
Abstract
The possibility of editing complex genomes in a targeted fashion has revolutionized basic research as well as biomedical and biotechnological applications in the last 5 years. The targeted introduction of genetic changes has allowed researchers to create smart model systems for basic research, bio-engineers to modify crops and farm animals, and translational scientists to develop novel treatment approaches for inherited and acquired disorders for which curative treatment options are not yet available. With the rapid development of genome editing tools, in particular zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the CRISPR-Cas system, a wide range of therapeutic options have been—and will be—developed at an unprecedented speed, which will change the clinical routine of various disciplines in a revolutionary way. This review summarizes the fundamentals of genome editing and the current state of research. It particularly focuses on the advances made in employing engineered nucleases in hematopoietic stem cells for the treatment of primary immunodeficiencies and hemoglobinopathies, provides a perspective of combining gene editing with the chimeric antigen receptor T cell technology, and concludes by presenting targeted epigenome editing as a novel potential treatment option.
Introduction
T
Designer nucleases enable therapeutic genome editing by cleaving the DNA at predetermined sites in the human genome. 1 The resulting DNA double-strand break activates the DNA repair machinery of the cell, which repairs the DNA cut by means of either non-homologous end-joining (NHEJ) or homology-directed repair (HDR). Both mechanisms can be harnessed for targeted genome editing. NHEJ repairs the double-strand break by ligating the two DNA strands directly without requiring a template for the repair. Although repair can be accurate, NHEJ often inserts or deletes a few nucleotides at the break site, resulting in so-called indel mutations. As a result, NHEJ is primarily used to inactivate target genes. HDR, on the other hand, requires the presence of a DNA repair template. In a natural context, this role is taken over by the sister chromatid. However, the HDR machinery also accepts exogenous DNA if it contains sufficient sequence homology to the target site. For HDR-mediated genome editing, an exogenous DNA “donor” template is introduced into the cell in the form of single- or double-stranded DNA and generally used to correct disease-underlying mutations in the genome.
The key to a successful gene editing approach is the effective introduction of the gene editing tools into the cell type of interest. 1 However, in a clinical context, not only transfer efficacy but also transfer-associated cytotoxicity and genotoxicity are critical. For cells of the blood and immune system, it became widely accepted to introduce designer nucleases by electroporation as either protein or its blueprint in the form of mRNA. This method has several advantages: on the one hand, the nuclease is rapidly available to the cell; on the other hand, long-term exposure of the genome to high nuclease concentrations is avoided by the relatively rapid degradation of the proteins or mRNAs. For HDR-based strategies, a donor must be co-introduced into the cell. Generally, viral vector systems based on adeno-associated virus type 6 (AAV6) or integrase-deficient lentiviral (IDLV) vectors have been used for this purpose.
In addition to its activity at the target sequence, designer nucleases can be active at so-called off-target sites, that is, at sites in the genome that share high sequence identity to the actual target site. 6 Ideally, the nuclease activity at off-target sites is minimal and will not have any adverse effects. However, in the worst case, a cell may undergo transformation if an off-target site is located in the coding region of a tumor suppressor gene or in a crucial cis-regulatory element. Therefore, specificity of an engineered nuclease must be determined thoroughly before any clinical translation. As the number of preclinical studies using ZFNs, TALENs, and the CRISPR-Cas system is steadily increasing, the number of clinical genome editing trials is expected to increase in the future as well, particularly if specificity of the designer nuclease platforms will be manageable.
Genome Editing Strategies to Treat Primary Immunodeficiencies
Primary immunodeficiencies (PIDs) are a heterogeneous group of disorders characterized by variable susceptibility to infections due to hereditary defects in the immune system. Even if the classification is cumbersome, PIDs are categorized in major disease groups based on the underlying defects and symptoms. The number of identified mutations responsible for PIDs is rapidly increasing thanks to the improvement of sequencing technologies, with >300 gene defects described thus far. 9 Severe combined immunodeficiencies (SCIDs) belong to the worst forms of PIDs and are characterized by deficient T cell function that can be coupled with a defect in the B cell and/or natural killer cell compartment. Since all immune cells derive from hematopoietic stem cells (HSC), a major therapeutic approach to treat SCID patients is allogeneic stem cell transplantation. Even if HSC transplantation protocols have substantially improved, there are still some limitations, such as the availability of matched donors and the risk of graft-versus-host disease (GvHD). Hence, the genetic correction of the patients' own HSCs is still an appealing alternative for patients who lack a suitable donor or where allogeneic HSC transplantation is still associated with high morbidity and/or mortality. Indeed, gene therapy involving cells that have an intrinsic ability of both self-renewal and the production of all blood lineages has the potential to cure numerous blood disorders, also beyond PIDs. 10 Many gene therapy trials for X-linked SCID (X-SCID), adenosine deaminase-deficient SCID (ADA-SCID), Wiskott–Aldrich syndrome (WAS), and X-linked chronic granulomatous disease (X-CGD) have been successfully completed. 10 While early clinical trials with first-generation gamma-retroviral vector mediated gene transfer led to severe adverse effects in quite a few patients in the X-SCID, 11 WAS, 12 and X-CGD 13 trials, the use of safer self-inactivating (SIN) gamma retroviral and lentiviral vectors led to the sustainable cure of many children suffering from PIDs. 14 Of note, the European Medicines Agency approved Strimvelis as the first ex vivo stem cell gene therapy to treat ADA-SCID in 2016. 15
While gene addition type gene therapy has been successfully implemented for a small number of PIDs, it might not work for disorders in which the underlying mutations are located in tightly regulated loci or in genes expressed in a temporal and lineage specific manner, such as mutations in CD40LG, STAT1, RAG1, and RAG2 genes. The main advantage of using targeted gene editing consists of the potential to correct the disease-causing mutation in a site-specific manner, hence leaving the amended genes under control of the endogenous regulatory elements. An in situ correction approach will also be appropriate to treat dominant negative mutations for which the traditional addition of a normal gene copy would not rescue the disease phenotype. For example, the design of programmable nucleases to disrupt the mutated sequence in an allele-specific manner will be helpful to treat autosomal-dominant hyper-IgE syndrome (AD-HIES), which is caused by mutations in the STAT3 gene.
In 2014, Naldini et al. were the first to demonstrate the functional repair of a defective gene in HSCs (Table 1). To this end, they inserted a corrective cDNA into the IL2RG locus in HSCs of an X-SCID patient using mRNA electroporation to deliver the ZFNs and IDLVs as a donor template. 16 Of note, targeting IL2RG gene was achieved in all CD34-positive subpopulations, but was least effective in the most primitive stem cell compartment, which contains the long-term repopulating HSCs. In 2016, Torgerson's group used TALENs to restore CD40L function. 17 Mutations in the CD40LG locus result in an inability to undergo immunoglobulin class switch and are associated with hyper-IgM (HIGM) syndrome. Previous attempts to perform gene therapy with retroviral delivery of a CD40L expression cassette to mouse HSCs were able to restore adaptive immunity but treated mice developed thymic lymphoproliferative disorder later on. 18 In their genome editing approach, the authors rescued CD40L function in T cells of X-HIGM patients through a combination of TALEN and AAV donor template, correcting some 30% of alleles. 17 The CRISPR-Cas9 system was used to treat X-CGD arising from mutations in the CYBB gene coding for GP91phox, a component of the microbicidal oxidase system in phagocytes. To this end, a CRISPR-Cas nuclease targeting a single base pair mutation was employed in HSCs together with single-stranded oligodeoxynucleotides (ssODNs) to revert the mutation, so restoring the generation of reactive oxygen species in neutrophils. The gene correction frequency detected in transplanted HSCs was in the range of 10%. 19
Overview: preclinical gene editing for PIDs and hemoglobinopathies
PID, primary immunodeficiency; HDR, homology-directed repair; NHEJ, non-homologous end joining; X-SCID, X-linked severe combined immunodeficiency; IL2RG, interleukin 2 receptor subunit gamma; ZFN, zinc finger nuclease; IDLV, integrase-deficient lentiviral vector; X-CGD, X-linked chronic granulomatous disease; CYBB, cytochrome B-245 beta chain; CRISPR, clustered regularly interspaced short palindromic repeats; ssODN, single-stranded oligodeoxynucleotide; X-HIGM, X-linked hyper IgM syndrome; CD40LG, CD40 ligand; TALEN, transcription activator-like effector nuclease; AAV6, adeno-associated virus type 6 vector; SCD, sickle cell disease; HBB, hemoglobin subunit beta; Thal, β-thalassemia; RNP, ribonucleoprotein; GATA1, GATA binding protein 1; BCL11A, B-cell CLL/lymphoma 11A; HBG1, hemoglobin subunit gamma 1; LV, lentiviral vector.
In conclusion, gene targeting in human HSCs to treat PIDs has become a reality in a preclinical context. Several improvements have been achieved in the last few years with regard to strategies for delivering the designer nuclease and the donor, as well as the ex vivo culturing of these delicate cells to maintain multipotency. Donors based on ssODNs will be useful for correcting disease-causing point mutations, considering that production of ssODN is easier and more economical than the manufacturing of viral vectors. However, it will be a challenge to assess the random integration frequency of ssODNs, as they are difficult to detect once integrated in the host genome. The use of non-pathogenic AAV6 vectors for donor delivery bears several advantages, including the low integration frequency, the—for most cases—sufficiently large packaging capacity (4.7 kb), and, possibly, its single-stranded nature. On the downside, transduction of HSCs with AAV6 vectors seems to be accompanied by some cytotoxicity. 20 Alternative AAV serotypes or nanoparticles that are able to transduce HSC with high efficacy without invoking cytotoxicity are sought after and might help to achieve the high gene targeting frequencies needed for clinical translation.
Genome Editing Strategies to Treat Hemoglobinopathies
Hemoglobinopathies are among the most frequent monogenic disorders. 21 Hemoglobin is a heterotetramer composed of two α-globin monomers and two β-globin monomers, and responsible for the transport of oxygen from lungs to all tissues. The β-globin locus is composed of five genes—HBE1 (ɛ-globin), HBG2 (γ-globin-G), HBG1 (γ-globin-A), HBD (δ-globin), and HBB (β-globin)—that encode the beta-like subunits of hemoglobin (Fig. 1). Expression of these five genes is controlled by the locus control region (LCR) and characterized by a known switch phenomenon during fetal development until the first months after birth. While ɛ- and γ-globins are expressed during embryonic and fetal stage, respectively, β-globin becomes prevalent after birth and persists during adult life. Dysregulation of globin gene expression or mutations in the α- or β-globin encoding genes result in mild to severe anemia, called α- or β-thalassemia, or in a disorder characterized by globin polymerization that leads to sickle-shaped red blood cells, called sickle cell disease (SCD). SCD shows various degrees of anemia and can potentially cause life-threatening vaso-occlusion. 21 In this context, it is worth noting a benign syndrome known as hereditary persistence of fetal hemoglobin (HPFH) that is genetically characterized by mutations in regulatory elements of the β-globin locus that lead to persistent fetal γ-globin expression throughout life. This syndrome was first described for a Greek family that harbored a 7.2 kb deletion encompassing the δ-globin transcriptional start site (Corfu deletion), resulting in a mild δβ-thalassemia. 22
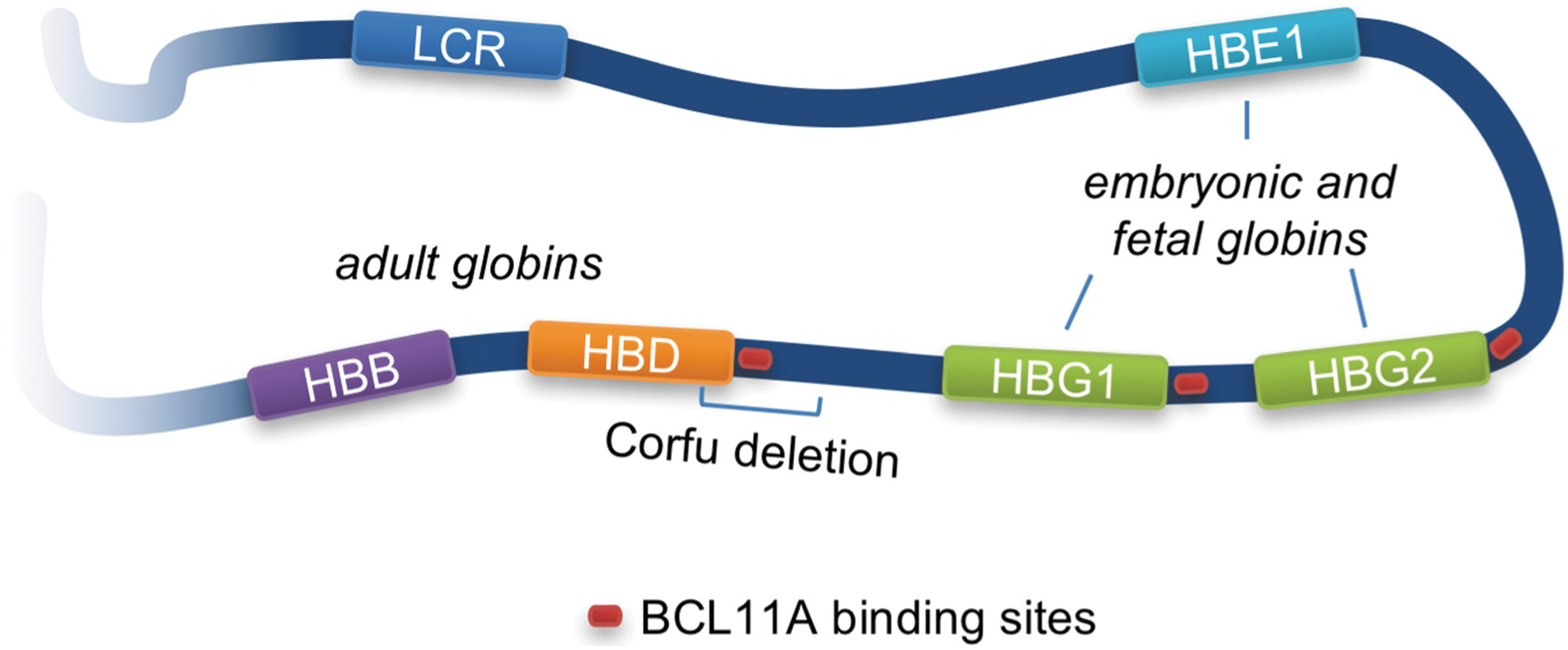
Schematic of β-globin locus. The β-globin locus on chromosome 11 harbors five genes that encode the β-like subunits of hemoglobin: HBE1 (ɛ-globin), HBG2 (γ-globin G), HBG1 (γ-globin A), HBD (δ-globin), and HBB (β-globin). Expression of these genes is controlled by the locus control region (LCR) and the cis-regulatory elements of each gene. ɛ-globin is expressed during the embryonic stage and replaced by γ-globin A and G during later stages of pregnancy. The complete switch to adult δ- and β-globin is completed within the first 6 months after birth. BCL11A is a major regulator of the globin switching process.
About 7% of the world population is estimated to be a carrier of the “sickle allele.” Mostly due to the protective effect of the sickle cell trait against malaria, those mutations can affect up to 80% of the population in some regions. 21 Current treatment for SCD is based on prophylactic administration of antibiotics, long-term transfusion regimens, and hydroxyurea administration, which was shown to recover fetal γ-globin expression partially, a treatment option that can also ameliorate the thalassemic phenotype. 23 Those treatment options improve quality of life and life expectancy but cannot fully protect from occasional strokes caused by vaso-occlusion through sickled red blood cells. Conventional care of thalassemic patients involves frequent blood transfusions and iron chelation therapy, depending on disease severity. Despite the good efficacy of those approaches, individualized therapy still requires life-long specialized disease management. Allogenic HSC transplantation is a suitable alternative in case of severe forms of anemia or SCD, although this option is not without risks due to GvHD and the variable success of HSC engraftment. 24 As a consequence, autologous HSC transplantation after gene therapy is considered a promising alternative to treat thalassemic and sickle cell phenotypes.
The first gene therapy approaches were based on the integration of a β-globin sequence via lentiviral transduction of HSC. 25 Basically, LCR sequences and the β-globin promoter were combined in those vectors to obtain a specific and sustained β-globin expression in the erythroid compartment. In some clinical gene therapy studies, a modified β-globin sequence was employed in order to decrease the likelihood of globin polymerization in the context of SCD. 26 Some of these clinical phase I/II trials showed promising results, with patients able to stop or significantly reduce the number of transfusions after gene therapy. Other clinical trials did not show a significant improvement of the phenotype, although they used a similar vector design but applied different conditioning regimens. 25 Those positive and negative results highlight the significance of fine-tuned β-globin transgene expression, leaving room for further improvements in gene addition type strategies and opening the space for approaches based on gene editing.
Basic studies performed on the β-globin locus defined important regulatory elements that are involved in the globin switch process. Some of these elements were characterized to be necessary for a sustained β-globin expression in adults and were defined as erythroid-specific strong enhancer and DNase I hypersensitive sites. 27 Other cis-regulatory elements were characterized as being necessary for binding of a repressor complex involving BCL11A. 25 In particular, the cis-regulatory elements surrounding the fetal globin genes, HBG2 and HBG1, are targeted by the BCL11A repressor complex. 28 BCL11A knockout studies confirmed the role of BCL11A in regulating fetal globin expression but also underlined its importance in other pathways related to the leukemogenesis and hematopoiesis, demanding a careful evaluation when manipulating its expression for clinical purposes. 29
A promising approach relies on reverting the globin-switch process from adult to fetal γ-globin expression, as observed in HPFH (Table 1). A natural HPFH genotype was recreated in HSCs by employing two CRISPR-Cas nucleases that produce a 13 kb deletion in the β-globin locus, deleting both HBD and HBB. Erythrocytes derived from the edited HSCs revealed a robust γ-globin expression. 30 Brendel et al. selectively knocked down BCL11A in HSCs through the erythroid-specific expression of a BCL11A targeting shRNA, resulting in successful reactivation of γ-globin. 31 A complementary approach was successfully employed using designer endonucleases to delete a GATA-1 enhancer element present in the BCL11A gene that is required for its robust expression in the erythroid compartment. 29,32 A further alternative includes the knockout of BCL11A regulatory elements in the fetal γ-globin genes, which led to a significantly reduced sickle phenotype in vitro using HSC derived from SCD patients. 33
Two studies demonstrated restored fetal-globin expression by harnessing the chromatin looping complex that acts in the globin LCR. An engineered zinc-finger protein that recognizes the HBG promoter was fused to the self-association domain of the LDB1 protein. 34,35 This chimeric protein was placed under the control of an erythroid-specific promoter and integrated via lentiviral gene transfer into the genome of HSCs of SCD patients. The SCD phenotype regressed significantly in vitro and the approach was more effective than hydroxyurea or other drugs. 34
Other laboratories focused on correcting the HBB gene in HSCs by means of designer nucleases together with an exogenous donor DNA template using the HDR pathway. Multiple examples have shown up to 40% of correctly edited “sickle alleles” when employing ssODN or IDLV donors in combination with ZFNs 36,37 or CRISPR-Cas9. 38 Dever et al. observed around 20–40% of edited HBB alleles by utilizing CRISPR-Cas9 together with AAV6-based delivery of the donor DNA template. 39 After xenotransplantation of the gene edited HSCs in immunodeficient mice, however, all studies reported a strong reduction of the fraction of edited cells (down to 1–3%), suggesting a low HDR frequency in long-term repopulating HSCs. A strategy to enrich gene edited HSCs was included in Dever et al., utilizing the truncated low-affinity nerve growth factor receptor (ΔLNGFR) as a selectable marker. 39 However, such an approach only seems meaningful if edited long-term repopulating HSCs can be expanded ex vivo. Two of these studies reported significant off-target activity of the used nucleases, raising concerns of genotoxicity. 36,39 Moreover, the high frequency of NHEJ-mediated disruption of the β-globin coding sequence observed in gene edited HSCs bears the risk of altering the SCD phenotype to a more severe β-thalassemia.
In conclusion, even though lentiviral-based conventional gene therapy approaches are successfully pursued for treating hemoglobinopathies, targeted editing of the β-globin locus will remain a promising alternative. In particular, the NHEJ-based knockout strategies aimed at reinstating fetal γ-globin expression could develop into effective therapies in the near future. On the other hand, the low HDR-mediated gene correction frequency in long-term repopulating HSCs and the ratio between gene correction and NHEJ-based gene disruption remains a major challenge for HDR-based gene targeting approaches in blood stem cells.
Genome Editing in T Cells to Fight Cancer
Adaptive immunotherapy using tumor-specific cytotoxic T cells or chimeric antigen receptor (CAR) T cells has been successfully applied in several clinical phase I/II trials to treat hematologic malignancies. 40 –42 The efficacy of engineered CAR T cells is mediated by the recognition of tumor-specific or tumor-associated antigens through the CAR in a human leukocyte antigen (HLA)-independent manner. Upon binding of an antigen, the CAR T cells are activated and eliminate the tumor cells. In particular, anti-CD19 CAR T cells have shown remarkable success in treating CD19-expressing B-cell malignancies, with up to 90% of patients suffering from acute lymphoblastic leukemia (B-ALL) going into complete remission. 43 Despite the great clinical success of CAR T cell therapy, serious side effects have been observed in patients, such as cytokine release syndrome (CRS), neurological toxicities, and the so-called “on-target-off-tumor” effect involving B cell aplasia in patients treated with anti-CD19 CAR T cells. These side effects are, at least in part, due to the lack of controlling CAR T cell activation and expansion. To improve the safety profile of CAR T cell therapy, several laboratories have developed safety switches to eliminate CAR T cells if necessary, including suicide genes such as iCasp9 or HSV thymidine kinase. 40 –42
While CAR T cells have been extremely effective in targeting B-ALL, the potency of those cells seems to be low in the context of solid tumors. 44 This is mainly due to the immunosuppressive microenvironment, which is known to inhibit the function of tumor-specific T cells, including CAR T cells. The use of antagonistic antibodies able to block check point inhibitory receptors, such as programmed death 1 (PD-1) or cytotoxic T lymphocyte antigen 4 (CTLA-4), was shown to enhance the anti-tumor effect 41 of tumor-infiltrating T cells significantly. Since these antagonistic antibodies can be associated with high toxicity, an attractive alternative approach is the genetic knockout of the loci encoding PD-1 and CTLA-4 to generate “enhanced CAR T cells.” 45 On the other hand, CAR T cells are mostly manufactured as an autologous T cell product, which has several limitations, including patient-to-patient variation in the quality of the starting T cell material, as well as the high manufacturing costs. Much effort has been undertaken to produce so-called “universal CAR T cells,” which can be produced from a healthy donor and then stocked until needed. Such off-the-shelf CAR T cells would improve the overall quality of the CAR T cell product and significantly reduce the manufacturing cost.
Designer nuclease technology will play a major role in the development of such “universal CAR T cells” and “enhanced CAR T cells” (Table 2). Several studies have used programmable nucleases to knockout the loci encoding the T cell receptor (TCR) alpha or beta chains, TRAC or TRBC, 46 –49 or B2M that codes for beta-2-microglobulin, a major component of the HLA-I complex. 50 While TRAC or TRBC knockout will abolish the expression of the TCR/CD3 complex at the cell surface, B2M knockout will prevent HLA-I expression. As a result, these knockouts will eliminate TCR-mediated autoimmunity and/or alloreactivity against HLA-mismatched cells without affecting the potency of these edited CAR T cells. 50 Of note, gene editing has also been successfully applied in T cells expressing a recombinant tumor-specific TCR. One of the limitations in these applications is the mispairing between the exogenous and the endogenous TCR chains, which can lead to unpredictable and harmful TCR hybrids that affect safety and efficacy of the generated T cell product. Indeed, Mastaglio et al. demonstrated reduced alloreactivity in a mouse tumor model when applying ZFNs to knockout the endogenous TRAC gene in T cells expressing a recombinant TCR targeted to the NY-ESO-1 antigen. 51
Overview: gene editing in CAR T cells
PDCD1, programmed cell death 1; LAG3, lymphocyte activation gene 3; TRAC, T cell receptor alpha constant; TRBC, T cell receptor beta constant; B2M, beta-2-microglobulin; CCR5, C-C-chemokine receptor type 5; HDR, homology-directed repair; NHEJ, non-homologous end joining; CRISPR, clustered regularly interspaced short palindromic repeats; RNP, ribonucleoprotein; ZFN, zinc finger nuclease; megaTAL, meganuclease fused to transcription activator-like effector domain; TALEN, transcription activator-like effector nuclease; LV, lentiviral vector; AAV6, adeno-associated virus type 6 vector; HIV, human immunodeficiency virus.
Manufacturing of CAR T cells generally involves the delivery of the CAR transgene using randomly integrating viral 40 and non-viral 52 vector systems. While genotoxicity in T cells does not seem to be a major concern, continuous overexpression of a CAR might be problematic because of accelerated effector T cell exhaustion, which could reduce the persistence of these engineered CAR T cells in vivo. To address this safety aspect, transient CAR expression was explored by transfecting T cells with CAR encoding mRNA. 53 However, such a short-term CAR expression might limit clinical applicability if multiple infusions will be needed to reach therapeutic effect. To realize a more physiological expression level of the CAR, designer nuclease technology was used to target the integration of the CAR transgene into various loci. HDR frequencies of up to 40% were achieved in primary T cells using CRISPR-Cas9 and AAV6 vectors to deliver the CAR donor template. 20,47,54 This strategy was used, for example, to generate human immunodeficiency virus (HIV)-specific CAR T cells that were resistant to HIV-1 infection by targeting the integration of the CAR into the CCR5 locus, which codes for the co-receptor used by HIV to enter the cells. 55 Of particular interest is the targeted integration of a CAR transgene into TCR encoding loci. Such a strategy has been employed to place the CAR under control of the TRAC promoter and thus ensure “physiological” expression. Indeed, when Sadelain et al. placed a CD19-specific CAR into the endogenous TRAC locus, the expression of the anti-CD19 CAR was considerably lower compared to expression from the retroviral vector. However, this “physiological” control of CAR expression proved to lead to a more uniform CAR transgene expression, which was accompanied by delayed effector T cell exhaustion and superior CAR T cell potency in a murine cancer model in vivo. 54 Whether “physiological” CAR expression will have an advantage in targeting solid tumors with CAR T cells remains an open question.
As mentioned above, CAR T cells with augmented antitumor activity can be produced by disrupting the genes coding for inhibitory immune checkpoints, such as PDCD1 that encodes PD-1, CTLA4, or LAG3 56 coding for lymphocyte activation gene 3. PD-1, for instance, is considered an exhaustion marker, which inhibits the function of tumor-infiltrating lymphocytes to eliminate tumors. Knockout of PDCD1 in tumor-reactive T lymphocytes has improved the cytokine release profile, 57 augmented cytotoxic activity in vitro, 57 and enhanced antitumor activity in a mouse model. 58 Several phase I/II clinical trials exploring PD-1 knockout (CAR) T cells have been initiated as a consequence. 1
Engineered nucleases can also be harnessed to knockout multiple genes simultaneously. For instance, CD19-specific CAR T cells were efficiently modified with TALENs at two loci that code for the TCR alpha chain and CD52, a protein targetable with alemtuzumab, which is used as an antibody in the treatment of leukemia. Recently, two infants suffering from relapsed B-ALL were successfully treated with such a CAR T cell product in combination with an anti-CD52 serotherapy. 59 The CRISPR-Cas system is particularly amenable to multiplexing. Ren et al. manufactured CD19-targeting CAR T cells in which the TRBC, B2M, and PDCD1 loci were knocked out. These universal CAR T cells did not express a TCR or any HLA-I molecules or PD-1, which resulted in high antitumor activity and longer persistence of the modified CAR T cells in a tumor mouse model. 50 Furthermore, in order to augment resistance to Fas-mediated apoptosis, Ren et al. also manufactured triple knockout CAR T cells by combining the genetic disruption of TRAC, B2M, and FAS. The generated CAR T cells demonstrated enhanced killing activity and longer persistence in mouse tumor models. 45 Multiplex gene editing with two or more nucleases that cleave multiple sites in the genome at the same time certainly has an advantage in reducing the number of manufacturing steps. However, the final product should be carefully assessed with regard to chromosomal translocations. Simultaneous cleavage at two genomic sites resulted in up to 1% of cells with translocation events. 48
In conclusion, the combination of CAR T cell technology with genome editing has heralded a new era in cancer immunotherapy, leading to the development of novel CAR T cell products to treat tumor entities for which currently no cure is available. Several clinical trials have been initiated with gene edited CAR T cells to treat patients suffering from various solid tumors by using designer nucleases to knockout PD-1 in order to enhance effector T cell function (NCT02793856, NCT02867345, NCT02863913, and NCT02867332). The data from these clinical trials will be extremely valuable to assess both the safety and applicability of genetically modified CAR T cells in a clinical setting.
Epigenome Editing
Genome editing aims to achieve therapeutic benefit by harnessing the cellular DNA repair machinery to introduce targeted genomic changes. As outlined above, such strategies can be explored to correct disease-causing mutations or to provide a cell with a new feature, such as the resistance to a pathogen through a targeted gene knockout. In the latter case, the reliance on designer nuclease induced NHEJ-based random mutagenesis to inactivate host genes, however, may hamper the general applicability of such an approach for safety reasons. Targeted editing of the epigenome, on the other hand, represents an alternative way through which a cell's phenotype and/or function can be altered without changing the underlying DNA sequence. 60,61 In the last decade, the molecular mechanisms that control gene expression through read, write, and erase of epigenetic marks and the correlation between aberrant epigenetic landscapes and disease states, such as imprinting defects, 62 neurological disorders, 63 and ultimately cancer, 64 have been better understood. Notably in this context, targeted epigenome editing has enabled researchers to rewrite the epigenome specifically in defined genomic locations in order to prove or disprove some of these hypotheses.
The ability to modulate gene expression in a specific fashion 65 has been attempted in the last decades using artificial transcription factors, which were shown to up- or downregulate target gene expression in a specific context. 66,67 If not continuously expressed, however, their activity is generally transient, and the effect is typically diluted during cell division. 68 To overcome this limitation, several groups have developed synthetic methyltransferases (SMTs) that can be used for inducing sustained control of gene expression via the modification of heritable epigenetic marks. This was achieved through the fusion of effector domains, capable of inducing epigenetic changes, with DNA targeting domains that bind to specific sites in the genome and define the region where the epigenome is altered. The first example of targeted epigenome editing dates back to a pioneering study in 1997. Xu and Bestor fused a bacterial DNA methyltransferase to a zinc-finger-based DNA binding domain (ZF) with the aim of establishing specific CpG methylation of an oligonucleotide in vitro. 69 It took another 10 years to establish a proof-of-concept in human cells. Jeltsch et al. generated a chimeric DNA methyltransferase by fusing the murine DNA methyltransferases 3a and 3b to an engineered ZF to target DNA methylation to the herpes-simplex-virus 1 (HSV1) IE175k promoter, resulting in reduced HSV1 replication in cell culture. 70 To improve the overall efficiency of SMTs, Siddique et al. mimicked the natural assembly of the DNA methylation complex and generated a single fusion protein consisting of the C-terminal portions of DNA methyltransferases 3A and 3L coupled to an engineered ZF targeting the VEGF-A promoter. 71 Transient delivery in immortalized cell lines led to strong repression of the target gene as result of targeted DNA methylation. Despite these first successes, the failure to maintain the introduced epigenetic marks represented a major drawback, with many studies reporting the loss of gene silencing after loss of SMT expression. This hurdle was overcome by combining the activities of transcriptional repressor domains with that of methyltransferases at the target locus. As recently reported, such systems were capable of sustained target gene silencing in immortalized human cell lines 72,73 with remarkable specificity. Importantly, gene expression can also be controlled by depositing synthetic marks on histones. For instance, selective histone methylation has been associated with reduced gene expression due to modification of chromatin architecture at the specified locus. 74 Moreover, it will be interesting to see whether the targeted integration of cis or trans-regulatory elements, as shown for the human X-inactivating gene, XIST, 75 can be employed to change the epigenetic state of a chromosomal region or the whole chromosome.
Although the efficiency of targeted epigenome editing has tremendously increased in the last decade, with successful examples of controlling target gene expression by modulating chromatin structure via deposition of synthetic epigenetic marks on DNA or histones, the challenge to translate this knowledge in the clinic still remains. 65 From a therapeutic perspective, the use of targeted epigenome editing to silence stably or reactivate the expression of a target gene remains a valuable option, in particular in a context where gene inactivation may provide a therapeutic benefit. It has recently been demonstrated that targeted epigenome editing can be explored to silence both co-receptors of HIV-1 in primary T cells after short-term exposure of the cells to designer epigenome modifiers (DEMs) that target the CCR5 and CXCR4 promoters. 73 The absence of direct off-target effects due to the action of these DEMs highlights the potential safety of such a strategy. On the other hand, the missing understanding of the dynamics of gene silencing at the single cell level, as well as the undocumented stability of the epigenetic marks in highly dynamic T cells that undergo continuous remodeling of their transcriptome and chromatin in response to intrinsic and extrinsic stimuli throughout their life-span, are still open questions. 76
Notwithstanding, the modification of the epigenome is currently explored to treat cancer, which, in many instances, was shown to originate from aberrant epigenetic regulation. Epigenetic drugs inducing hypomethylation, such as Azacitidine, have been explored to normalize blood cell count in individuals suffering from myelodisplastic syndrome. 77 Even though the observed positive effects in patients suggest that modulation of the epigenome offers a new concept to treat cancer, the nonspecific action of these epigenetic drugs can lead to severe adverse effects. 78 Targeted epigenome editing may provide an approach to understand better the correlation of epigenetic aberrations with the onset of malignancies. However, two major hurdles have to be considered when contemplating targeted epigenome editors as cancer therapeutics: (1) the efficiency in delivering the editors to a sufficiently high number of malignant cells, which is particularly challenging in the case of solid tumors; and (2) the selective growth advantage of non-targeted cells. With this respect, approaches combining chemotherapy and targeted epigenetic drugs may offer new solutions to reduce cancer progression, augment the sensitivity of cancer cells to chemotherapeutic agents, and eventually increase a patient's life-span. 79
In summary, in the last few years, we have learnt that genome editing can suffer from promiscuous binding of the designer nucleases to so-called off-target sites. 6 Even a small number of off-target cleavage events in any region of the genome has the potential to induce translocation events with deleterious consequences. Conversely, off-target epigenome editing is only of concern if introduced in cis-regulatory regions of the genome, hence greatly reducing the potential risks. Moreover, because epigenome editing does not involve any alteration of the genomic DNA sequence, the effect can be reversed. Although in its infancy, target epigenome editing may offer novel therapeutic opportunities in the future. To this end, it will be instrumental to understand better the basic mechanisms that link the epigenome to cellular functions, and to develop novel and more efficient epigenome editors capable of controlling gene expression in a sustained manner.
Conclusions
Classical gene therapy approaches have been successfully applied in patients for roughly 10 years, and two gene therapeutics, Strimvelis and Glybera, have been approved as drugs in Europe. Genome editing using designer nucleases has also made the first steps into the clinic. 1,2 Although few data from these clinical trials are available, it is safe to assume that ZFNs, TALENs, and CRISPR-Cas will be tested in numerous additional trials over the next few years. Among the most promising applications in the field of hematological disorders are ex vivo genome editing strategies for the therapy of cancer using “enhanced” and “universal” CAR T cells and the transplantation of genetically corrected blood stem cells for the treatment of hereditary hemoglobinopathies and primary immunodeficiencies. Although ZFNs are still the most frequently used platform in the clinic, it is foreseen that this platform will get strong competition from TALENs and in particular from the CRISPR-Cas system. The better availability of these platforms combined with their high activities and specificities make TALENs and CRISPR-Cas nucleases suitable candidates for clinical translation. Novel approaches to control gene expression through epigenome editors offer additional opportunities to develop therapeutics based on new principles. Even though we are still at the dawn of targeted epigenome editing, a better understanding of the function of the different epigenetic marks, as well as the dynamics driving gene expression and remodeling of the chromatin structure, will generate new impetus to the field. Over the next decade, many of the preclinical approaches described in this review will find their way to patients and fundamentally and sustainably change clinical medicine.
Footnotes
Acknowledgments
We thank the members of the laboratory for helpful discussions. Current research in the lab is supported by the Federal Ministry of Education and Research (IFB—01EO0803; iMACnet—01EK1602B; HBV-TALE—01DG15005), the German Research Foundation (SFB1160—TP17), the European Commission's Horizon 2020 Program (SCIDNET—666908; CARAT—667980), the Marie Skłodowska-Curie Innovative Training Networks (IMGENE—765269), Casebia Therapeutics (CAU-950-20140702), Cellectis (ZVS20170614), and the German Academic Exchange Service.
Author Disclosure
T.C. is a consultant for TRACR Hematology. Research of the lab is supported by Casebia Therapeutics, Cellectis, and Miltenyi Biotec. None of the other authors declare any conflict of interest.