
Abstract
Heart disease is a major health threat for Duchenne/Becker muscular dystrophy patients and carriers. Expression of a 6–8 kb mini-dystrophin gene in the heart holds promise to change the disease course dramatically. However, the mini-dystrophin gene cannot be easily studied with adeno-associated virus (AAV) gene delivery because the size of the minigene exceeds AAV packaging capacity. Cardiac protection of the ΔH2-R19 minigene was previously studied using the cardiac-specific transgenic approach. Although this minigene fully normalized skeletal muscle force, it only partially corrected electrocardiogram and heart hemodynamics in dystrophin-null mdx mice that had moderate cardiomyopathy. This study evaluated the ΔH2-R15 minigene using the same transgenic approach in mdx mice that had more severe cardiomyopathy. In contrast to the ΔH2-R19 minigene, the ΔH2-R15 minigene carries dystrophin spectrin-like repeats 16 to 19 (R16–19), a region that has been suggested to protect the heart in clinical studies. Cardiac expression of the ΔH2-R15 minigene normalized all aberrant electrocardiogram changes and improved hemodynamics. Importantly, it corrected the end-diastolic volume, an important diastolic parameter not rescued by ΔH2-R19 mini-dystrophin. It is concluded that that ΔH2-R15 mini-dystrophin is a superior candidate gene for heart protection. This finding has important implications in the design of the mini/micro-dystrophin gene for Duchenne cardiomyopathy therapy.
Introduction
Loss of dystrophin results in Duchenne muscular dystrophy (DMD). While skeletal muscle presentations start at the toddler age, symptoms of myocardial involvement are rarely seen before the teenage years. Despite the later onset, up to 40% of patients may die from heart failure and/or sudden cardiac death. 1 –3 Currently, there is no etiology-based treatment for Duchenne cardiomyopathy. Gene therapy may solve the fundamental problem of dystrophin deficiency. 4 –6 Some serotypes of adeno-associated virus (AAV) have the intrinsic property of reaching all body muscles (including the heart) after a single intravascular delivery. 7 –11 This makes AAV a favored vector for DMD gene therapy. Unfortunately, the dystrophin gene is one of the largest genes in the genome, and AAV is one of the smallest viruses. As a matter of fact, the size of the full-length dystrophin coding sequence is about three times the size of the AAV genome. Development of minimized dystrophin may open the door for AAV-mediated DMD gene therapy.
Dystrophin is a 427 kD rod-shaped protein encoded by 79 exons. It has four major domains. The N-terminal domain interacts with filamentous cytoskeletal γ-actin. Immediately following the N-terminal domain is the rod domain, which accounts for >70% of the molecular weight of dystrophin. The rod domain can be further divided in 24 spectrin-like repeats (R) and four intervening hinges (H). The rod domain contains the second actin-binding domain and neuronal nitric oxide synthase (nNOS) binding domain. Toward the carboxyl end is the cysteine-rich (CR) domain and the C-terminal domain. The CR domain interacts with the transmembrane protein dystroglycan. The C-terminal domain binds to syntrophin and dystrobrevin. Abbreviated dystrophins have been generated largely based on our understanding of the structure–function relationship of dystrophin in skeletal muscle. Of particular interest is the notion that most of the rod domain can be deleted without significant consequences on function. 12,13 A 6.2 kb Δ17-48 minigene was found in a 61-year-old ambulant patient. 13 The Chamberlain lab optimized this 6.2 kb minigene into the 6-kb ΔH2-R19 minigene. Transgenic expression of the ΔH2-R19 minigene yielded better skeletal muscle protection than the Δ17-48 minigene in dystrophin-null mdx mice. Importantly, ΔH2-R19 mini-dystrophin fully restored muscle force. 12,14 In light of these encouraging skeletal muscle data on the ΔH2-R19 minigene, 12,14 cardiac specific ΔH2-R19 mini-dystrophin transgenic mdx mice were generated. 15 Heart protection of ΔH2-R19 mini-dystrophin was studied in 21-month-old male mdx mice that showed moderate cardiomyopathy. 15 –18 In contrast to what was seen in skeletal muscle, 12,14 expression of ΔH2-R19 mini-dystrophin in the heart only partially corrected electrocardiogram (ECG) and cardiac hemodynamic deficiencies. 15 This unexpected result suggests that ΔH2-R19 mini-dystrophin may lack domain(s) important for heart function.
On reviewing the clinical literature, we found that patients with deletion mutations in the region of R16 to R19 often display early-onset and/or more severe heart disease. 19 –33 This raised the question of whether inclusion of R16–19 might lead to better cardiac protection. To this end, R16–19 was inserted into ΔH2-R19 mini-dystrophin to make ΔH2-R15 mini-dystrophin (Fig. 1A). Then, cardiac-specific ΔH2-R15 mini-dystrophin transgenic mice were generated. 15 To increase the stringency of the study, we focused on a more severe model that showed classic end-stage dilated cardiomyopathy found in human patients. 16 –18,34 Despite the fact that the ΔH2-R15 minigene was tested in mice with more severe heart disease, surprisingly, several physiological parameters that were not corrected by the ΔH2-R19 minigene in the less severe model were now normalized by ΔH2-R15 mini-dystrophin. The results suggest that inclusion of R16–19 in synthetic mini-dystrophins can enhance cardiac rescue.

Heart-specific expression of ΔH2-R15 mini-dystrophin ameliorated cardiac but not skeletal muscle pathology.
Materials and Methods
Experimental animals
All animal experiments were approved by the institutional animal care and use committee and were in accordance with guidelines of the National Institutes of Health. FVB mice were used as the wild-type control mice. They were generated in a barrier facility using breeders purchased from The Jackson Laboratory. Congenic FVB background mdx mice were generated, as described before.
35
The transgenic founder lines were generated on the FVB background at the University of Missouri transgenic core. These mice expressed the ΔH2-R15 mini-dystrophin gene under the transcriptional control of the cardiac muscle specific α-myosin heavy chain (αMHC) promoter. Mini-dystrophin transgenic mdx mice were generated by crossing transgenic founder mice with FVB background mdx mice. Two founder lines (line 271 and line 272) were generated. In the studies, the data from both lines were combined in the results because (1) it has previously been shown that 5- to 50-fold transgenic overexpression of mini-dystrophin in the heart yielded similar protection, and (2) a statistically significant difference was not detected between these two lines (Supplementary Table S1; Supplementary Data are available online at

ΔH2-R15 mini-dystrophin completely rescued electrocardiogram (ECG) abnormalities.
ΔH2-R15 mini-dystrophin improved the left ventricular hemodynamics.
Evaluation of calcium handling proteins and cardiac hypertrophy-related signaling proteins.
Weights and weight ratios
Significantly different from other two groups.
Significantly different from FVB.
BW, body weight; HW, heart weight; VW, ventricle weight; TL, tibia length; TW, anterior tibialis muscle weight.
Morphological studies
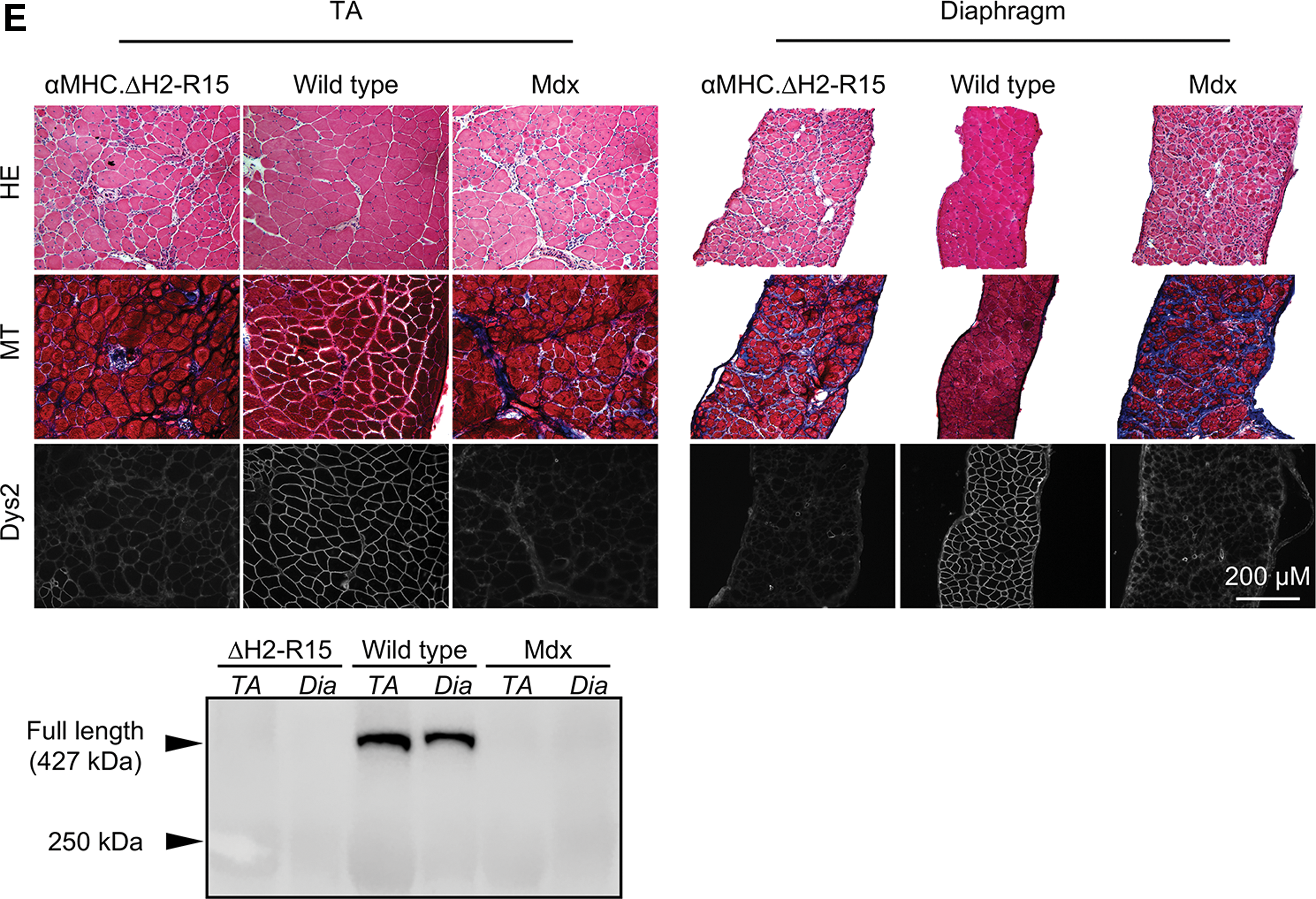
Dystrophin expression was evaluated by immunofluorescence staining using three independent dystrophin monoclonal antibodies including Dys2 (1:30; Vector Laboratories), DysB (1:80, clone 34C5, IgG1; Novocastra), and Mandys8 (1:200; Sigma–Aldrich). Dys2 and DysB react with ΔH2- R15 mini-dystrophin. Mandys8 recognizes an epitope in dystrophin repeat 11, which is absent in ΔH2-R15 mini-dystrophin. 36,37 General histology was examined by hematoxylin and eosin (H&E) staining. Fibrosis was examined by Masson trichrome staining, as described before. 38 Slides were viewed at the identical exposure setting using a Nikon E800 fluorescence microscope. Photomicrographs were taken with a QImage Retiga 1300 camera. 38 Fibrotic tissue deposition was quantified using photomicrographs of Masson trichrome stained images using Adobe Photoshop (Supplementary Fig. S1). Briefly, the measurement scale was set up for the relevant magnification for the microscope, and using the lasso tool in Photoshop, the fibrotic area was marked in individual images. The fibrotic area from multiple images was averaged per individual animal, and the total area of fibrosis was calculated for three to four animals for each strain.
Western blot
Whole heart and muscle lysate was prepared, as described before. 39 Briefly, the tissues were snap frozen in liquid nitrogen. The frozen tissue samples were ground to a fine powder in liquid nitrogen followed by homogenization in a buffer containing 10% sodium dodecyl sulfate, 5 mM of ethylenediaminetetraacetic acid, 62.5 mM of Tris-HCl at pH 6.8, and the protease inhibitor cocktail (Roche). The crude lysates were heated at 95°C for 3 min, chilled on ice for 2 min, and then centrifuged at 20,800 g for 2 min. The supernatant was collected as the whole muscle lysate. Protein concentration was measured using the DC protein assay kit (Bio-Rad). Dystrophin was detected with the Dys2 antibody (1:100; Novocastra). The calcium handling proteins were detected using antibodies against sarcoplasmic/endoplasmic reticulum calcium ATPase 2a (SERCA2a; 1:2,500; Badrilla) and phospholamban (1:2,500; Badrilla). Proteins involved in cellular signaling were detected using antibodies for c-jun N-terminal kinase (JNK1; 1:1,000; BD Pharmingen), p38α (1:500; Santa Cruz Biotechnology), Akt (1:1,000; Cell Signaling Technology), and endothelin-A receptor (ET-A, 1:5,000; Abcam). For the loading control, antibodies against glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:3,000; Millipore) and vinculin (1:2,000; Abcam) were used.
Western blot quantification was performed using LI-COR Image Studio v5.0.21 (
ECG and hemodynamic assay
Cardiac functions were evaluated using published protocols, as described in the standard operating procedure in the Cardiac Protocols for Duchenne Animal Models (
Statistical analysis
Data from individual experimental subject are presented using scatter plots. Data from the experimental group are presented as mean ± standard error of mean. One-way analysis of variance with Bonferroni's multiple comparison analysis was performed using GraphPad Prism v7.0 for Mac OSX (GraphPad Software). A p-value of <0.05 was considered statistically significant.
Results
Generation of αMHC.ΔH2-R15 mini-dystrophin transgenic mdx mice
To study the cardiac benefit of ΔH2-R15 mini-dystrophin, cardiac-specific αMHC.ΔH2-R15 minigene transgenic mice were generated (Fig. 1A). Cardiac-specific expression was regulated by the αMHC promoter and the bovine growth hormone gene polyadenylation sequence. Two founder lines (line 271 and line 272) were generated on the FVB background and subsequently crossed to the FVB background mdx mice.
The progression of Duchenne cardiomyopathy undergoes several distinctive phases from pre-symptomatic stage to compensatory hypertrophic cardiomyopathy and eventually dilated cardiomyopathy. 16,17 It was recently discovered that only ≥21-month-old female mdx mice display severe end-stage dilated cardiomyopathy. 18 Hence, the current study focused on ≥21-month-old female mdx mice. Cardiac expression of the ΔH2-R15 minigene was confirmed by dystrophin immunofluorescence staining in both founder lines using Dys2, DysB, and Mandys8 antibodies. DysB and Dys2 recognize H1-R2 and the C-terminal domain, respectively, while Mandys8 reacts with R11, which is absent in ΔH2-R15 mini-dystrophin (Fig. 1B). Heart lysate Western blot revealed the right size band at levels 7.5- and 13-fold higher than that of full-length dystrophin in a normal heart (Fig. 1C). It was previously shown that transgenic mini-dystrophin expression in the mdx heart at 5- to 50-fold higher than that of full-length dystrophin in wild-type mice yielded similar levels of histological and physiological rescue. 45 Hence, quantification data presented in the rest of the manuscript are from both lines. On H&E staining and Masson trichrome staining, nominal inflammation and fibrosis were detected in the heart of αMHC.ΔH2-R15 mice (Fig. 1D and Supplementary Fig. S1). On quantification, the fibrotic area in the heart of αMHC.ΔH2-R15 mice was significantly lower than that of mdx mice and was actually comparable to that of wild-type mice (Supplementary Fig. S1). As expected, skeletal muscle of αMHC.ΔH2-R15 mice had no dystrophin expression and displayed characteristic degeneration and fibrosis comparable to those of mdx mice (Fig. 1E). 35
ΔH2-R15 mini-dystrophin normalized aberrant ECG changes
To determine therapeutic benefits on cardiac electrophysiology, the 12-lead ECG assay was performed. 40,41 Except for the lack of tachycardia, all other characteristic ECG abnormalities were observed in transgene-negative mdx mice (Fig. 2). Specifically, the PR interval was significantly reduced, QRS duration and Mitchell's corrected QT (QTc) interval were significantly prolonged, and the absolute value of the Q wave amplitude and the cardiomyopathy index were significantly increased (Fig. 2). These abnormal changes were completely corrected in αMHC.ΔH2-R15 mice (Fig. 2).
ΔH2-R15 mini-dystrophin prevented heart dilation
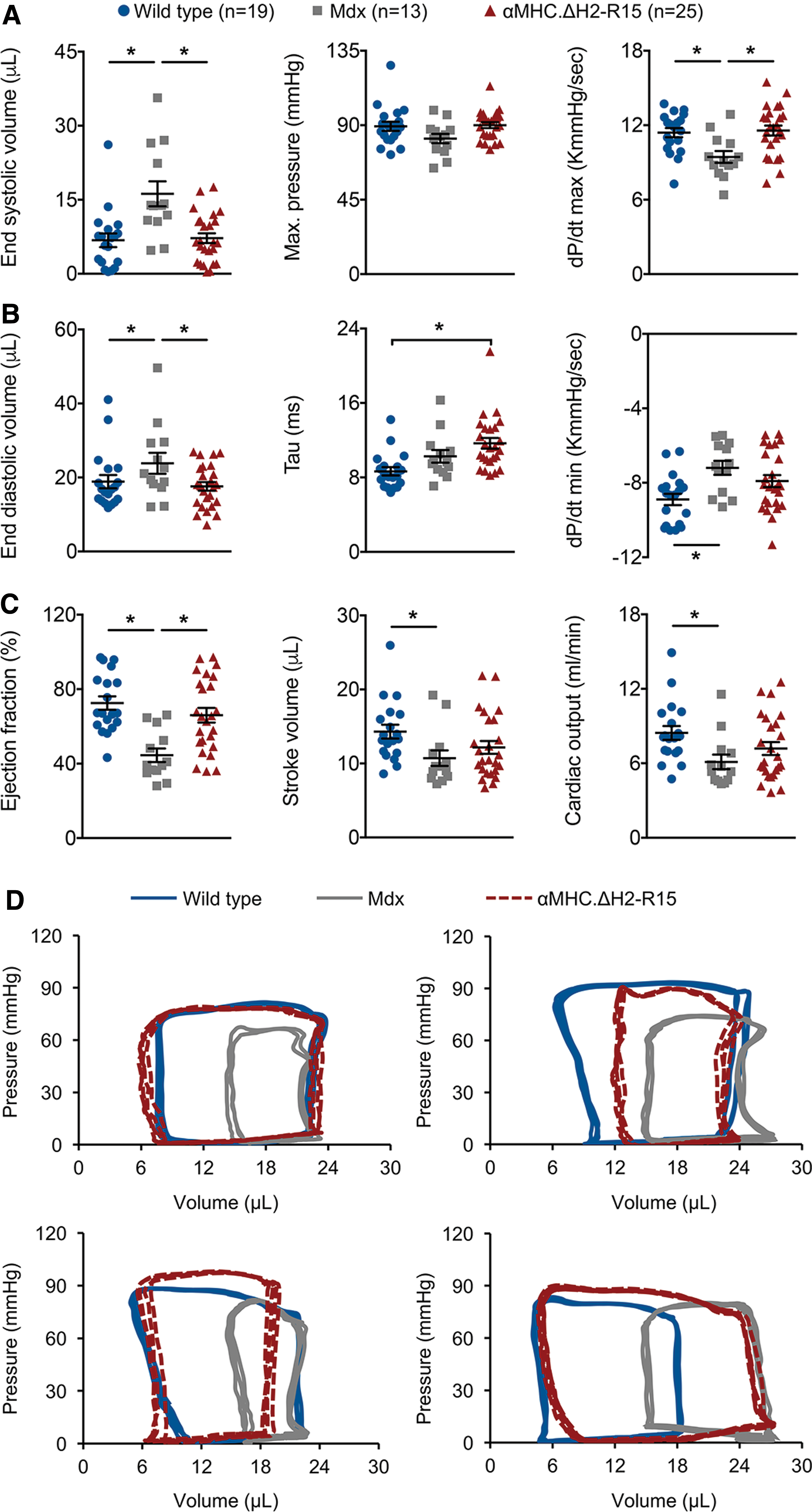
Hemodynamics was examined using left ventricular catheterization (Fig. 3 and Supplementary Table S1). 15,42 Similar to a previous report, 18 characteristic signs of dilated cardiomyopathy were observed, such as a significant increase of the end-systolic volume and end-diastolic volume in transgene-negative mdx mice (Fig. 3). Transgenic expression of ΔH2-R15 mini-dystrophin in the heart normalized the end-systolic volume, dP/dt maximum, end-diastolic volume, and ejection fraction. A trend of improvement was also seen in maximal pressure, dP/dt minimum, stroke volume, and cardiac output (Fig. 3). Nevertheless, the isovolumetric relaxation time constant tau was not corrected (Fig. 3). In contrast, the mean tau value was significantly increased compared to that of wild-type controls.
Anatomic examination revealed cardiac hypertrophy in αMHC.ΔH2-R15 mice
At the end of the in-life study, body weight (BW), heart weight (HW), ventricular weight (VW), tibia length (TL), and TA muscle weight (TW) were measured (Table 1). Compared to that of wild-type controls, the TW was significantly reduced in αMHC.ΔH2-R15 mice, consistent with skeletal muscle disease-related limb muscle atrophy. Interestingly, the HW and VW of αMHC.ΔH2-R15 mice were significantly (∼14%) higher than those of wild-type controls. The HW/TW and VW/TW ratios of αMHC.ΔH2-R15 mice were also significantly increased compared to those of wild-type control mice, suggesting cardiac hypertrophy in αMHC.ΔH2-R15 mice. Since the TW was affected by skeletal muscle disease, we evaluated TL-normalized HW and VW. 34,46 Compared to those of wild-type control mice, the HW/TL and VW/TL ratios of αMHC.ΔH2-R15 mice were significantly higher, thus confirming cardiac hypertrophy in αMHC.ΔH2-R15 mice.
Evaluation of calcium handling proteins and cardiac hypertrophy related signaling proteins
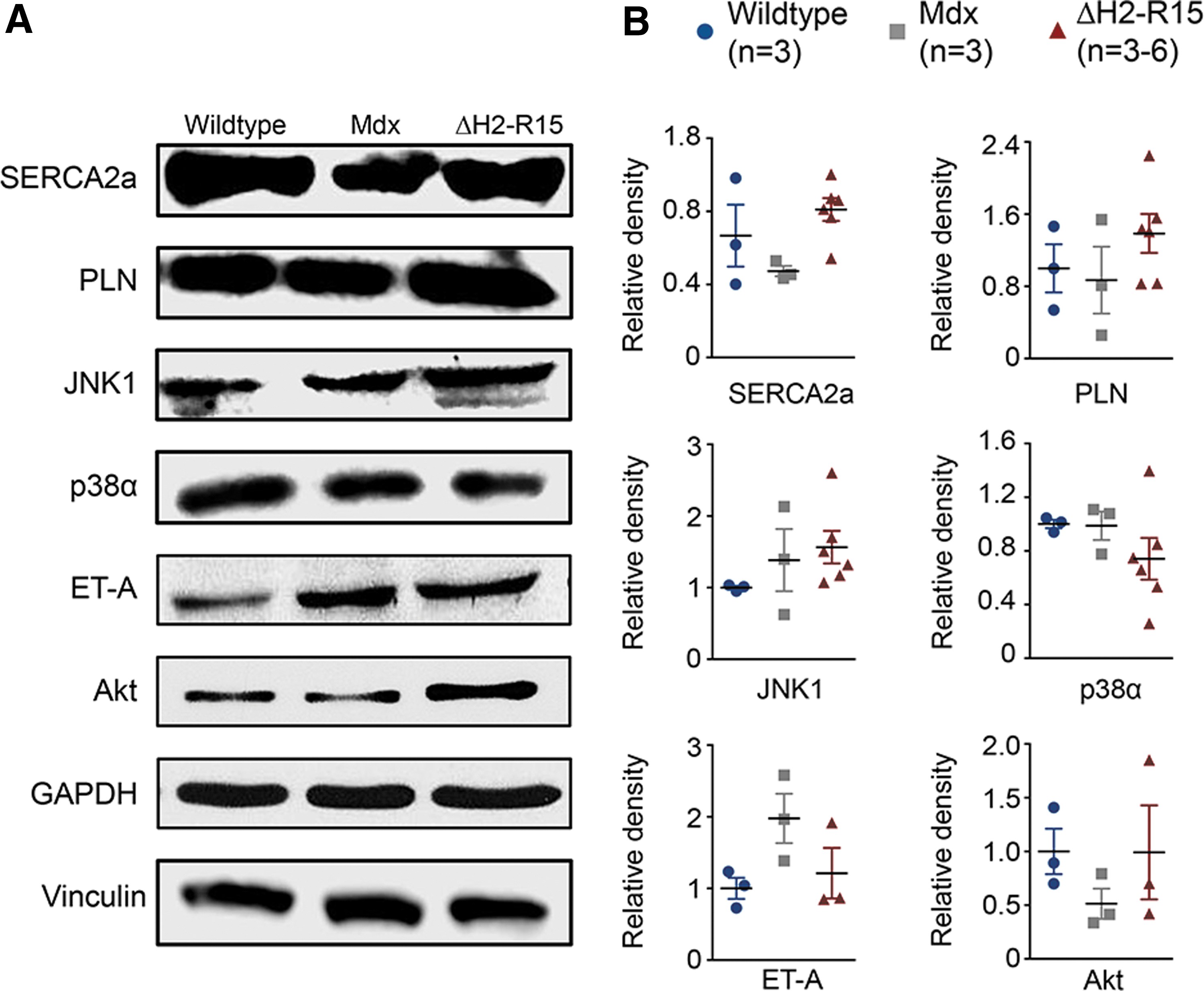
To begin to understand the mechanisms of heart protection by ΔH2-R15 mini-dystrophin, the expression of SERCA2a and phospholamban, two important calcium handling proteins that are known to regulate heart contractility (Fig. 4), were quantified. Compared to wild-type control mice, both SERCA2a and phospholamban appeared reduced in mdx mice. Their levels were increased in αMHC.ΔH2-R15 mice but did not reach statistical significance.
Many signaling pathways have been implicated in cardiac hypertrophy (reviewed in Rohini et al., 47 Balakumar and Jagadeesh, 48 Barry et al., 49 Heineke and Molkentin, 50 and Kehat and Molkentin 51 ). As the first step toward elucidation of the molecular processes underlying myocardial hypertrophy seen in αMHC.ΔH2-R15 mice, several proteins were examined in the mitogen-activated protein kinase (MAPK) pathway, Akt signaling, and G-protein coupled receptor signaling (Fig. 4). 51 –60 These pathways have been implicated in either pathological or physiological cardiac hypertrophy. JNK1 and p38α are two important branches of the MAPK signaling cascade. A statistically significant difference was not detected (Fig. 4). Endothelin receptor A (ET-A) is a G-protein coupled receptor. Western blot on ET-A and Akt did not show noticeable trends (Fig. 4). In light of the substantial individual differences (especially for Akt) and the small sample size (n = 3 for αMHC.ΔH2-R15 mice), a solid conclusion could not be drawn on the involvement of the Akt signaling and G-protein receptor signaling.
Discussion
Two distinctive strategies have been used to study the structure–function relationship of dystrophin: the transgenic approach and AAV-mediated gene transfer. Systemic AAV delivery results in simultaneous transduction of both skeletal muscle and heart. This creates a challenge for sorting out the cardiac-specific effect because it is controversial whether treating skeletal muscle can reduce or aggravate heart disease in mdx mice. 38,61,62 To obtain a definitive answer on the cardiac-specific effect, the transgenic approach was used instead of AAV delivery to avoid confounding influences from skeletal muscle dystrophin expression.
Dystrophin is essential for the survival and function of both skeletal muscle and cardiac muscle. However, recent studies suggest that there may be important differences between skeletal muscle dystrophin and cardiac dystrophin. For example, dystrophin directly binds to nNOS in skeletal muscle, and this interaction is critical to the sarcolemmal localization of nNOS in skeletal muscle. 14,63 However, in cardiac muscle, dystrophin dose not interact with nNOS, and nNOS is localized in the sarcoplasmic reticulum and mitochondria in the heart. 64 –67 Cytosolic nNOS compromises muscle function in mdx mice by inducing nitrosative stress, but cytosolic overexpression of nNOS in the heart improves cardiac function in aged mdx mice. 39,67 Cardiac dystrophin directly associates with myofibrils at the Z-disk, but skeletal muscle dystrophin does not interact with myofibrils. 68 Recent proteomic studies have further identified cellular proteins that selectively interact with dystrophin in the heart but not in skeletal muscle. 66 Collectively, these observations suggest that dystrophin may play overlapping but distinctive roles in the heart and skeletal muscle. It is thus important to determine whether a therapeutic candidate dystrophin gene that can protect skeletal muscle can also protect the heart.
The ΔH2-R19 minigene and ΔH2-R15 minigene have both been shown to rescue skeletal muscle contractility fully. 12,14,69 Interestingly, cardiac-specific expression of the ΔH2-R19 minigene in transgenic mice failed to correct ECG abnormalities completely. In the hemodynamic assay, the ΔH2-R19 minigene also did not correct the end-diastolic volume, an important parameter in the context of Duchenne cardiomyopathy. It was unclear whether the ΔH2-R15 minigene can lead to better rescue. To address this question, αMHC.ΔH2-R15 minigene transgenic mice were generated (Fig. 1), and then cardiac histopathology, anatomy, and function were compared among normal, transgene-positive, and transgene-negative mdx mice when they reached ≥21 months of age. In the absence of the minigene, mdx mice showed ECG and hemodynamic features that are characteristic for dilated cardiomyopathy (Figs. 2 and 3). All aberrant ECG changes were normalized in αMHC.ΔH2-R15 minigene transgenic mice (Fig. 2). On the cardiac catheter assay, the enlarged end-diastolic and end-systolic volumes were normalized by ΔH2-R15 mini-dystrophin, the reduced dP/dt max was returned to the wild-type level, and ejection fraction was normalized (Fig. 3). Collectively, these results suggest that the ΔH2-R15 minigene is an outstanding candidate gene for treating Duchenne dilated cardiomyopathy.
The exact molecular mechanisms underlying superior cardiac rescue by the ΔH2-R15 minigene will have to wait for future in-depth studies. Nevertheless, it is speculated that it may at least be partially due to the presence of R16–19 in this minigene. Nigro et al. studied 284 patients and found that the deletion of exons 48 and 49 (R19) correlates with severe cardiac disease. 23 In a more recent study, Kaspar et al. analyzed 78 patients and discovered the N-terminal domain and R17–19 might protect the heart. Deletion of one these two regions often results in early-onset heart disease. 33 Numerous clinical studies on Becker muscular dystrophy (a mild form of DMD) and X-linked dilated cardiomyopathy (a disease caused by selective dystrophin deficiency in the heart) from other groups also pointed toward a potential cardiac protective role of R16–19. 19 –33 The present results align well with these patient-oriented studies. Collectively, these data suggest that R16–19 may represent a putative heart protection domain in dystrophin.
It is worth noting that the existence of the tissue-specific domain in dystrophin has been well documented in the literature. For example, repeats 16 and 17 are essential for anchoring nNOS to the sarcolemma in skeletal muscle. 14 The C-terminal domain appears to be required for normal electroretinography, and mutations in the C-terminal domain often associate with cognitive deficiency. 70,71 However, except for the nNOS-binding R16/17 domain for skeletal muscle, dystrophin tissue-specific domains have rarely been studied and/or validated in animal models. The study described here is the first to try to determine experimentally whether certain regions of dystrophin can result in better heart rescue. While the results are encouraging, additional studies are needed before a solid conclusion can be drawn. Some of these studies may include exploration of cardiac-specific dystrophin interacting proteins such as cavin-1 and αB-crystalline. 66 Cavin-1 is particularly interesting because several recent studies suggest that cavin-1 deficiency contributes to the pathogenesis of cardiomyopathy and muscular dystrophy. 72 –74
An intriguing finding of this study is the presence of myocardial hypertrophy in αMHC.ΔH2-R15 mice (Table 1). While the molecular trigger for cardiac hypertrophy in αMHC.ΔH2-R15 mice remains elusive (Fig. 4), it certainly suggests a reverse of the disease course from heart failure associated dilated cardiomyopathy to compensatory hypertrophic remodeling. 16,17
It should be pointed out that the transgenic approach has been used in the hope of (1) comparing with previously published data from αMHC.ΔH2-R19 transgenic mdx mice, and (2) more clearly delineating the cardiac-specific effect of dystrophin R16–19. However, the transgenic approach cannot be directly applied to gene therapy. While the size of the ΔH2-R15 minigene (∼7 kb) exceeds the packaging capacity of a single AAV vector, it can be expressed using various dual AAV vector strategies. Specifically, the minigene expression cassette can be split into two parts and separately packaged with two independent AAV virions. Co-delivery and intermolecular recombination would allow expression of the ΔH2-R15 minigene. Future studies with a set of ΔH2-R15 minigene dual AAV vectors will be necessary to corroborate transgenic findings described here further. Alternatively, the therapeutic benefit of R16–19 may be investigated using AAV-mediated expression of a micro-dystrophin gene. In this case, one will have to engineer a novel synthetic microgene (≤4 kb) that contains R16–19.
In summary, our study suggests that the inclusion of R16–19 in a therapeutic candidate gene could be beneficial for treating Duchenne cardiomyopathy.
Footnotes
Acknowledgments
This work was supported by Jackson Freel DMD Research Fund, Jesse's Journey—The Foundation for Gene and Cell Therapy and the Margaret Proctor Mulligan endowment to the University of Missouri. Supplementary funding was provided through grants from the National Institutes of Health (HL-91883, NS-90634). N.W. was partially supported by the life science fellowship, University of Missouri. F.M. is supported by a Marie Curie senior fellowship from the EU H2020 program and by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London. We thank the University of Missouri Transgenic Animal Core for the help with generating the founder transgenic mice.
Author Disclosure
D.D. is a member of the scientific advisory board for Solid Biosciences LLC and an equity holder of Solid Biosciences LLC. The Duan lab has received research supports from Solid Biosciences LLC.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.