
Abstract
Microtubule actin crosslinking factor 1 (MACF1) is a large spectraplakin protein known to have crucial roles in regulating cytoskeletal dynamics, cell migration, growth, and differentiation. However, its role and action mechanism in bone remain unclear. The present study investigated optimal conditions for effective transfection of the large plasmid PEGFP-C1A-ACF7 (∼21 kbp) containing full-length human MACF1 cDNA, as well as the potential role of MACF1 in bone formation. To enhance MACF1 expression, the plasmid was transfected into osteogenic cells by electroporation in vitro and into mouse calvaria with nanoparticles. Then, transfection efficiency, osteogenic marker expression, calvarial thickness, and bone formation were analyzed. Notably, MACF1 overexpression triggered a drastic increase in osteogenic gene expression, alkaline phosphatase activity, and matrix mineralization in vitro. Mouse calvarial thickness, mineral apposition rate, and osteogenic marker protein expression were significantly enhanced by local transfection. In addition, MACF1 overexpression promoted β-catenin expression and signaling. In conclusion, MACF1 overexpression by transfecting the large plasmid containing full-length MACF1 cDNA promotes osteoblast differentiation and bone formation via β-catenin signaling. Current data will provide useful experimental parameters for the transfection of large plasmids and a novel strategy based on promoting bone formation for prevention and therapy of bone disorders.
Introduction
M
MACF1 has been found to be expressed in bone-forming osteoblasts, partly co-localizes with F-actin and microtubules, 6,13 –15 and regulates osteoblast proliferation and cell cycle progression. 13,16 Nonetheless, the role of MACF1 in osteoblast differentiation and bone formation remains unclear. Furthermore, MACF1 has been found to regulate the Wnt/β-catenin signaling pathway positively 17 ; its deficiency not only blocks Axin translocation, but also inhibits downstream β-catenin/T-cell transcription factor transcriptional activation. 17 However, the potential action mechanisms of MACF1 in skeletal cells have not been explored previously. Since β-catenin signaling has been implicated in regulating multiple stages of osteoblast differentiation and bone formation, 18,19 it is likely that MACF1 may affect osteoblast differentiation and bone formation via β-catenin.
In the current work, the functions and mechanisms of MACF1 in osteoblast differentiation and bone formation in vitro and in vivo were investigated. Notably, gene transfection is critical to the regulation of gene expression and investigation of gene function(s) in gene therapy studies. However, this is extremely challenging for MACF1 (620 kDa) due to the enormous size of the plasmid (PEGFP-C1A-ACF7[P-ACF7]), which contains full-length MACF1 cDNA(∼21 kbp), and technologies for efficient large plasmid transfection have yet to mature. 20,21 At present, methods for transfecting cells with plasmids include lipidosomes, nanoparticles, electroporation, microinjection, and calcium phosphate coprecipitation. 22 –24 Previous studies have indicated that transfection efficiency is inversely proportional to plasmid size, and plasmids >12 kbp do not transfect into cells well, if at all, 21,25 especially in osteogenic cells. To carry out gene transfection studies to investigate MACF1 functions in osteogenic cells, it is imperative to define the optimum conditions for transfecting the large plasmid P-ACF7 (∼21 kbp). Herein, novel conditions for transfecting P-ACF7 were explored, and then the function of MACF1 in osteoblast differentiation, bone formation, and β-catenin signaling was examined in vitro and in vivo.
Materials and Methods
Plasmids, cell cultures, animals, and treatments
P-ACF7 was generously provided by Dr. Xiaoyang Wu (University of Chicago, Chicago, IL). The PEGFP-C1A plasmid (P-C1A, 4,731 bp; a negative control plasmid) was purchased from GeneChem Intl. (Shanghai, China). The TOPflash reporter and RenillapRL-TK plasmids were generously provided by Dr. Pengsheng Zheng (Xi'an Jiaotong University, Xi'an, China). The murine osteoblast MC3T3-E1 cell line was generously provided by Dr. Hong Zhou (University of Sydney, Sydney, Australia). The MC3T3-E1 cell line was cultured in α-Modified Eagle's Medium (α-MEM; Gibco, Carlsbad, CA) with 10% fetal bovine serum (FBS; Biological Industries, Kibbutz BeitHaemek, Israel), 1% L-glutamine (Sigma–Aldrich, St. Louis, MO), and 1% penicillin/streptomycin (Amresco, Solon, OH). Cell cultures were maintained at 37°C with 5% CO2. Cells were passaged after digestion with 0.25% trypsin containing 0.02% EDTA (Life Technologies, Carlsbad, CA). Four-month-old C57BL/6 mice were obtained from the Animal Center of Fourth Military Medical University (Xi'an, China).
Ten male mice were randomly divided into two groups. In the experimental group, five mice were subcutaneously injected over the calvarial surface with 40 μL of P-ACF7 plasmid mixed with Entranster™ in vivo transfection reagent (nanoparticles; Engreen Biosystem Co. Ltd., Beijing, China) twice per day for five consecutive days, according to the manufacturer's instructions. The transfection condition with this reagent was non-optimized. In the control group, the calvarial surface of five mice received the same volume of normal saline. They all received the same standard diet during the experimental period. All mice were euthanized 2 weeks after treatment. In the current work, all efforts were made to reduce the number of the mice used and their suffering. All animal experiments were performed in accordance with the Guiding Principles for the Care and Use of Laboratory Animals, and all experimental procedures were approved by the Institutional Experimental Animal Committee of Northwestern Polytechnical University (Xi'an, China).
Plasmid transfection in vitro
MC3T3-E1 cells (1 × 107 per well) were transfected by electroporation (1,800 V, 30 ms) with the large plasmid P-ACF7 or control plasmid P-C1A with a Neon Transfection System (Invitrogen, Carlsbad, CA). Next, cells were seeded onto a six-well plate with 10 mL of α-MEM. Adherent cells were washed with 2 mL of α-MEM twice, and the medium was changed to antibiotic-free growth medium 6 h after electroporation. For reagent transfection, cells were transfected with a mixture of the plasmid and Entranster™ H4000 (the main component being nanoparticles; Engreen Biosystem Co. Ltd), Micropoly Transfecter™ (Micropoly, Nantong, China), or Lipofectamine 2000 (Invitrogen), according to each manufacturer's instructions.
Quantitative real-time polymerase chain reaction
To examine levels of MACF1 mRNA expression and osteogenesis- and β-catenin signaling-related genes, total RNA was extracted from mouse calvarial tissues or MC3T3-E1 cells using TRIzol reagent (Invitrogen), according to the manufacturer's instructions. RNA (1 μg) was used for cDNA synthesis using a one-step PrimeScript RT reagent kit (TaKaRa, Dalian, China). Quantitative real-time polymerase chain reaction (qRT-PCR) amplification was performed using a Thermal Cycler C-1000 Touch system (Bio-Rad, Hercules, CA) with the SYBR Premix Ex TaqIIkit (TaKaRa). Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was used as the internal control. qRT-PCR reaction conditions included initial denaturation at 95°C for 30 s, followed by 44 cycles at 95°C for 10 s, 60°C for 30 s, and 72°C for 5 s. Data were analyzed using the comparative Ct method (2–ΔΔCt) and expressed as fold changes compared to the corresponding control (GAPDH). Primers (for sequences, see Supplementary Table S1; Supplementary Data are available online at
Western blot analysis and immunohistochemical, hematoxylin, and eosin staining
To examine levels of protein expression, Western blotting analyses were carried out using primary antibodies recognizing rabbit polyclonal MACF1 (1:500; Abcam, Cambridge, United Kingdom), rabbit polyclonal β-catenin (1:1,000; Cell Signaling Technology, Danvers, MA), and mouse monoclonal GAPDH (1:1,000, Calbiochem, Darmstadt, Germany) primary antibodies, as previously described. 13 Cells or tissues were lysed in lysis buffer (Beyotime, Shanghai, China) on ice. Proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10% stacking and 6% separating gels for MACF1; 5% stacking and 12% separating gels for other proteins) and transferred onto nitrocellulose membranes (PALL Corp., Port Washington, NY). Membranes were blocked with 5% nonfat milk and incubated with primary antibodies at 4°C overnight. After primary antibodies were rinsed off, membranes were incubated with a horseradish peroxidase-labeled secondary antibody (CWBio, Beijing, China) for 2 hours at room temperature. Afterward, secondary antibodies were washed off, and blots were visualized using a chemiluminescence detection system (Promega, Madison, WI). Protein bands were exposed to X-ray film (Kodak, Rochester, NY), and band intensities were analyzed and normalized to GAPDH levels. To localize protein expression, mouse calvaria were dissected, fixed in 4% paraformaldehyde, decalcified in 17% EDTA for 2 days, embedded in paraffin, and sliced into 5-μm-thick sections for immunohistochemistry. After dewaxing and rehydration, sections were blocked in 5% goat serum in phosphate-buffered saline (PBS) and then incubated overnight at 4°C with primary antibodies against MACF1 (1:100; Abcam), osteoprotegerin (OPG; 1:200; R&D, Minneapolis, MN), runt-related transcription factor (Runx)2 (1:200; R&D), osteocalcin (OCN;1:200; Santa Cruz Biotech, Dallas, TX), and collagen type I (Col Iα1; 1:100; Santa Cruz Biotech). A non-immune serum was used to replace the first antibody as a negative control. After antibodies were rinsed off, sections were incubated with a horseradish peroxidase-labeled secondary antibody (CWBio) for 1 h at room temperature and processed for color development using 3,3′-diaminobenzidine (CWBio) and hematoxylin counterstain. For HE staining, dewaxed sections were rehydrated, and then nuclei were stained with the alum hematoxylin for 5 minutes. After rinsing in running tap water, sections were differentiated with 1% acid alcohol 30s, stained with eosin 2 minutes more, dehydrated by graded ethanol, and cleaned by dimethylbenzene. Stained sections were scanned with a Aperio AT2 Digital Pathology Scanner (Leica, Wetzlar, Germany).
Bone histomorphometric analyses
The thickness of calvaria was measured by a 3D Super Depth Digital Microscope (Hirox, Tokyo, Japan). To measure the mineral appositional rate (MAR), double calcein labeling was performed by intraperitoneal injection of calcein green (20 mg/kg body weight; Sigma–Aldrich) 10 and 2 days before euthanasia for specimen collection. Dissected calvarial samples were fixed in 75% ethanol and embedded in methyl methacrylate (Sigma–Aldrich). Undecalcified and unstained transverse sections (10 μm thick) were examined with a fluorescent microscope (Nikon 80i; Nikon, Tokyo, Japan). Bone dynamic histomorphometric analyses for MAR were performed using Image J software (National Institutes of Health, Bethesda, MD).
Alkaline phosphatase and Alizarin red staining
To examine transfection effects on osteogenic differentiation, cells were washed and seeded onto 96-well plates (1.5 × 104 cells/well) in triplicate 24 h after electroporation. Osteoblasts were induced by culturing in osteogenic medium with α-MEM, 10% FBS, 1% β-glycerophosphate, 1% ascorbic acid, and 1% L-glutamine (Sigma–Aldrich). The cultures were maintained at 37°C with 5% CO2, and the medium was replaced every 2 days. Sixty hours (2.5 days) after electroporation, alkaline phosphatase (ALP) staining was performed with a 5-bromo-4-chloro-3-indolyl-phosphate (BCIP)/nitro blue tetrazolium (NBT) ALP color development kit (Beyotime). Briefly, cells were gently rinsed with PBS three times and fixed with 10% neutral buffered formalin (Sigma–Aldrich) at room temperature for 15 min. After washing in PBS, the fixed osteoblasts were then incubated with 500 μL/well of BCIP/NBT substrate for about 6 h in the dark. After washing with double-distilled water, plates were scanned with a CanoScan 9000F Mark II scanner (Canon, Tokyo, Japan). Alizarin red staining of mineralized osteoblast nodules was carried out on day 15. Briefly, cells were fixed and washed as above and then stained in 0.5% Alizarin red S (pH 4.0; Sigma–Aldrich) for 30 min. After washing, the plates were scanned with a CanoScan 9000F Mark II scanner.
Luciferase reporter gene assays
To analyze MACF1 regulation of β-catenin activity, the TOPflash luciferase reporter plasmid and internal control pRL-TK plasmid were mixed and co-transfected into MC3T3-E1 cells together with PEGFP-C1A or PEGFP-ACF7 plasmids using the Neon Transfection System. The TOPflash luciferase reporter plasmid has a lymphoid enhancer binding factor/T-cell transcription factor binding site that recognizes endogenous activated β-catenin in transfected cells. The luciferase reporter assay was performed 72 h after transfection. Firefly and Renilla luciferase activity were analyzed using the dual-luciferase reagent assay kit (Promega), according to the manufacturer's instructions; TOPflash luciferase activity was normalized with Renilla activity.
Statistical analyses
All cell experiments were independently repeated at least three times with cells established in triplicate for each single assay, and the data were reported as the mean ± standard error of the mean. Statistical analyses of the data were performed using GraphPad Prism v6 (GraphPad Software, Inc., La Jolla, CA), and a Student's t-test was used to compare differences. A p-value of <0.05 was considered statistically significant.
Results
Transfection efficiency of P-ACF7 in vitro
The large plasmid P-ACF7 carries an enhanced green fluorescent protein (EGFP) label and contains full-length human MACF1 cDNA (Supplementary Fig. 1A). Electrophoresis analyses suggested it is much larger size than the control plasmid P-C1A (Supplementary Fig. 1B). Further, BLAST analysis of the sequence inserted in plasmid P-ACF7 showed that this sequence is highly homologous with those of human and mouse MACF1 (SSupplementary Fig. 1C).
To examine optimal conditions in which to promote MACF1 expression in vitro, the P-ACF7 was transfected into mouse MC3T3-E1 osteoblast cells using different transfection reagents or electroporation systems (Fig. 1 and Supplementary Fig. S2). Fluorescence microscopy results showed green fluorescence in MC3T3-E1 cells after transfecting P-ACF7. The transfection efficiency is the number of cells with green fluorescence divided by the number of surviving cells. The transfection efficiencies of P-ACF7 (as analyzed by fluorescent microscopy measurement of EGFP expression) in MC3T3-E1 cells were 2%, 1.8%, and 17% using lipidosomes, liquid transfectants, and nanoparticles, respectively (Fig. 1A). In addition, electroporation was also implemented to transfect P-ACF7, with the electroporation transfection efficiencies and cell survival rates being detected by different parameters. The transfected cell survival rates by electroporation were inferior to those by lipidosome and nanoparticle methods, and the electroporation condition used (1,800 V, 30 ms) was chosen based on optimal results obtained for both electroporation transfection efficiency and cell survival rate (Fig. 1B and Supplementary Fig. S2).

Transfection efficiency of the large plasmid PEGFP-C1A-ACF7 (P-ACF7) in vitro.
The transfection efficiencies of plasmids in MC3T3-E1 cells were compared using nanoparticles and electroporation. The green fluorescence intensity in MC3T3-E1 cells after treatment with P-ACF7 was obviously superior using electroporation relative to that with nanoparticles. The control group (treated with the small P-C1A plasmid) showed the opposite (Fig. 1C). Moreover, MACF1 expression at both mRNA and protein levels was significantly increased by electroporation in P-ACF7 groups (p < 0.01), but not by nanoparticles, compared to the corresponding control groups (Fig. 1D). These results suggested that electroporation efficiently transfected the large P-ACF7 plasmid, which promoted MACF1 expression in MC3T3-E1 cells in vitro, indicating this method was feasible for transfecting large plasmids >20 kbp in size into osteoblasts.
MACF1 overexpression promotes osteoblast differentiation in vitro
To investigate the effect of MACF1 overexpression on osteoblast differentiation in vitro, MC3T3-E1 cells were treated with P-ACF7. The results showed that mRNA expression of osteogenic marker genes Alp, Runx2, and Col Iα1was dramatically upregulated by 390% (p < 0.05), 156% (p < 0.05), and 396% (p < 0.001) in the P-ACF7 group, respectively, compared to the control group (Fig. 2A). In addition, the ALP-positive blue-violet complexes and Alizarin red-stained mineralized nodules in the P-ACF7 group were significantly increased compared to the control group, confirming the positive effect of transfection with P-ACF7 on osteoblast differentiation (Fig. 2B). These findings indicate that MACF1 overexpression with P-ACF7 notably promotes osteoblast differentiation.

MACF1 overexpression promotes osteoblast differentiation.
Transfection efficiency of large plasmid P-ACF7 in vivo
To enhance MACF1 expression in vivo, P-ACF7 with nanoparticles was subcutaneously injected into mouse calvaria. On day 10, green fluorescence in calvaria of plasmid-transfected mice was not observed. Therefore, the time to harvest samples was doubled from 10 to 19 days. By day 19, distinct green fluorescence was observed in craniofacial bones of mice (Fig. 3A). Furthermore, EGFP (carried by P-ACF7) was only found expressed in the calvarial skin and calvaria, which verified the successful transfection and expression of P-ACF7 (Fig. 3B and C). Meanwhile, immunohistochemical analyses showed the MACF1 protein was strongly expressed on the membrane surface of calvaria and bone marrow after subcutaneous injection of P-ACF7. Further, integral optical density (which assesses the bone formation area) of MACF1 staining in the P-ACF7 group was approximately 3.6-fold higher (567 ± 95.1) than in the control group (155 ± 41.8; p < 0.01, Fig. 3D and E). These results indicate that transfection of P-ACF7 can effectively increase MACF1 expression in vivo and that this transfection method is feasible and practicable.

Transfection efficiency of the large plasmid P-ACF7 in vivo.
MACF1 overexpression promotes local bone formation in mouse calvaria
To verify the role of MACF1 overexpression in bone formation further, transfection of P-ACF7 was implemented for five consecutive days in calvaria of mice (Fig. 4A). Results showed the average calvarial thickness of the P-ACF7 group was 649.5 ± 63.86 μm, which was about 1.5 times higher than that of the control group (424.2 ± 23.07 μm; p < 0.05; Fig. 4B and C). Consistently, the MAR (an assessment of bone formation) of P-ACF7-treated mice (1.521 ± 0.05512 μm/day) was greater than that of control mice (1.271 ± 0.02756 μm/day; p < 0.001; Fig. 4D and E). Furthermore, hematoxylin and eosin staining showed that calvarial bones of all mice were morphologically intact, with clear cortical bone structures and bone marrow, and no abnormalities were observed in either P-ACF7 or control groups (Fig. 4F). In addition, ALP was strongly expressed on the mouse calvaria in the P-ACF7 group (Fig. 4G).

MACF1 overexpression promotes bone formation in mouse calvaria.
Protein products of osteogenic marker genes (Runx2, OCN, and OPG) on the mouse cranium were observed by immunostaining. In the P-ACF7 group, the integral optical density values of Runx2, OCN, and OPG were nearly 6.3-, 2.2-, and 2.8-fold higher than that of the control group, respectively (Fig. 5A–C and E–G). However, the integral optical density of Col Iα1, an early osteogenic marker gene, was suppressed in the P-ACF7 group compared to the control (Fig. 5D and H). A non-immune serum was used to replace the first antibody as a negative control, which produced no staining (Supplementary Fig. S3). These results revealed MACF1 overexpression clearly promoted local bone formation in mouse calvaria.

Effects of P-ACF7 transfection on immunohistochemical staining of bone formation markers in mouse calvaria.
MACF1 overexpression promotes β-catenin expression and activity
MACF1 has been reported to play a vital role in Wnt/β-catenin signaling that is critical in regulating osteoblast differentiation. 17,26 Therefore, β-catenin was hypothesized to mediate the effects of MACF1 overexpression in facilitating osteoblast differentiation. qRT-PCR results showed that β-catenin mRNA expression was obviously upregulated (by 138%) in MC3T3-E1 cells transfected with P-ACF7 (p < 0.01; Fig. 6A). Additionally, β-catenin protein expression in the P-ACF7 group was distinctly enhanced versus the control group (Fig. 6B).
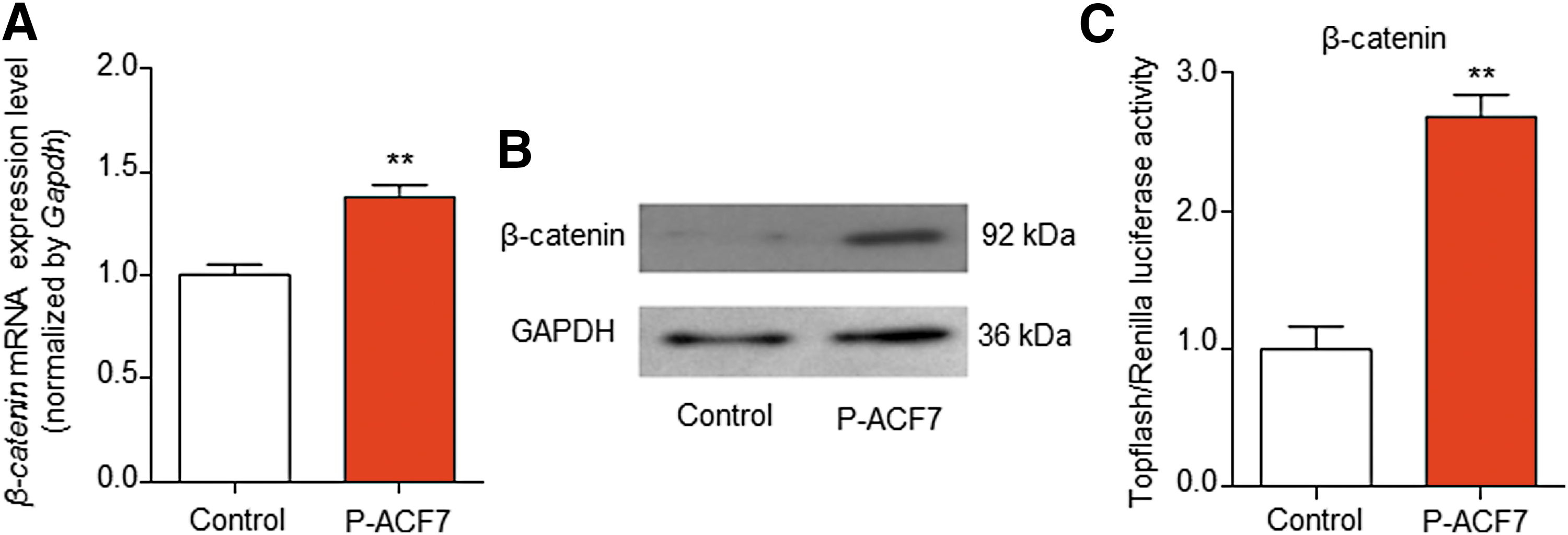
MACF1 overexpression promotes β-catenin expression and activity.
To clarify the relationship between MACF1 expression and β-catenin signaling activation further, P-ACF7 and the β-catenin-lymphoid enhancer binding factor/T-cell transcription factor activation luciferase reporter plasmid (Renilla plasmid as negative control) were co-transfected into MC3T3-E1 cells. The results showed that β-catenin activity was evidently increased (∼269%) after P-ACF7-overexpression of MACF1 (p < 0.001; Fig. 6C), suggesting that MACF1 overexpression may promote osteoblast differentiation via β-catenin signaling activation.
Discussion
MACF1 is a large molecule and a crucial member of the spectraplakin family that is widely expressed in different cells. This protein also takes part in multiple cellular processes, such as cellular polarization and migration. 4,7,12,27,28 Though MACF1 has been shown to affect osteoblastic proliferation, 13,14 the effect and mechanism of MACF1 on osteoblast differentiation and bone formation are not yet clear. This study explored optimal conditions for transfecting the large plasmid P-ACF7 (∼21 kbp), which carries full-length MACF1 cDNA, and then investigated the effects and action mechanism of MACF1 overexpression on osteoblast differentiation and bone formation. After optimizing transfection parameters and conditions, P-ACF7 was found to transfect effectively into osteogenic cells using local electroporation (1,800 V, 30 ms) both in vitro and in vivo at calvaria with nanoparticles. Current results indicate that MACF1 is a key positive regulator of osteoblast differentiation and bone formation, and MACF1 overexpression promotes osteoblast differentiation via β-catenin signaling.MACF1's role in β-catenin signaling will provide a useful experimental parameter and novel strategy for transfection of large plasmids and promotion of bone formation for prevention and therapy of bone disorders.
Gene transfection is a key genetic approach for studying gene function and expression modulation. However, transfection of large plasmids to induce overexpression has been a great technical challenge. Notably, the size of the plasmid is a critical factor for transfection efficiency 21,25,29 ; while small plasmids (3.5 kbp) have higher transfection efficiency and cell viability in mesenchymal stem cells under the same experimental conditions, transfection of larger plasmids (6–16 kbp) results in very low cell viability and transfection efficiency. 30,31 As the most common gene carrier, viruses (e.g., lentiviruses and adeno-associated viruses) offer generally good delivery efficiencies. 32 However, the viral delivery method is limited by the size of gene fragment being packaged. Nonviral strategies, such as lipidosomes and cationic polymers, are limited by packaging capacity and poor delivery. 33 In addition, it has been reported that small plasmids (pEGFP-IRESneo, 5,981 bp) can be transfected into human marrow stromal cells by electroporation (600 V, 100 ms), 34 and electroporation is capable of delivering large plasmids into cells in vitro but with a low transfection efficiency. 20,31 Kretzmann 33 used a nonviral delivery methodology to deliver larger EGFP-encoding pDNA (plasmid, 10.3 kbp) successfully into MCF-7 cells. However, little research has been reported that aims to optimize transfection conditions for large plasmids, including the viral and nonviral strategies. In the current study, the size of plasmid P-ACF7 reaches up to 21 kbp, and three different nonviral delivery strategies were implemented to transfect it into osteoblasts(electroporation, lipidosomes, and nanoparticles; Fig. 1). After repeated testing, continuous improvement, and comparison with different transfection reagents, P-ACF7 was transfected effectively into MC3T3-E1 cells by electroporation (1,800 V, 30 ms). These gene and protein expression studies showed that MACF1 expression was significantly enhanced by transfecting P-ACF7 in MC3T3-E1 cells using electroporation (Fig. 1). This indicates that the electroporation conditions were effective and feasible for large plasmid transfection in vitro, and this strategy is well-suited for the cloning and expression of huge DNA fragments into osteoblasts.
In primary osteoblasts, the transfection technology available for large plasmids is still immature. The difficulty in transfecting primary osteoblasts is mainly due to a lower cell survival rate in transfected cells and that transfection experiments of primary cells have so far only focused on delivering small molecules, such as small interfering ribonucleic acid 35,36 and small plasmids. 34 The large size of the MACF1 recombinant plasmid (∼21 kb) makes it difficult to transfect. Therefore, to overcome this difficulty, the authors have begun to generate a MACF1-conditional knock-in mouse model with specific high expression of MACF1 in osteoblastic cells. With this tool, the aim is to verify the effects of MACF1 on osteoblastic differentiation in primary MACF1-conditional knock-in osteoblastic cells in future studies.
Although the viral method shows great transfection efficiency, 32 viruses have a finite capacity 37 for in vivo plasmid transfection. As a nonviral vector, nanoparticles provide an alternative that overcomes the side effects of viral vectors. 38 Moreover, gene delivery with nanoparticles exhibits low toxicity and flexible control in vivo. 39 –41 For example, p (plasmid) bone morphogenic protein-2/CaPi microspheres can be injected into the tibialis anterior muscles of Wistar rats, 42 and the CMVGFP c-CPE plasmid with nanoparticles can be transfected in mice via intraperitoneal injection. 43 Notably, these previous studies using nanoparticles focused on carrying small molecules in vivo, such as small interfering RNA, micro RNA, and small plasmids. 38,44 –46 Herein, the in vivo transfection results suggest the large P-ACF7 plasmid encapsulated in nanoparticles can be successfully transfected into calvaria of mice by local injection (Fig. 3). In P-ACF7-transfected mice, the craniofacial bones showed obvious green fluorescence and MACF1 overexpression (Fig. 3). This is the first time transfection of a large plasmid into osteogenic cells has been shown in vitro and in vivo, and this work will lay the foundation for further studies of large plasmid transfection. In future, transfection conditions for large plasmids will be explored further and optimized in vitro and in vivo.
MACF1 knockdown was previously shown to have an impact on cellular metabolic activity and induce large cells with a binuclear/multinuclear structure. 13 The absence of MACF1 clearly inhibits cell proliferation in MC3T3-E1 osteoblastic cells. 13 These data indicate that MACF1 plays an important role in osteoblasts. Here, the data demonstrated that transfection of P-ACF7 significantly increased expression of osteoblast differentiation marker genes (Alp, Runx2, and Col Iα1), ALP activity, and the formation of mineralized nodules in MC3T3-E1 cells (Fig. 2). These findings implied that MACF1 overexpression remarkably promotes osteoblast differentiation in vitro. The current study also explored the function of MACF1 in bone formation in vivo. Craniofacial bones are formed via intramembranous ossification by osteoblasts derived from cranial neural crest cells. 47 In MACF1-overexpressing mice, immunohistochemical results verified strong expression of osteogenic marker proteins Runx2, OCN, and OPG on the membrane surface of calvaria and in bone marrow(except for Col Iα1;Fig. 5); this result is also in line with work by Andersen et al. 48 and Yan et al. 49 The lack of strong Col Iα1 expression is connected with bone toughness, as reduced collagen deposition is linked to increased calcium deposition to a certain degree at a particular moment. 48,50,51 Additionally, the calvaria thickness and MAR were also distinctly increased in MACF1 overexpressing mice (Fig. 4). Overall, these results indicate that MACF1 overexpression significantly promotes local bone formation in the calvaria of mice.
During the process of osteoblast differentiation and bone formation, many molecules are actively involved in Wnt/β-catenin signaling. 52 Furthermore, expression of β-catenin is reduced in parallel with decreased MACF1 expression. 13,15 The current results suggest that MACF1 overexpression markedly promoted β-catenin expression and obviously improved β-catenin signaling activation in osteoblasts (Fig. 6). All of this suggests that MACF1 overexpression may promote osteoblast differentiation via promoting β-catenin signaling, indicating that MACF1 may play a vital role in the Wnt/β-catenin signaling pathway.
Taken together, the present study optimized transfection parameters and conditions for large plasmid (∼21 kbp) transfection into osteogenic cells were optimized by electroporation (1,800 V, 30 ms) in vitro and were tested for calvaria local transection with nanoparticles in vivo. Data from this study will be beneficial to future studies of large plasmid transfection and gene therapy. In addition, they study revealed that MACF1 may promote osteoblast differentiation and bone formation via enhancing β-catenin signaling. The findings suggest that MACF1 may be a novel target or approach for prevention and treatment of bone disorders. Future work is necessary to enhance transfection efficiency and subsequentMACF1 expression in osteogenic cells to stimulate bone formation. Furthermore, the functional fragment ofMACF1 will be explored, and an attempt will be made to transfect it in vitro and in vivo.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31570940, 31400725, and 81671928), China Postdoctoral Science Foundation (2015T81051), Shenzhen Science and Technology Project (JCYJ20160229174320053), and Fundamental Research Funds for the Central Universities (3102016ZY037). L.W. was supported by an Australian National Health and Medical Research Council grant (1094606) and the National Natural Science Foundation of China (81671928), and C.J.X. was supported by an Australian National Health and Medical Research Council Senior Research Fellowship (1042105) and the National Natural Science Foundation of China (81671928).
Author Disclosure
The authors declare no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.