA synthesis and also causes intracellular accumulation of toxic levels of S-adenosylhomocysteine, SCID is its leading manifestation, with pronounced lymphopenia. HSCT is curative for the SCID but not for the nonhematopoietic toxicity associated with ADA deficiency. Enzyme replacement therapy (ERT) is available and seems to target and protect also non-hematopoietic cells, but is costly and may lose efficacy over time. While initial gene therapy attempts were directed at T-cells, 128 shortly thereafter several HSC-GT protocols were independently developed, one of which has become the first commercial hematopoietic stem cell gene therapy (Strimvelis). The critical breakthrough for ADA gene therapy was not advances in vector or transduction technology, but rather the decision to introduce a nonmyeloablative course of dose busulfan conditioning. 5 The protocol is quite successful in that all 18 patients treated with this protocol between 2000 and 2011 remained alive in 2016 (however, 20-year survival with ERT is also >80%) without a single case of insertional leukemia. 5,16 Besides absence of iatrogenic damage, the impression of a clinical benefit was reported for most patients, of note, compared with the presumed spontaneous course of the disease, not compared with standard of care, ERT. Similarly promising outcomes were reported by several other groups which all together comprise more than 40 patients, all alive, none with insertional mutagenesis-related leukemias, and many with at least somewhat functional adaptive immune systems. 14,15 The question of whether successful HSC-GT makes ERT redundant, as most groups have concluded, is not self-evident, given that stem cell-directed gene therapy addresses only the hematopoietic system. Indeed, poor growth continues to characterize ADA patients after HSC-GT, although children develop along their calculated growth curve. 129 Despite use of classical gammaretroviruses with transgene expression driven by viral LTRs, no untoward genetic events have occurred, even though detailed analyses show insertion at the same hotspots as implicated in the insertional leukemias observed after other stem cell-directed gene therapies. Expressing the enzyme apparently provides a survival advantage that at least balances out the burden of expressing an extra gene. Pools of gene corrected stem cells, albeit small, are maintained, and are obviously sufficient to provide approximately normal numbers of (largely gene-corrected, reasonably polyclonal, and somewhat functional) lymphocytes.
X-linked deficiency of the common gamma chain of the receptors for six nonredundant interleukins causes classical SCID. Survival hinges on early HSCT or, if no donor is available, HSC-GT. In clinical studies, the Paris (Fischer/Cavazzana) and London (Thrasher) groups transplanted a series of 20 young boys, with autologous CD34+ cells transduced with similar vectors as for ADA deficiency. 4,6 Even without conditioning, long-term engraftment of gene-corrected cells was most often observed, giving rise to a numerically normal and largely functional, polyclonal adaptive immune system in the majority of patients. By contrast, a similar attempt failed in a cohort of older patients. Sadly, with variable delay from transplantation a quarter of the children went on to develop acute leukemia, caused by insertional mutagenesis, to which at least one succumbed. 9,10 All leukemias were characterized by further mutations (NOTCH1 gain-of-function mutation, deletions in the CDKN2A locus). These unexpected occurrences triggered a flurry of studies aiming to predict and prevent genotoxicity which are discussed above. Consequently, a multinational trial of gene therapy in X-linked SCID used one of those new tools, a SIN γ-retrovirus vector devoid of strong viral enhancers; 7 patients benefited and none suffered genotoxicity. 17 Specific analyses suggest, although for definitive conclusions it is too early, that indeed SIN gammaretroviral vectors with mammalian promoters may predispose less to catastrophic genetic events.
Chronic granulomatous disease (CGD) is caused by a heritable deficiency for a phagocytic oxidase protein of the NADPH oxidase complex; inability to generate reactive oxygen species impairs the ability to kill phagocytosed pathogens. Allogeneic HSCT is curative, but inherently dangerous because of lingering infections. In CGD, expression of the therapeutic gene does not entail a selective advantage, necessitating full conditioning and very high transduction rates. Selection of suitable patients (who, when) is also highly challenging since the course of CGD is unpredictable, as well as chronic inflammation jeopardizes the ability of BM stroma to support engraftment. In spite of these challenges, about two dozen patients have received HSC-GT for CGD, albeit never with protracted transgene expression and development of MDS in three patients, to which one succumbed.
Wiskott-Aldrich Syndrome (WAS) is due to hemizygous deletion of the hematopoietic-specific WAS protein (WASp), which activates actin polymerization. WAS presents with thrombocytopenia and/or immunodeficiency. Autologous HSC-GT with an essentially similar virus as in the first X-SCID trial, was performed. 7/9 developed acute leukemia to which several succumbed. Subsequent trials with a lentiviral vector and the WASp promoter driving expression of the therapeutic gene have enrolled at least 11 patients, thus far apparently without adverse genetic events, with sustained high levels of transgene expression in polyclonal populations in all hematopoietic lineages, and with strong evidence of clinical efficacy, albeit not full correction of the phenotype. 17,18,130
Inborn errors of metabolism
Inborn errors of metabolism can be effectively treated with HSC-GT if their phenotype is due to accumulation of toxic metabolites in plasma, which metabolically competent hematopoietic cells can take up and break down into nontoxic metabolites. The “HSCT test” applies: if it can't be cured with allogeneic HSCT, it's no target for HSC-GT either. Several potential targets have been identified, but with the exception of X-linked adreno-leuko-dystrophy (X-ALD) and MLD, not matured beyond pre-clinical studies.
X-linked adreno-leuko-dystrophy is the result of any of >600 different loss-of-function mutations in a gene encoding for a peroxisomal transport protein, ABCD1. One consequence is accumulation of very long chain fatty acids in tissues anywhere in the body, but specifically in adrenal glands, testes, and brain white matter. Progressive neurodegeneration being the chief cause for morbidity and ultimately death, it was hypothesized that replacement of ABCD1-deficient by ABCD1-competent phagocytes might alleviate (progression of) symptoms. The hypothesis that this might be achieved by HSCT was in part confirmed: Specifically, boys with minimal disability and modest MRI changes were protected long-term (although possibly not indefinitely) from disease progression. Recent studies indicate a long-term symptom-free survival in these boys of 95% compared to natural disease progression which will see almost 50% patients die by 5 years. With this in mind, the idea to aim for phenotype control with HSC-GT is a bar set high. HSC-GT (using lentiviral vectors) was successfully attempted by the Paris group 21 and triggered the commercially sponsored (bluebird bio) Starbeam trial; it has enrolled 17 of the targeted 25 patients. Treatment was with myeloablative conditioning followed by lentivirally transduced autologous CD34+ cells. According to data presented at the 2016 American Academy of Neuology (AAN) meeting, 16/17 patients seem to have benefitted in that neurological deterioration was largely arrested; insertional events were not observed. 131 Further updates are expected in the near future.
An essentially similar disease is metachromatic leukodystrophy which is inherited in an autosomal recessive fashion; deficiency of arylsulfatase A leads to accumulation of pathological (lyso)sulfatides which cause degeneration of myelin sheaths. The impression is that pre-symptomatic stages of MLD are responsive to HSCT, altering the trajectory of neurodegeneration, although not providing a cure. The Milan group is performing a clinical trial of HSC-GT, using their established platform with lentiviral gene transfer into autologous HSCs for transplantation after busulfan-conditioning of presymptomatic patients with early-onset disease. 23 An interim report after treatment of 9/20 patients gives the impression of safety and delayed or arrested disease progression/onset of symptoms. 22 In this study, the cross-correction of nonhematopoietic cells in the nervous system by migrating transduced hematopoietic progenitors was demonstrated.
Miscellaneous
The relative resistance to malaria provided by a heterozygous (carrier) status for certain ß-globin mutations is responsible for the high prevalence of the ß-globin diseases ß-thalassemia and sickle cell disease. For the most part, both can be very successfully treated conservatively, with frequent red blood cell transfusion combined with iron depletion, albeit requiring high patient compliance and associated with considerable cumulative costs. 132 Thus, allogeneic HSCT, although obviously curative if successful, is indicated in a highly select subgroup of patients. ß-thalassemia patients with residual production of ß-like globins (termed ß+) rarely need HSCT. In high-risk ß-null thalassemia and sickle cell anemia patients, the unpredictable clinical course bedevils identification of the ideal timing of allogeneic HSCT: Inherent risks of complications restrict the indication to patients who cannot be managed conservatively, but late transplantation is often limiting due to toxicity. 133 Autologous HSC harvesting in ß-globin disease patients has its own challenges, myeloablative conditioning is required to achieve engraftment of gene-corrected HSCs and for lack of a selective advantage, very high doses of transduced cells are required. The assumption that HSC-GT will be safer than allogeneic HSCT remains to be tested. Thalassemia syndromes are in principle straightforward to treat with HSC-GT, by expressing ß-(like) globin under the control of an erythroid-specific promoter. For sickle cell disease, a more sophisticated approach is required, since the disease is caused by a qualitative defect in ß globin, but anti-sickling mutants of ß globin have been generated. Alternatively, induction of γ globin by “editing” (gene editing, small hairpin (sh)RNA, etc.) motifs switching transcription from the ß to the γ locus (e.g., BCL11A), can potentially result in sufficient levels of fetal hemoglobin to cure ß-globin disease. 134,135 Gene correction of sickle-ß-globin with gene editing tools will be an interesting challenge (for thalassemia, there are too many different mutations). The lentiviral vectors for ß-like globin have proven very safe. Clinical benefit from HSC-GT was derived by patients with ß+ thalassemia, but less so by patients with ß-null disease, and even then, benefits may not be consistent. An excellent and very recent article by Zhi et al. 3 provides detailed review of the status of HSC-GT in ß-globin disease; suffice it to say that significant inroads have been made, major hurdles been overcome, and clinical benefit been demonstrated, but the role of this approach relative to conservative therapy and allogeneic HSCT as well as the selection of patients and the timing for HSC-GT and many more issues remain to be addressed.
Fanconi anemia is a family of disorders of DNA repair, the hematopoietic aspects of which—marrow failure and predisposition for leukemiaare treatable with allogeneic HSCT, despite the particular toxicity of conditioning therapy for Fanconi patients. These will equally apply to conditioning for HSC-GT, with the added complications of HSC extraction, especially from patients with advanced marrow failure, and difficulties of transduction because of the chromosomal fragility. The occasional cases of mosaicism or spontaneous genetic correction of Fanconi anemia clearly indicate a stark selective advantage of gene corrected cells, 136 predicting that even if initially a minority, over time the gene corrected cells will replace all mutated cells, although this expectation is not universally confirmed by the limited body of clinical data available. 137 Several studies plan to offer HSC-GT to Fanconi patients, including one at the Fred Hutchinson Cancer Research Center (open since 2012) and one at the Vall d'Hebron University Hospital (open since 2013). If either has recruited any patients—and if so, what has become of them—has apparently not been reported. Instead, a recent paper summarizes two decades' worth of failures bringing HSC-GT to Fanconi patients. 138
Chemoresistance genes have been popular tools for in vivo selection of gene modified cells in preclinical models. At least one study has attempted to explore chemoresistance gene transfer as an avenue toward reducing off-target toxicity of (in this case, glioblastoma-directed) chemotherapy. 139 The authors report that an increased cumulative dose of temozolomide was tolerated as a result of the gene therapy, although only a minuscule fraction of hematopoietic cells was gene corrected, thus whether these conclusions are justified is not entirely clear, as clearly the temozolomide tolerance of the nontransduced cells equally strongly exceeded expectations.
The famous (and famously irreproducible 140 ) experience of the “Berlin patient” who achieved cure from HIV from a CCR5-null allogeneic HSCT led to attempts to knock out CCR5 by means of gene engineering tools. Gene knock-out was successful per se, CCR5 gene engineered T-cells repopulated HIV patients' immune system, eventually contributing one seventh of PB T-cells, but viral recrudescence was seen in all, albeit with variable kinetics, upon discontinuation of expanded highly active antiretroviral therapy. 141 Not discouraged by these findings, gene editing in HSCT was attempted, and in NOD mice engrafted with the product thereof. When challenged with C5-tropic HI virus CCR5-engineered cells were spared from the virus-induced destruction of lymphocytes. 142 Several clinical trials of autologous HSCT with CCR5 gene editing or knock-down with short hairpin RNA are listed as recruiting, but no results have come forth.
What overarching conclusions can be gleaned from the cumulative data on HSC-GT?
Thus far, successfully performed HSC-GT approaches have certain commonalities with respect to the genetic aberrations causing the targeted disease. Most successful gene therapies were for diseases caused by loss-of-function mutations, which were additionally characterized by the following criteria: tight temporal, spatial, or reactive regulation of the gene product was not required; small or modest amounts of gene expression sufficed to achieve meaningful disease modification; and the therapeutic gene expression cassette did not exceed the limitations to genetic payload of currently available vector systems. If the therapeutic gene provides a selective survival effect, ideally already at HSC level, this will be useful, although even without an apparent selective advantage HSC-GT can be effective, then requiring high transduction rates and patient conditioning prior to transplantation (Fig. 2). Use of lineage-specific promoters can direct gene expression to specific lineages, with the potential advantages of (i) no deselection of lineage-undetermined stem cells and progenitors due to metabolic burden of transgene expression for which they have no use in the first place, (ii) absence of toxicity of an ectopically expressed transgene, and (iii) reduced risk of malignant transformation by activation of transgenes only after lineage specification (i.e., in cells devoid of stemness). Lineage-specific promoters will be particularly relevant in diseases where very high transgene expression is required, such as in hemoglobinopathies.
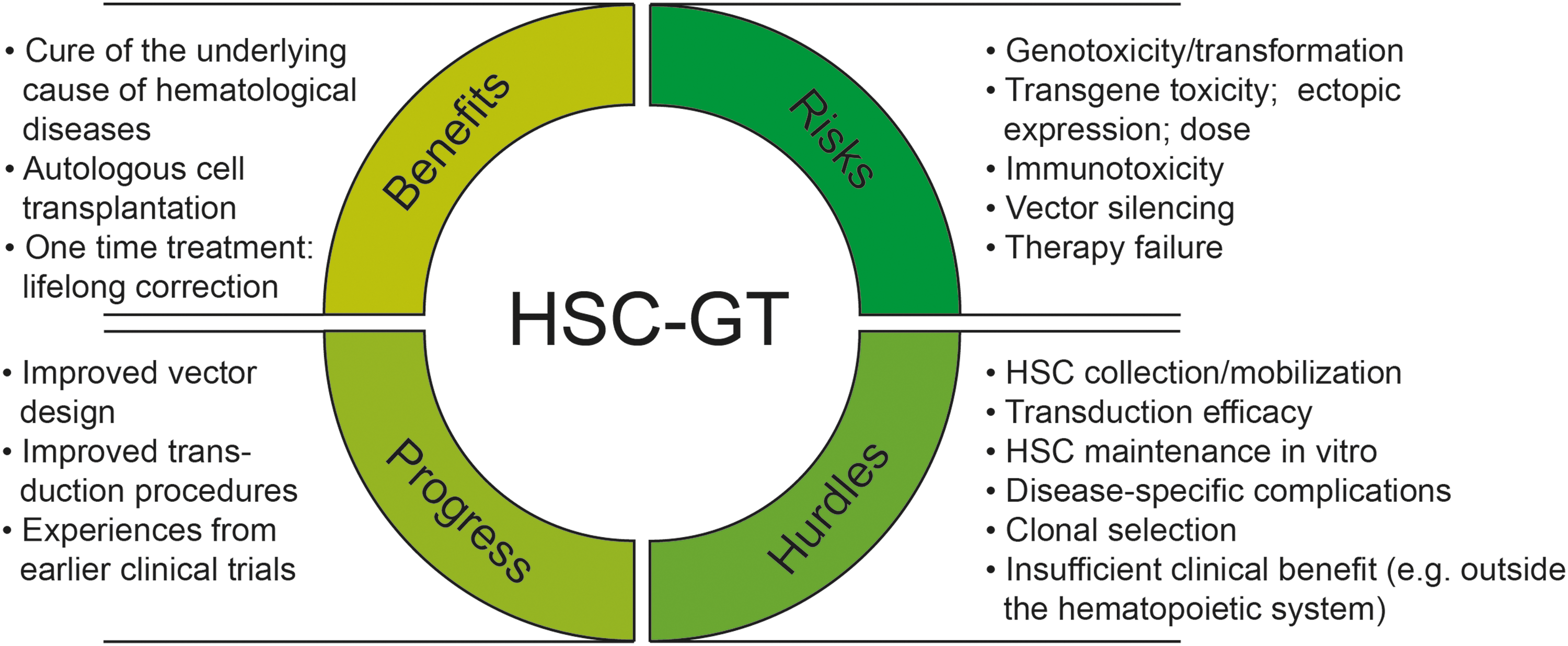
Hematopoietic stem cell gene therapy (HSC-GT): Benefits, risks, and hurdles.
A different class of genetic disorders that could in theory benefit from gene therapy are those diseases where instead of a knock-out/knock-down a pathological gene product is generated which interferes with normal gene function, for instance in a dominant-negative fashion. Novel gene editing techniques, even if not for a while sufficiently specific to actually elicit gene repair without causing too much collateral damage, can potentially be utilized to destroy the pathological gene to induce a state of haplo-insufficiency, which is often asymptomatic. Similarly, activation of genes that might be able to compensate for the deficient gene is a promising strategy, as, for instance, applied to ß-globin disease where re-expression of γ promises to cure any ß-globin disease irrespective of the mutation in the ß-globin gene and its pathophysiological consequences. For both, novel gene editing tools will be very useful.
HSC-GT: how much further?
In light of the above-summarized treatment failures and even casualties of HSC-GT, the occasional claim that somatic gene therapy is “safe and effective” and ready to enter main stage may appear premature, but very significant advances have been made. Novel vector generations—SIN gammaretroviral or lentiviral vectors with nonviral promoters—have been developed for both of which no transformative effects have been observed, even including very sensitive assays. Importantly, the relevant comparator for the efficacy of HSC-GT should not be their impending death if left untreated, but the risk associated with standard of care – often ERT (or transfusion in the ß-globin diseases) or allogeneic stem cell transplantation. Clearly the major enemies of personalized medicine lure elsewhere: Currently laws and regulations governing drug licensing are based on cohort studies (i.e., the exact opposite of personalized medicine). We posit that there should be a selection of licensed gene therapy tools that could be readily combined with the relevant expression cassette. Manufacturing authorizations could be for vector types and transduction protocols irrespective of cargo. Another impediment to widespread availability of HSC-GT has been that thus far, the cost for gene therapy has been born almost exclusively by charities and funding agencies, and the prospect of scientific reputation has been the fuel driving these activities. The inevitable consequence thereof is that for each disease, with each vector and conditioning regime, very small cohorts are treated—because academia's “currency” is publications, hence novelty is paramount. If the aim is to cure many patients, the community must devise ways to keep costs low and to secure reimbursement. Here the two major obstacles tie together: It is in the vested interest of society as a whole, as well as that of patients afflicted with these rare genetic diseases, to keep the regulatory bar surmountable, thus development costs affordable, in order to enable general access to such innovative therapies.
An immeasurable amount of funding has been infused into HSC-GT research over at least the last thirty years. What is the direct return? It is reasonable to assume that all successful gene therapy recipients (as well as many of the unsuccessful ones) have been published at least once. But today's tally—a couple hundred patients at best, of whom many never engrafted or lost the transgene and at least 15 suffered treatment-associated leukemia—is deceiving. Thanks to novel vector technology, we have not seen genotoxicity in several years, although this is purely empiric, since reliable preclinical assays for human transformation are still missing. Insulators might help to circumvent position effects leading to transgene silencing. Despite large screens seeking to identify HSC maintaining and expanding agents, decades later we still rely on the classical cytokine cocktail (SCF, THPO, FLT3-L, IL-3) for cultivation and transduction, which seems to be quite acceptable also in terms of clonal diversity for the recently reported HSC-GT trials (Fig. 3). Some more refinement is needed for gene-editing applications. Together with improved patient conditioning and the quality of starting materials, all these achievements have added to the fact that marketing authorizations can be supported, and indeed patient numbers seem to be snowballing of late. Then again, alternative therapeutic avenues have been concurrently developed for several of the targets for HSC-GT, namely ERT and ever-improved protocols for HSCT. Thus, transplantation from alternative donors has reached success rated deemed inconceivable not long ago. Eligibility for HSC-GT will therefore, be defined by a tight and increasingly tightening margin but may become first line therapy in the future for some of the diseases.

Bulletin board of take-home messages describing the state of HSC-GT. FLT3, fms-like tyrosine kinase 3; IL-3, interleukin-3; SCF, stem cell factor; SIN, self-inactivating; THPO, thrombopoietin.
Footnotes
Acknowledgments
H.B. and U.M. are members of the LOEWE Cell and Gene Therapy Frankfurt faculty funded by Hessian Ministry of Higher Education, Research, and the Arts (reference number III L 4 518/17.004. S.K. is funded by the German Research Foundation.
Author Disclosure
No competing financial interests exist.