
Abstract
The administration of ex vivo expanded natural killer (NK) cells as potential antitumor effector cells appears to be suitable for effector cell-based immunotherapies in high-risk cancer patients. However, good manufacturing practice (GMP)-compliant manufacturing of clinical-grade NK cells at sufficiently high numbers represents a great challenge. Therefore, previous expansion protocols for those effector cells were improved and optimized by using newly developed culture medium, interleukin (IL)-21, and autologous feeder cells (FCs). Separation of primary human NK cells (CD56+CD3−) was carried out with the CliniMACS Prodigy® in a single process, starting with approximately 1.2 × 109 leukocytes collected by small-scale lymphapheresis or from buffy coats. Enriched NK cells were adjusted to starting cell concentrations within approximately 1 × 106 effector cells/mL and cultured in comparative expansion experiments for 14 days with IL-2 (1,000 IU/mL) in different GMP-compliant media (X-VIVO™10, CellGro®, TexMACS™, and NK MACS®). After medium optimization, beneficial effects for functionality and phenotype were investigated at the beginning of cell expansion with irradiated (25 Gy) autologous FCs at a ratio of 20:1 (feeder: NK) in the presence or absence of IL-21 (100 ng/mL). Additionally, expanded NK cells were gene modified to express chimeric antigen receptors (CARs) against CD123, a common marker for acute myeloid leukemia (AML). Cytotoxicity, degranulation, and cytokine release of transduced NK cells were determined against KG1a cells in flow cytometric analysis and fluorescent imaging. The Prodigy manufacturing process revealed high target cell viabilities (median 95.4%), adequate NK cell recovery (median 60.4%), and purity of 95.4% in regard to CD56+CD3- target cells. The process in its early phase of development led to a median T-cell depletion of log 3.5 after CD3 depletion and log 3.6 after the whole process, including CD3 depletion and CD56 enrichment steps. Manually performed experiments to test different culture media demonstrated significantly higher NK cell expansion rates and an approximately equal distribution of CD56dimCD16pos and CD56brightCD16dim&neg NK subsets on day 14 with cells cultivated in NK MACS® media. Moreover, effector cell expansion in manually performed experiments with NK MACS® containing IL-2 and irradiated autologous FCs and IL-21, both added at the initiation of the culture, induced an 85-fold NK cell expansion. Compared to freshly isolated NK cells, expanded NK cells expressed significantly higher levels of NKp30, NKp44, NKG2D, TRAIL, FasL, CD69, and CD137, and showed comparable cell viabilities and killing/degranulation activities against tumor and leukemic cell lines in vitro. NK cells used for CAR transduction showed the highest anti-CD123 CAR expression on day 3 after gene modification. These anti-CD123 CAR-engineered NK cells demonstrated improved cytotoxicity against the CD123pos AML cell line KG1a and primary AML blasts. In addition, CAR NK cells showed higher degranulation and enhanced secretion of tumor necrosis factor alpha, interferon gamma, and granzyme A and B. In fluorescence imaging, specific interactions that initiated apoptotic processes in the AML target cells were detected between CAR NK cells and KG1a. After the fully automated NK cell separation process on Prodigy, a new NK cell expansion protocol was generated that resulted in high numbers of NK cells with potent antitumor activity, which could be modified efficiently by novel third-generation, alpha-retroviral SIN vector constructs. Next steps are the integration of the manual expansion procedure in the fully integrated platform for a standardized GMP-compliant overall process in this closed system that also may include gene modification of NK cells to optimize target-specific antitumor activity.
Introduction
N
For cellular immunotherapies, NK cells have to be isolated and may be expanded and activated ex vivo. However, it is still a challenge to generate large numbers of good manufacturing practice (GMP)-compliant NK cells for patient treatment. Satisfactory results were achieved using the CliniMACS device for CD3 T-cell depletion, which was followed by CD56 cell enrichment. 7 These isolated NK cells can subsequently be activated and expanded ex vivo by interleukin (IL)-2 and/or irradiated autologous peripheral blood mononuclear cells (PBMCs) as cytokine secreting feeder cells (FCs) for a limited period of time. 8 –11 As well as the use of whole PBMCs, T cells can be depleted before merging residual PBMC with NK cells in a defined ratio. 12 This may prevent overgrowth of T cells during cultivation and decreases the risk of graft-versus-host disease (GVHD) by using expanded NK cells in allogeneic settings.
Ex vivo expansion of NK cells can also be used as a tool to modulate NK cell receptor expression and to trigger NK cells for enhanced killing of target cells. Most culture conditions include IL-2, 11 which induces cytokine secretion in NK cells and upregulation of NKG2D surface receptor and various NCRs. 13,14 IL-15 also seems to be a suitable cytokine to maintain antitumor cytotoxic activity, 15 as well as increased expression of NCRs and CD69. 16 For NK cell cultivation, IL-21 can be used as an initial stimulus following expansion with IL-2 and/or IL-15. 17 IL-21 enhances cytotoxic NK cell functions and viability without demonstrating direct effects on NK cell proliferation. 18,19 First results in combining FCs, IL-21, and IL-2 showed a sustained proliferation of human NK cells. 12,19
To overcome limitations such as tumor immune escape mechanisms (TIEMs) 20 and to improve cellular immune control, NK cells can be engineered to express chimeric antigen receptors (CARs). These artificial surface receptors consist of a variable external recognition domain (single-chain variable fragment [scFv]) that is linked to a transmembrane domain and one or more intracellular signaling domains. 21 The formation of immune synapses between modified NK cells and antigen-presenting target cells results in NK cell activation, enhanced release of IFN-γ, and increased effector cell degranulation. 6
The IL-3 receptor alpha chain (CD123) has been described as a marker that is frequently overexpressed by acute myeloid leukemia (AML) cells. 22,23 Therefore, genetic manipulation of activated NK cells to generate anti-CD123 CAR-engineered NK cells holds potential to improve retargeted killing activity against CD123pos leukemia cell lines and native AML blasts.
In order to improve future NK cell manufacturing protocols, the aims of this study were (1) to establish a fully automated NK cell separation protocol, (2) to optimize NK cell expansion, and (3) to demonstrate the proof of principle using CAR-engineered NK cells redirected AML cells. Based on a previous GMP-compliant two-step immune-magnetic purification protocol from a clinical Phase I/II study, 7,24 this study shifted to the fully automated closed systems of CliniMACS Prodigy®. Next, novel developed culture media, especially NK MACS® basal medium (Miltenyi Biotec), irradiated autologous FCs, IL-21 as an initial “starter cytokine,” and repeated stimulations of IL-2 were evaluated. Distributions of NK cell surface and degranulation markers involved in NK cell activation were monitored over a period of 14 days. Additionally, expanded NK cells were modified to express an anti-CD123 CAR to retarget NK cell-dependent cytotoxicity against AML cells, and immunofluorescence time-lapse microscopy experiments revealed insight to the killing mechanisms of these anti-CD123 CAR-expressing NK cells.
Materials and Methods
Source of cell material
Peripheral blood samples (leukapheresis [LA], buffy coats [BC]) from healthy donors were collected at the Institute for Transfusion Medicine of Hannover Medical School (MHH). Written informed donor consent was collected and approved by the ethics committee of MHH (protocol #2159-2014).
Alpha-retroviral SIN vectors
Detailed composition of the vector and the transduction protocols have been described previously. 25 –27 Briefly, alpha-retroviral self-inactivating (SIN) vectors were equipped with an internal MPSV promoter to drive expression of the transgene cassettes (i.e., EGFP alone or anti-CD123 CAR followed by an internal ribosomal entry site for EGFP expression). The third-generation anti-CD123 CAR was codon optimized for human codon usage and included an anti-CD123 scFv, CD28 transmembrane domain, CD28 and 4-1BB (CD137) co-stimulatory signaling endodomains, and the CD3ζ signaling domain. 27 NK cell transduction (a multiplicity of infection [MOI] of 1) with RD114/TR-pseudotyped alpha-retroviral SIN vectors was accomplished on Retronectin, as previously described. 25
Cell lines
K562 was purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ) and maintained in Roswell Park Memorial Institute (RPMI) 1640 medium (Biochrom AG) supplemented with 10% fetal bovine serum (FBS; Biochrom AG). KG1a was obtained from the American Type Culture Collection (ATCC) and maintained in Iscove's modified Dulbecco's medium (IMDM; Biochrom AG) supplemented with 20% FBS.
Thawed primary human AML cells
Proof of principle for engineered NK cells was performed using AML blasts from three patients (kindly provided by Prof. M. Heuser, Medical School, Hannover) that were classified as M0 (sample T110) or M5 (samples T427 and T600) following the French–American–British (FAB) system. After thawing, samples were washed in phosphate-buffered saline (PBS) containing 10% FBS and treated with DNase I (0.1 mg/mL) for 15 min. Cells were cultivated in IMDM supplemented with 10% FBS, penicillin/streptomycin, L-glutamine, and IL-3, IL-6, stem cell factor, granulocyte colony stimulating factor (CSF), and granulocyte macrophage CSF (final cytokine concentration each 20 ng/mL, except CSF with 50 ng/mL) at 37°C and 5% CO2. Before using the AML cells in cytotoxicity assays, AML blasts were washed with PBS and re-suspended to 2.5 × 105 cells/mL in TexMACS medium supplemented with 5% human serum albumin (HSA). CD123 surface expression was determined for each patient sample by flow cytometry using an anti-CD123-APC antibody (BD Biosciences). 7-AAD (Beckman Coulter) was used for viability estimation of the AML blasts.
Fully automated one-step separation of NK cells using the Prodigy system
NK cells were isolated from LA or BC, respectively, with the CliniMACS Prodigy® device (Miltenyi Biotec). These effector cells were enriched from LA by the two-step procedure for CD3 depletion followed by CD56 enrichment using CD3 and CD56 MicroBeads without changing the tubing set TS 310, as directed by the user manual (Miltenyi Biotec), which comprises a single manufacturing process. The negative fraction (NF), positive fraction (PF), and fraction of reapplication bag (RB) were then analyzed. For NK cell isolation from buffy coats, PBMCs were harvested after standard density-gradient centrifugation using Ficoll (Ficoll-Paque™; GE Healthcare Biosciences) and TS730 (Miltenyi Biotec) prior to enrichment on the CliniMACS Prodigy®.
NK cell expansion
Primary human NK cells with an initial cell concentration of 2.5 × 106 NK cells/mL were cultivated in T-flasks at 37°C and 5% CO2 in various media for up to 14 days to determine the most promising NK cell expansion conditions. Therefore, CellGro® (CG; CellGenix GmbH), X-Vivo 10™ (X-VIVO; Lonza Group AG), TexMACS™ (TM; Miltenyi Biotec), and NK MACS® (NK MACS; Miltenyi Biotec) were tested. Each expansion medium was supplemented with 5% human serum type AB (Biochrom AG) and 1,000 IU/mL of IL-2 (Proleukin S; Novartis Pharma GmbH). In addition, NK MACS contained 2% NK MACS Supplement® (Miltenyi Biotec). Cell cultivation was initiated in 50 cm3 flasks and expanded to 250 cm3 flasks on day 8. Growth medium was exchanged every 2–3 days. After media optimization, a further experimental design in the presence and absence of 25 Gy-irradiated autologous FCs, which were derived from splitting the original sample and performing a standard density-gradient centrifugation (Ficoll-Paque™ Premium; GE Healthcare), was performed for cultivation of resting NK cells (starting cell concentration: 2.5 × 106 NK cells/mL). For these experiments, NK cells were expanded up to 14 days in NK MACS, including 5% human serum type AB, 2% NK MACS supplement, and 1,000 IU/mL of IL-2 with or without supplementation of irradiated FCs at a ratio of 1:20 (NK cells:FCs). Human IL-21 (1 IU/mL; Miltenyi Biotec) was initially added to some FC batches. Different expansion approaches were performed as follows: 1. NK MACS medium containing 1,000 IU/mL of IL-2 and supplemented with 5% AB serum (control batch). 2. NK MACS medium containing autologous FCs (FCs:NK ratio: 20:1), 1,000 IU/mL of IL-2, and supplemented with 5% AB serum. 3. NK MACS medium containing autologous FCs (FCs:NK ratio: 20:1), 1,000 IU/mL of IL-2, 1 IU/mL of IL-21 (initial), and supplemented with 5% AB serum.
Cultivation was initiated in 250 cm3 flasks, and medium was changed every 2 days. On day 7 of expansion, the cells were split and cultivated in two 550 cm3 flasks until harvest.
Flow cytometric analysis
Cells from peripheral blood samples and NK cells were stained with monoclonal antibodies (mABs) for specific surface markers and subsequently analyzed on a Navios flow cytometer (Beckman Coulter) in a no-wash, single-platform procedure using Flow-Count™ fluorospheres (Beckman Coulter). Dead cells were excluded by 7-aminoactinomycin D (7-AAD) staining. The cell phenotypes were assessed with the following mABs: NKG2D (NK group 2 member D; CD314)-PE (Phycoerythrin), NKp30 (CD337)-PE, NKp44 (CD336)-PE, NKp46 (CD335)-PC7 (Phycoerythrin-Cyanin-7), CD69-ECD (Phycoerythrin-Texas Red®-x), CD137-APC (Allophycocyanin), CD178 (Fas ligand; FasL)-APC, CD253 (TNF-related apoptosis inducing ligand; TRAIL)-APC, CD3-PB (Pacific blue), CD19-FITC (Fluorescein Isothiocyanate), CD14-ECD, CD56-PC7, CD16-APC or -PB, CD107a-APC, CD137-APC, CD3-PB, and CD45-KO (Krome Orange). All antibodies were purchased from Beckman Coulter, except CD137-APC, CD178-APC, and CD253-APC (BD Biosciences).
Cytotoxicity assay
The cytotoxicity of NK cells was assessed against cell lines K562 for 4 h or against KG1a for up to 24 h at various effector-to-target ratios (E/T ratios 0.5:1, 1:1, and 5:1) of cell suspension co-cultures in TM containing 5% HSA. Co-cultivation of NK cells and primary AML cells was done at a 1:1 ratio. Detection of viable effector and target cells after cytotoxic interaction was based on a no-wash, single-platform flow cytometry (Navios; Beckman Coulter; FCM), including Flow-Count™ fluorospheres, and depended on surface expression of specific antigens such as CD16-APC or -PB, CD56-PC7 or -APC, and CD45-KO for NK cells, CD15-FITC for K562, and CD34-PC7 for KG1a. Dead cells were excluded by 7-AAD staining. All mABs were obtained from Beckman Coulter.
Lytic activity of the effector cells was calculated as the loss of viable target cells:
Cytotoxicity = [1 – concentration (co-cultured target cells/μL)/concentration(target control cells/μL)] × 100%
CD107a degranulation assay
In addition to cytotoxicity measurements, NK cells were co-incubated with K562 (E/T ratios of 0.5:1, 1:1, and 5:1) or KG1a (E/T ratio of 1:1) in the presence of CD107a (Phycoerythrin; PE) antibody (Beckman Coulter) for 1 h at 37°C and 5% CO2. Subsequently, Monensin and GolgiPlug (both 1:1,000; BD Biosciences) were added. After a total incubation time of 4 h, cells were washed in PBS, stained, and analyzed as described above.
CAR detection
In order to detect anti-CD123 CAR molecules at the NK cell surface, modified NK cells were incubated with the c-terminally polyhistidine-labeled antigen CD123 (final concentration 1 μg/mL; Sino Biological, Inc.) for 30 min at 4°C. After removing unbound CD123 antigen by a PBS washing step, transduced NK cells were stained with 7-AAD, CD45-KO, and anti-HIS APC (R&D Systems) and analyzed by FCM.
Blocking of anti-CD123 CAR
For blocking experiments, anti-CD123 CAR NK cells were pre-incubated with antigen CD123-polyHIS (1 μg/mL final concentration; 30 min at 4°C) following co-cultivation with KG1a or primary blasts at an E/T ratio of 1:1. Target cells were pre-incubated for 30 min at room temperature with anti-CD123 monoclonal antibody (Becton Dickinson). FCM analysis started after incubation of 0–24 h using 7-AAD, CD34-PC7, CD56-APC, CD16-PB, and CD45-KO (all monoclonal antibodies purchased from Beckman Coulter).
Cytokine analysis
Cytokine production of NK cells was assessed in supernatants by using the 13-plex flow assay kit LEGENDPLEX™ Human CD8 Panel (BioLegend®). Flow cytometric measurements of soluble cytokines and pro-apoptotic markers such as TNF-α, IFN-γ, perforin, granzyme A (GraA), granzyme B (GraB), and granulysin were done according to the manufacturer's instructions.
Time-lapse microscopy
Interactions between single cells of the acute myelogenous leukemia cell line KG1a and transduced primary human NK cells were monitored by fluorescence microscopy using IX81 microscope (Olympus). All transduced NK cells expressed EGFP and were additionally stained with monoclonal antibody CD56-PE (Miltenyi Biotec). KG1a cells were labeled intracellularly with the cell proliferation dye eFluor™ 450 (Affymetrix eBioscience™). NK cells were co-incubated with KG1a cells (E/T ratio of 1:1) on chamber slides at 37°C and 5% CO2 for 8 h after starting time-lapse recording.
Statistical analyses
Statistical analysis was performed using GraphPad Prism v6.05 (GraphPad Software, Inc.). Two-way analysis of variance was used to identify statistically significant differences between various cytotoxicity experiments or expansion results. This method assesses the significance of NK cell mediated killing activity and degranulation and NK cell expansion rates and evaluation of CD3+ cell contaminations, cytokine concentrations and secretions [fg/cell], transduction frequencies, and receptor surface expressions of effector and target cells. The same statistical test was performed to compare anti-CD123 CAR/EGFP- or EGFP-expressing NK cells, respectively. Differences were indicated as the median with range and were considered significant when p ≤ 0.01.
Results
Separation of primary NK cells using the fully automated Prodigy
The feasibility of a GMP-compliant single manufacturing procedure including two-step immunomagnetic separations for human primary NK cells using the Prodigy device was successfully demonstrated.
Initial materials contained 1.2 × 109 (range 0.94–3.0 × 109) viable white blood cells (WBCs), including 10.7% (range 8.5–18.1%) CD56+CD3− NK cells. Starting with a median of 0.38 × 109 (range 0.34–1.12 × 109) total CD3+ T cells in the LA/BC fractions, the immunomagnetic depletion of CD3+ cells led to a median T-cell decrease of log 3.5 (range 3.0–3.9), corresponding to a median frequency of 0.05% (range 0.01–0.06%) among total viable CD45+ cells detected in RB fraction. After CD56+ enrichment, the total T-cell numbers in final PF fractions decreased to a cell count median of 0.1 × 106 (range 0.05–0.2 × 106; Table 1).
Fully automated immunomagnetic bead selection of CD56+CD3− NK cells with the Prodigy system
Absolute numbers ( × 106) of viable CD45+ WBC, CD3+ T, and CD56+CD3− NK cells in leukapheresis or buffy coat (LA/BC) samples, reapplication bag (RB), and positive fractions (PF) derived from three independent manufacturing runs. The process performance was evaluated by the purity (%), recovery (%), and viability (%) of separated NK cells in target cell bag (PF) for three different clinical-scale runs, including CD3 depletion and CD56 enrichment with the Prodigy instrument. Cell viability was assessed by 7-AAD staining and FCM.
NK, natural killer; WBC, white blood cells.
Evaluation of PFs revealed a median yield of 0.07 × 109 (range 0.06–0.36 × 109) of total viable CD45+ WBCs and 70.5 × 106 (range 56.0–331.0 × 106) of total viable CD56+CD3− NK cells for all process runs. Correspondingly, the purity calculated as the percentage of CD56+CD3− NK cells of total viable CD45+ cells showed a median of 95.4% (range 93.1–97.5%), whereas the median recovery of CD56+CD3− NK cells in the final PF product compared to NK cells in LA/BC fractions was 60.4% (range 56.9–70.0%; Table 1). The immunoregulatory (CD56brightCD16dim&neg) and cytotoxic (CD56dimCD16pos) NK subsets in the final products ranged from 6.1% to 12.8% and 87.2% to 93.7% among total NK cells, respectively. Additional parameters comprising LA/BC, RB, and PF of the three Prodigy processes are compiled for individual comparison in Table 1. For quality control, a novel no-wash, single-platform 10-color FCM protocol was developed in order to enumerate CD56+CD3− NK cells and various CD45+ cell subsets in the differently produced fractions (LA, RB, NF, and PF) during the process manufacturing.
Medium optimization of in vitro expansion of human NK cells for cell therapeutic approaches
In order to improve the previously established X-VIVO 10–based expansion protocol from a previous clinical Phase I/II study, 7,24 a comparison of different GMP-compliant expansion media was carried out in small-scale laboratory-based experiments. Therefore, the enriched CD56+CD3− NK cells (median of purity 95.4%; range 93.1–97.5%; Table 1) from the Prodigy instrument were cultured manually for 14 days simultaneously at small-scale laboratory levels in different expansion media (X-VIVO, CG, TM, or NK MACS) supplemented with 5% AB serum and 1,000 IU/mL of IL-2 under cell culture conditions (37°C; 5% CO2). The density of those non-activated NK cells was adjusted to a median cell concentration of 2.1 × 106/mL (range 2.0–2.3 × 106/mL). Starting with a median of 1 × 106 (range 0.8–1.3 × 106) NK cells (Fig. 1A), the cell expansion in presence of NK MACS led to an absolute median cell number of 34.8 × 106 (range 23.6–41.9 × 106) viable NK cells on day 14 of cultivation (Fig. 1A), corresponding to a median NK cell fold expansion of 31.7 (range 24.2–45.9; Fig. 1B). Compared to the NK proliferation in the NK MACS medium, cell expansion rates in the other cell cultures containing X-VIVO, CG, or TM media were significantly lower (Fig. 1B) on day 14 of cultivation, with total median cell numbers of 4.9 × 106 (range 2.9–6.1 × 106 [X-VIVO]), 5.8 × 106 (range 3.2–21.4 × 106 [CG]), and 6.9 × 106 (range 2.4–18.0 × 106 [TM]; Fig. 1A). The well-known NK cell decrease in CD56+CD3− NK cells within the first 4 days of culture, which was published previously, 7,13 was observed for all tested media (Fig. 1A and B). Consequently, in all expansion batches, a median cell number loss of 0.43 × 106 (range 0.14–1.4 × 106) was determined, corresponding to negative expansion rates determined on the third day of expansion. Thereafter, NK cells started to proliferate in all media (Fig. 1A and B). Increased expression levels of surface NKp30, NKp44, NKp46, and NKG2D receptors were comparable on all expanded NK cells (day 14) cultured in different medium approaches (data not shown). Moreover, the medians of viability of expanded CD56+CD3− NK cells were similar after 14 days of cultivation in NK MACS and TM: median of viable NK cells in NK MACS = 93.3% (range 79.2–97.9%), and in TM = 95.0% (range 92.8–97.9%; Fig. 1C). Otherwise, the viability of NK cells cultured in X-VIVO and CG media for 14 days revealed lower medians: 86.0% (range 79.5–88.3% [X-VIVO]) and 88.5% (range 75.5–95.7% [CG]; Fig. 1C). Contaminating CD3+ T cells among total CD45+ cells in the final expansion products after 14 days ranged from 0.2% to 4.1% (median 2.6% [X-VIVO]), from 0.02% to 3.0% (median 0.3% [CG]), and from 0.1% to 1.6% (median 0.13% [TM]) compared to a lower T-cell contamination median of 0.01% (range 0.01–0.6%) in NK MACs–expanded NK cells (Fig. 1D). In order to compare the cytotoxic functionalities of those ex vivo activated NK cells derived from the different cultivation batches (X-VIVO, CG, TM, or NK MACS), FCM-based cytotoxicity assays against the reference target cell line K562 were performed after completion of expansion on day 14 and compared to killing rates of non-stimulated NK cells before starting cell expansions. Similar increases in the killing activities against K562 cells were determined at E/T ratios of 0.5:1, 1:1, and 5:1 for all differently media-cultured activated NK cells compared to significant lower killing rates of non-activated NK cells before expansion (Fig. 1E).
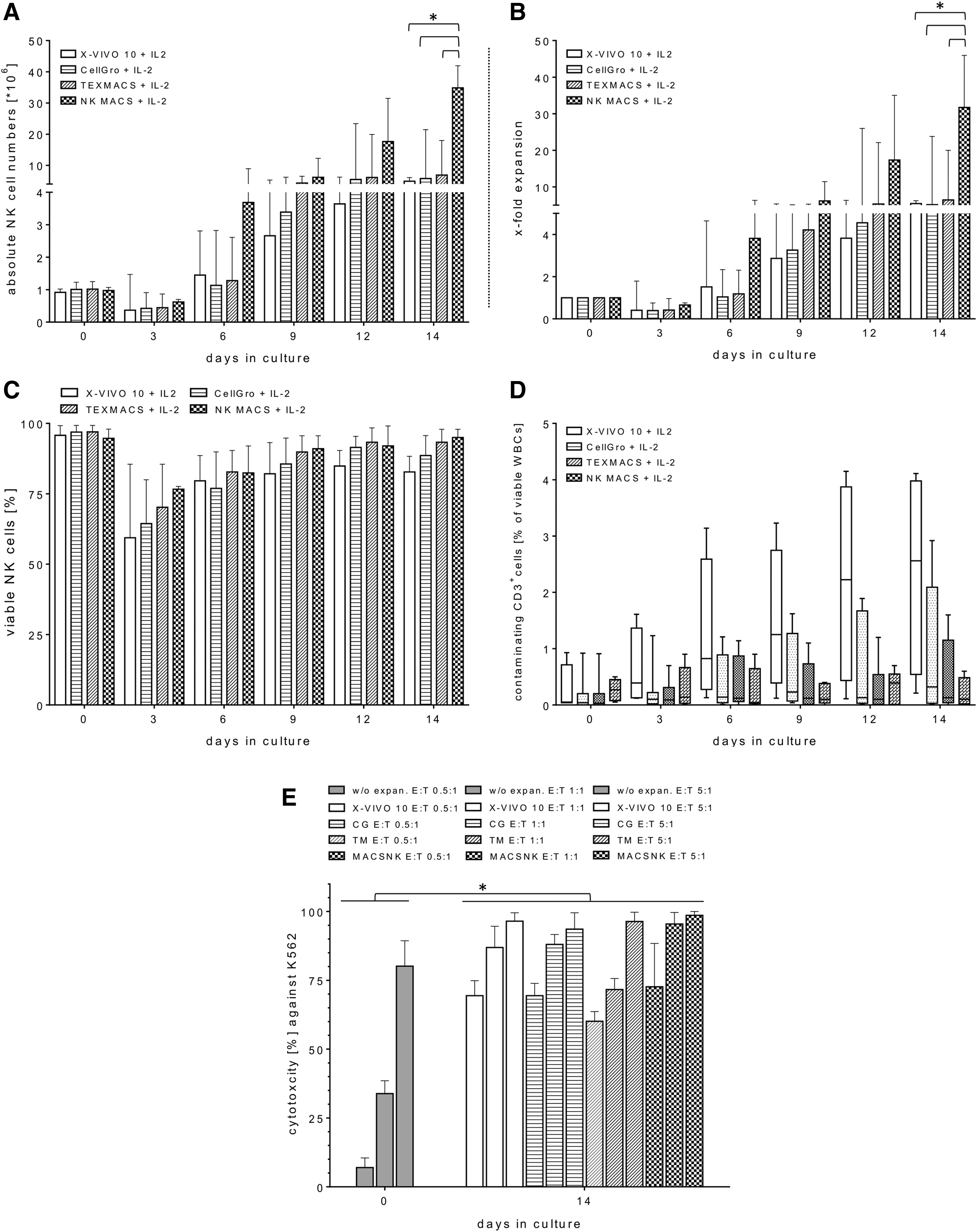
Expansion kinetics and effector functions of primary natural killer (NK) cells grown ex vivo in different media containing interleukin (IL)-2. Highly purified CD56+CD3− NK cells (range 93.1–97.5%) were adjusted to an approximate starting cell number of 1 × 106 NK cells and expanded for 2 weeks with different media (X-Vivo 10™ [X-VIVO], CellGro® [CG], TexMACS™ [TM] or NK MACS®, respectively), containing 5% AB serum and 1,000 IU/mL of IL-2 at 37°C and 5% CO2. Absolute NK cell numbers
Impact of IL-21 and irradiated autologous FCs on manual expansion of primary NK cells
A previous publication demonstrated that autologous PBMCs deployed as FCs were most effective for improved primary NK cell expansions after irradiation (25 Gy) and at a ratio of 1:20 (NK:FCs) with 500 IU/mL of IL-2. 28 Recently, Granzin et al. showed a beneficial synergism of FCs (irradiated EBV-LCLs), repeated IL-2 stimulations, and an initial administration of IL-21 to cell culture that resulted in a highly effective ex vivo expansion of human NK cells. 12 Therefore, the impact of IL-21 and autologous FCs was investigated in combination with NK MACS medium on the NK cell expansion profile. The primary NK cells from the Prodigy were expanded from a median starting cell number of 1.0 × 106 (range 0.9–1.1 × 106) in the presence and absence of irradiated autologous PBMCs, which were used as FCs at an absolute median cell number of approximately of 21 × 106 in different culture batches over a period of 14 days. In the presence of initial IL-21 (day 0) and irradiated FCs, the IL-2-driven NK cell expansion showed high median expansion rates of 84.8 (range 66.2–91.9), which resulted in an absolute median cell number of 81.4 × 106 (range 58.9–85.9 × 106) viable NK cells after 14 days (Fig. 2A and B). In comparison, lower expansion rates were seen with irradiated FCs in the absence of IL-21, with a median of 50.9 (range 38.5–58.4), while the control batch (w/o FCs/IL-21) showed a median of 31.7 (range 24.2–45.9; Fig. 2B). Similar to previous medium optimization experiments (Fig. 1A and B), a decreasing total median cell number of 0.6 × 106 (range 0.1–0.7 × 106) cells among all enriched CD56+CD3− NK cells could be observed in all three cultivation batches (day 3) within the early expansion time period (Fig. 2A). However, this cell decrease was overcome beginning on the fourth expansion day as a slightly rising median NK cell number of 0.8 × 106 (range 0.2–1.2 × 106). The viability of expanded CD56+CD3− NK cells was also comparable in the final expansion products from the different culture batches after 2 weeks (Fig. 2F). Interestingly, the proportion of immunoregulatory (CD56brightCD16dim&neg) and cytotoxic (CD56dimCD16pos) NK cell subsets was altered in all culture batches containing NK MACS medium. Thus, cultured NK cells had a median expression level of CD56brightCD16dim&neg and CD56dimCD16pos of 49.6% (range 42.2–51.5%) and 50.4% (range 48.6–57.2%), respectively, and showed no expression levels for CD56negCD16pos subsets after the 14 days.
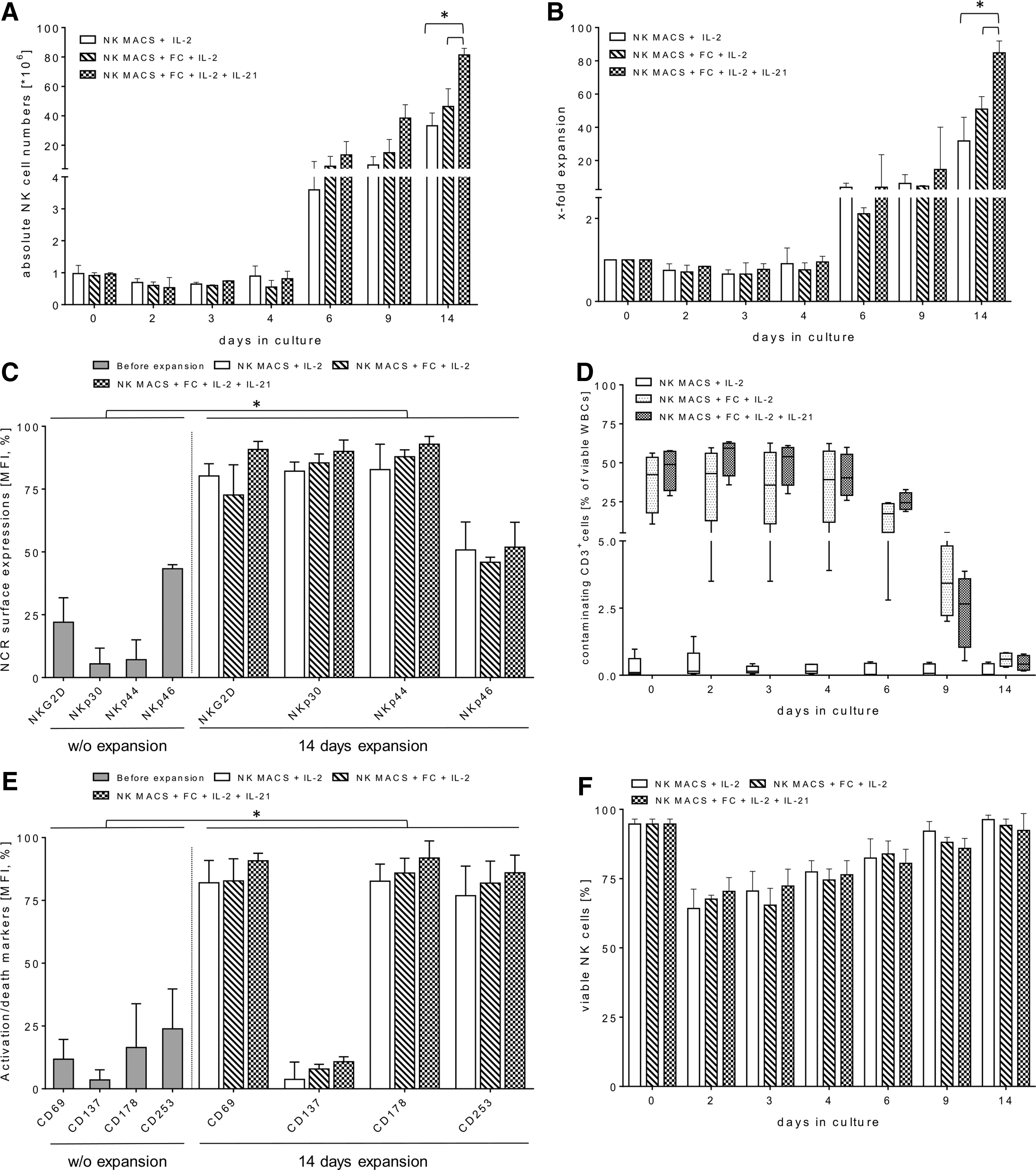
Combinations of irradiated autologous feeder cells (FCs), initial IL-21, and repeated IL-2 for long-term ex vivo expansions of highly purified NK cells. After immunomagnetic separation of primary NK cells via a one-step closed manufacturing process by the Prodigy device, effector cells were expanded in NK MACS medium containing 5% AB serum with an approximate starting NK cell number of 1.0 × 106 in the presence of 25 Gy-irradiated autologous FCs (NK:FCs ratio: 1:20), initial IL-21 (1 IU/mL), and repeated IL-2 (1,000 IU/mL) stimulation every 2–3 days over 14 days. FCs/IL-21/IL-2-expanded NK cells were monitored for absolute cell numbers
Residual FC leukocyte subpopulations were analyzed in the final products as a quantitative control of recovery in both cultivation approaches containing irradiated FCs over the entire expansion period. A significant decrease of irradiated FCs within 6 days of NK cell expansions was observed in both approaches. Contaminating CD3+ T cells ranged from 0.3% to 0.8% (FCs/IL-2) and 0.2% to 0.8% (FCs/IL-21/IL-2) among total CD45+ cells compared to the control batch without FCs (median 0.03%; range 0.01–0.5%; Fig. 2D). Moreover, both FC-incubated culture batches contained ≤1.5% residual CD56+CD3+ (NK-T) cells, CD14+ monocytes, and CD19+ B cells when gated on viable CD45+ cells (data not shown). Accordingly, the NK cells expanded via the different culture approaches all exhibited high purity (median 98.2%; range 98.0–99.2%). Phenotypic alterations of freshly separated versus 14-day-expanded NK cells were analyzed from the different culture batches by FCM. Freshly isolated CD56+CD3− NK cells expressed low-to-moderate levels of NKp30, NKp44, NKp46, and NKG2D in analogy to marginal surface levels of activations marker (CD69, CD137) and death receptors (CD178, CD253; Fig. 2C and E). NK cells from final products of all culture conditions had considerably higher amounts of surface NKG2D, NKp30, and NKp44 but in the highest expression levels were observed on FCs/IL-21/IL-2-expanded NK cells with a median expression of 90.8% (range 82.5–94.0% [NKG2D]), 89.9% (range 79.9–94.5% [NKp30]), and 92.8% (range 84.9–95.0% [NKp44]; Fig. 2C). Correspondingly, expression levels of CD69, CD137, CD178, and CD253 on FCs/IL-21-expanded NK cells ranged from 79.5% to 93.6% for CD69, 2.0% to 12.8% for CD137, 80.0% to 98.6% for CD178, and 72.9% to 93.0% for CD253 (Fig. 2E). However, NKp46 showed marginal gains in expression levels on activated NK cells from all batches compared to the resting NK cells before expansion (Fig. 2C).
Cytotoxicity, degranulation, and cytokine secretion of FCs/IL-21/IL-2-expanded NK cells
Killing rates of expanded NK cells derived from NK MACS culture batches in the presence and absence of FCs and/or IL-21 were analyzed in different E/T ratios (0.5:1, 1:1, and 5:1) against K562 target cells and compared to basal cytotoxicity of non-expanded NK cells. Increased lytic activity was most pronounced in the 0.5:1 E/T ratios. Moreover, in the 5:1 ratios, substantial increases in cytotoxic capacities were found in all ex vivo cultured NK cells from different expansion conditions. A maximum increase of killing rates were identified in activated NK cells expanded in the presence of FCs/IL-2 and initial IL-21, with median cytotoxicity levels of 84.6% (range 66.6–98.4% [E/T 0.5:1]), 95.5% (range 82.5–99.1% [E/T 1:1]), and 97.7% (range 89.4–99.8% [E/T 5:1]) compared to lower median killing activities by non-cultured NK cells of 4.5% (range 0.1–10.5% [E/T 0.5:1]), 37.1% (range 27.8–46.6% [E/T 1:1]), and 76.3% (range 60.5–81.7% [E/T 5:1]; Fig. 3A). Activated NK cells from different expansion approaches were co-cultured with K562 for 4 h (E/T: 0.5:1, 1:1, and 5:1) to detect surface lysosomal-associated membrane protein-1 (LAMP-1/CD107a) on NK cells as a marker for subsequent stimulation-induced granule exocytosis. In accordance to cytotoxic data, degranulation assays showed higher CD107a surface expression on activated NK cells derived from different culture batches compared to non-expanded NK cells with degranulation medians of 9.7% (range 6.8–16.8% [E/T 0.5:1]), 8.4% (range 7.8–17.8% [E/T 1:1]), and 4.9% (range 2.9–10.9% [E/T 5:1]; Fig. 3B). However, FCs/IL-21/IL-2-expanded NK cells showed highest CD107a-degranulation levels, which ranged from 28.8% to 38.8% (E/T 0.5:1), from 28.9% to 34.8% (E/T 1:1), and from 17.9% to 25.7% (E/T 5:1) after co-cultivation with K562 targets (Fig. 3B). Correspondingly, the analysis of pro-inflammatory cytokines (e.g., TNF-α and IFN-γ) in supernatants of cytotoxic experiments also showed enhanced secretion levels for all NK cell expansion batches after 14 days. The highest cytokine secretion levels were generated by FCs/IL-21/IL-2-expanded NK cells, which had median TNF-α release of 0.2 fg/cell (E/T 0.5:1), 0.3 fg/cell (E/T 1:1), and 0.03 fg/cell (E/T 0.5:1), and for IFN-γ of 0.9 fg/cell (E/T 0.5:1), 1.5 fg/cell (E/T 1:1), and 0.04 fg/cell (E/T 0.5:1; Fig. 3C). Similar increases were determined for apoptotic markers (granzyme A/B, granulysin) with maximum secretion levels released from FCs/IL-21/IL-2-expanded NK cells compared to expanded NK cells from the other culture approaches and to non-expanded NK cells (control batch). However, no significant alterations were detected for perforin release during cytotoxic reactions between NK cells and the cell line K562 (Fig. 3C). Interestingly, lower amounts of degranulation and cytokine secretion levels were detected from expanded NK cells at higher (5:1) compared to lower E/T ratios (0.5:1, 1:1). Similarly, lower degranulation and cytokine release levels were detected from non-activated NK cells (control batch) at higher E/T ratios (5:1; Fig. 3B and C).
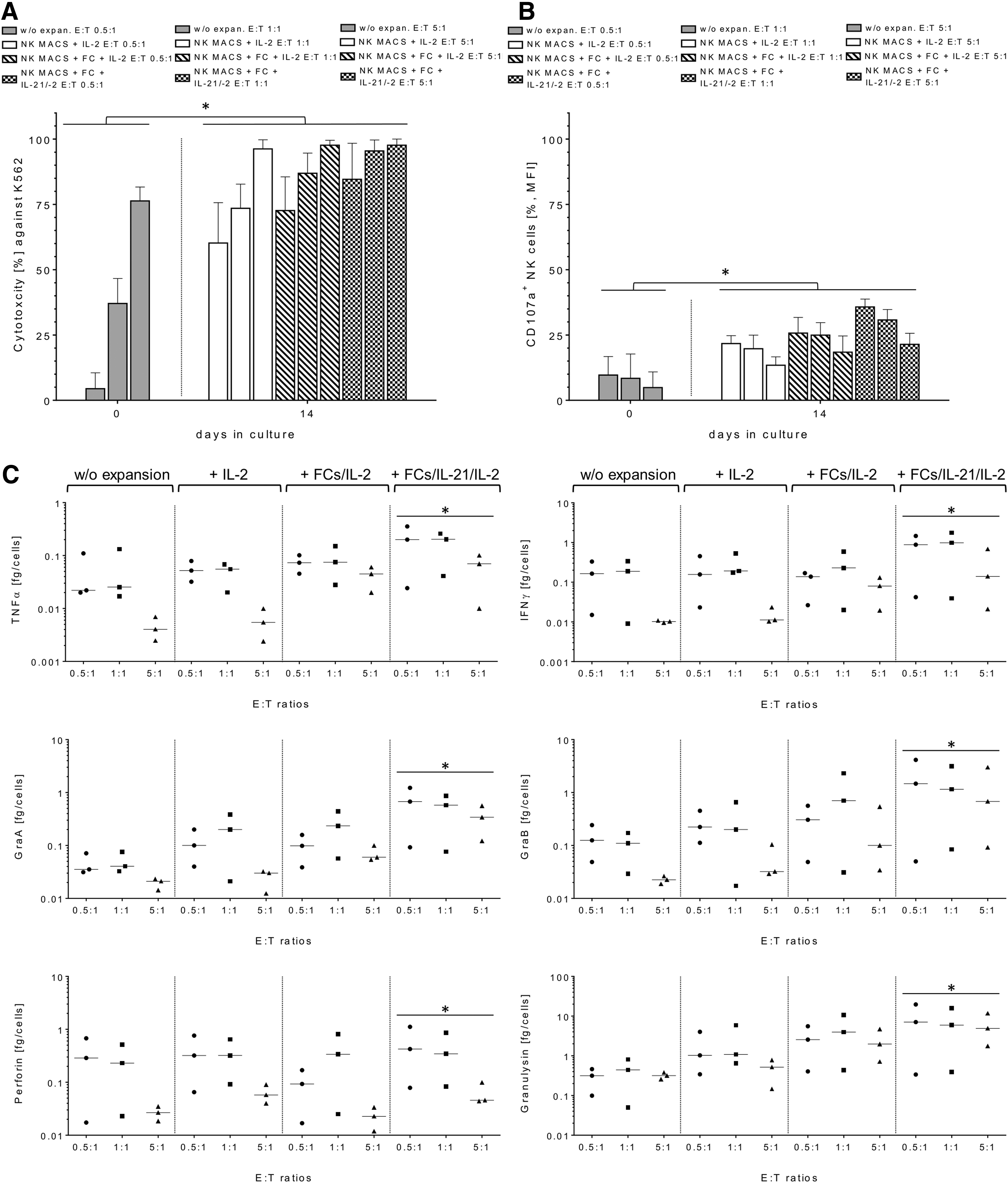
Comparison of cytotoxic functions of differently expanded NK cells after 2 weeks. NK cell-mediated cytotoxicity
Transduction efficiency and characterization of FCs/IL-21/IL-2-expanded NK cells
FCs/IL-21/IL-2-cultured NK cells were transduced with RD114/TR-pseudotyped alpha-retroviral vectors (a MOI of 1) encoding either the anti-CD123 CAR (CD123CAR/EGFP) or, as a control, only EGFP constructs (EGFP). Three days after gene-modification, CD123CAR/EGFP-engineered NK cells showed maximum surface CD123CAR levels of 22.9% (range 17.8–25.7%), but median expression decreased to 11.5% (range 5.9–18.8%) on day 9 post transduction (Fig. 4B). EGFP- and CD123CAR/EGFP-modified NK cells exhibited similarly high levels of cytotoxic NK cell receptors (data not shown), NK cell-mediated cytotoxicity at different ratios (0.5:1, 1:1, and 5:1; Fig. 4C), and CD107a-degranulation (Fig. 4D) as non-modified NK cells. Modified NK cells efficiently eliminated K562 cells, which do not express CD123 on their surface.
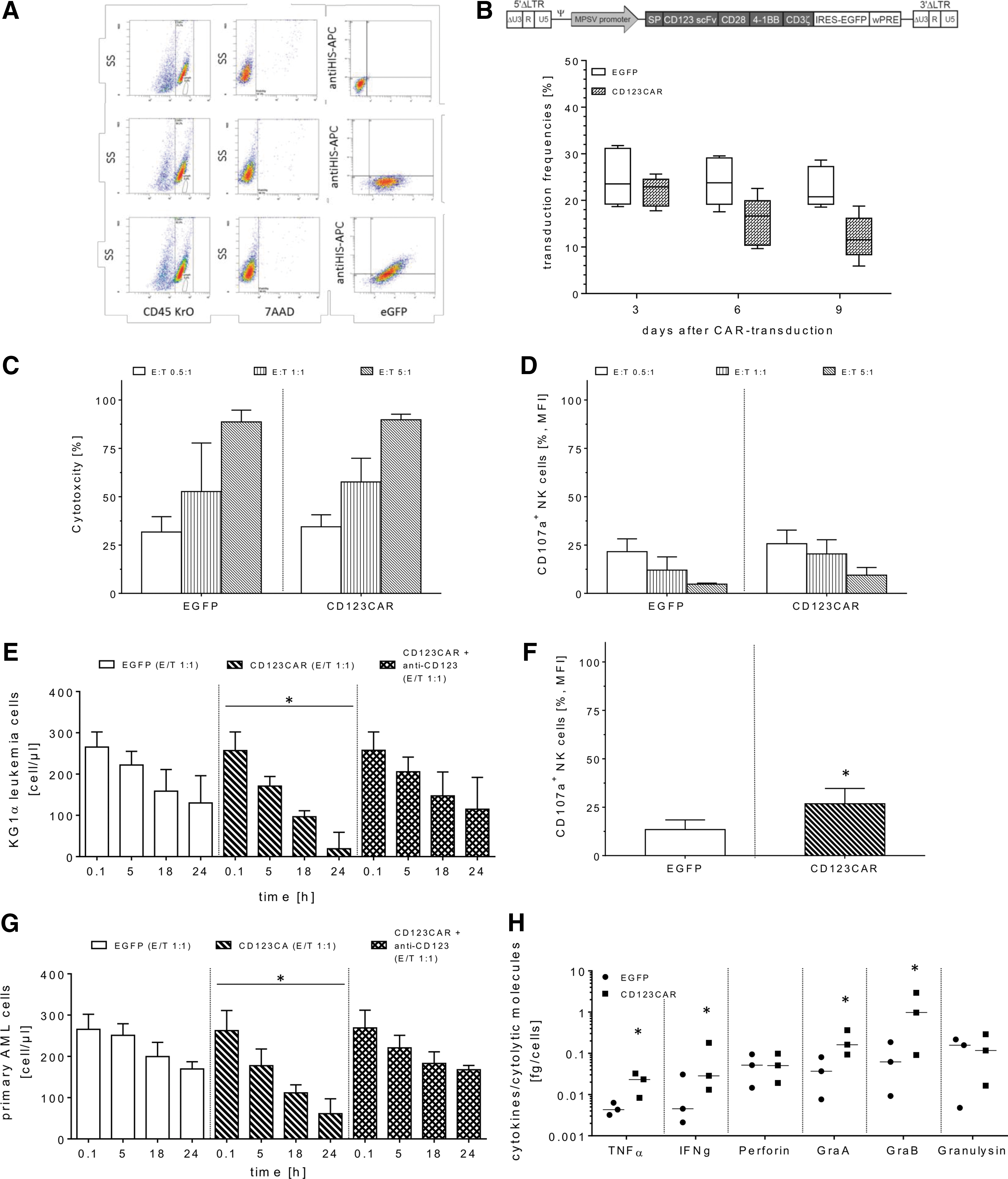
Analysis of transduction efficacy and cytotoxic function of FCs/IL-21/IL-2-expanded NK cells. After cell expansion, modification of FCs/IL-21/IL-2-cultured NK cells was accomplished with RD114/TR-pseudotyped alpha-retroviral vectors encoding either the CD123CAR/EGFP or EGFP w/o CAR constructs (EGFP). Detection of surface CD123CAR on expanded NK cells was determined with the recombinant CD123 peptide conjugated with C-terminal HIS-TAG and anti-HIS monoclonal antibodies (mABs) conjugated with APC
Retargeted cytotoxicity, degranulation, and cytokine secretion analyses with CD123CAR/EGFP transduced primary human NK cells
Transduced NK cells were co-incubated with the CD123+ KG1a leukemia cell line or three patient samples containing primary AML blasts (CD123+), respectively, at an E/T ratio of 1:1. These co-cultures were monitored for 24 h using the no-wash, single-platform FCM assay. Compared to EGFP-transduced NK cells (controls), CD123CAR/EGFP-modified NK cells exhibited improved CD123-specific killing activity, as determined by decreased target cell numbers of both KG1a cells and primary leukemia blasts (Fig. 4E and G). While the primary blasts cultivated without any effector cells showed only a slight increase in cell growth, KG1a cells expanded more than two- to threefold over the same time period of simultaneous cytotoxicity experiments (data not shown). In comparison, the CD123CAR/EGFP-modified NK cells elicited the greatest elimination of KG1a cells after 24 h with a median of 19 remaining KG1a cells/μL (range 1–59 cells/μL). In contrast, the more resistant AML blasts displayed a median of 61 remaining target cells/μL (range 44–97 cells/μL; Fig. 4E and G). In addition, the CD123CAR-specific cytotoxicity of transduced NK cells was efficiently blocked by pre-incubation (30 min at 4°C) of CD123CAR NK cells with recombinant poly-HIS peptides as well as pre-incubation of KG1a or primary AML blasts with monoclonal anti-CD123 antibodies (1 μg/mL, 30 min at RT; Fig. 4E and G). In support of retargeted eradication of KG1a cells, CD123CAR/EGFP-NK cells produced higher extracellular CD107α levels (median 26.8%; range 19.5–34.7%) and showed enhanced production of TNF-α and IFN-γ as well as granzyme A and B in response to KG1a cells compared to lower CD107a-degranulation (median 13.4%; range 7.0–18.4%) and cytokine/degranulation marker secretions of EGFP-expressing NK cells. However, no marked changes were found for the release of perforin or granulysin between CD123CAR/EGFP- and EGFP-engineered effector cells upon co-incubation with KG1a cells (Fig. 4H).
Time-lapse fluorescent microscopy and imaging were used to monitor specific CD123-retargeting of transduced NK cells in response to CD123+ KG1a cells. Therefore, 6 days after transduction, CD123CAR/EGFP- and EGFP-transduced NK cells were additionally labeled with anti-CD56 (PE), whereas target cells were intracellularly stained with cell proliferation dye (eFluor™ 450; see Methods). Subsequently, modified NK and KG1a cells (E/T ratio: 1:1) were co-cultured over 8 h under standard culture conditions. Transmission and fluorescent images were quantitatively evaluated by Olympus scanR acquisition analysis. A high amount of clustered single cells and increased specific E/T interactions were discovered in the presence of co-incubated CD123CAR/EGFP-NK cells (Fig. 5A) in contrast to low short-term-contact between EGFP-NK cells and KG1a cells (Fig. 5B). Moreover, retargeted KG1a elimination was detected as a consequence of target cell blebs that appear as multiple balloon-like, quasi-spherical membrane protrusions 29 in association of effector-mediated target cell killing tracked over a period of up to approximately 170 min, an example of which is shown in Fig. 5A. In comparison, non-specific cell–cell interactions mediated by EGFP-transduced NK cells did not induce apoptotic and/or necrotic blebs on KG1a cells resulting in a stable viability of the monitored target cells (Fig. 5B).
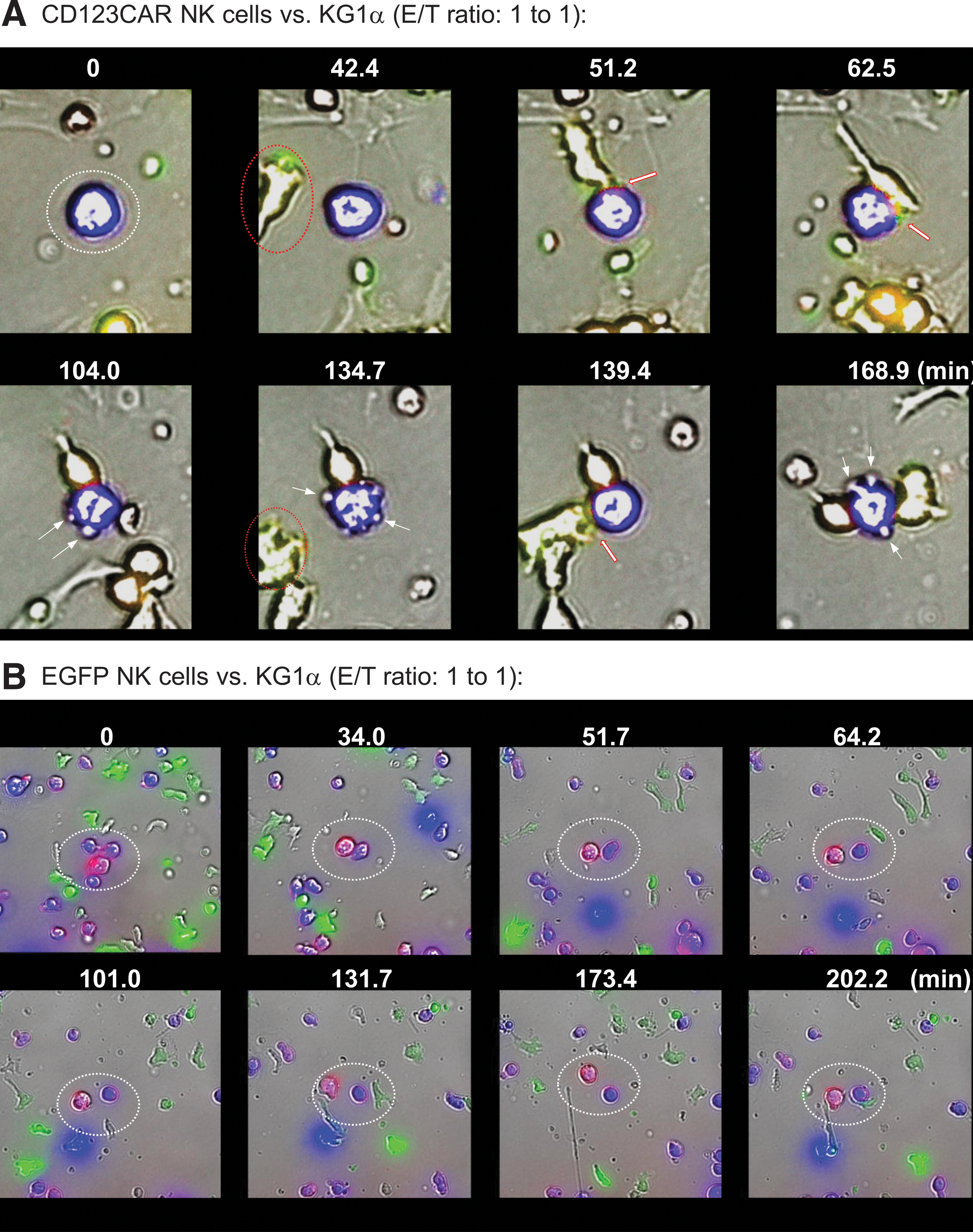
Comparative analyses of retargeted E/T cell interactions of gene-modified NK cells. KG1a were intracellularly stained with eFluor™ 450 (blue, target cells) and extracellularly labeled with anti-CD34 PE (red/pink), and co-cultured with CD123CAR/EGFP- or EGFP-engineered NK cells, respectively (green, effector cells) on chamber slides at a 1:1 E/T ratio for 8 h (37°C, 5% CO2). Exemplary results are shown for three independent experiments accomplished in duplicate. Specific E/T interactions were detected by scanR analysis, which allowed the time-limiting tracking of cell migration (0–168.9 min for CD123CAR/EGFP-NK cells
Discussion
The ability to separate primary donor NK cells derived from LA or BC fractions at a high clinical purification grade was demonstrated with a closed immunomagnetic manufacturing single process using the fully automated Prodigy device, which seemed to be safe and feasible. However, the subsequent expansion remains a great hurdle for NK cell-based immunotherapeutic applications because of the need of high effector cell numbers for multiple donor lymphocyte infusions (DLI), especially for adult high-risk patients. Similarly, overcoming potent TIEMs shown in several studies, which lead to a deactivation, apoptosis, and/or anergy of activated donor NK cells in the presence of an immunosuppressive tumor environment, remains a major challenge. Therefore, this study developed and analyzed a GMP-compliant strategy to generate high numbers of highly purified and activated donor NK cells for subsequent transduction using RD114/TR-pseudotyped alpha-retroviral vectors encoding anti-CD123 CAR constructs (CD123CAR).
Within the GMP-compliant development of this one-step manufacturing process for donor NK cells on Prodigy, the system for CD3 depletion and subsequent CD56 separation was automated. Here, an acceptable CD3+ depletion median rate of log 3.5 (range 3.0–3.9) and a high purity median of 95.4% in the end product was achieved, with regard to CD56+CD3− NK cells among all CD45+ cells containing an enhanced potential for cell proliferation and with a basal cytotoxicity against K562 target cells before expansion. Slightly above average target cell recovery (60.4%) and a high median viability of 96.3% for separated NK cells was achieved compared to previously described variable ranges for viability, purity, and recovery of primary NK cell products after two-step manufacturing processes using MACS systems. 7 –9,17,28,30,31
In order to optimize the current X-VIVO 10 and IL-2-based cultivation protocol from a closed clinical Phase I/II study, 7,24 a new GMP-compliant expansion protocol was developed using a culture medium specific for NK cells (NK MACS) containing 1,000 IU/mL of IL-2 in the presence of 25 Gy-irradiated autologous FCs (PBMNCs, 20:1, FC:NK ratio). It was shown that the use of irradiated autologous FCs and repeated IL-2 stimulation (every 2–3 days) in NK cell cultures strongly increased NK cell numbers due to a higher median expansion rate of approximately 51-fold. However, the presence of FCs did not prevent the well-known drop of cell growth of cultured NK cells within the early expansion period (1–4 days). This was also shown by others when irradiated autologous PBMCs were utilized as potent FCs to expand and activate freshly purified NK cells. 9,30,32 For example, Kim et al. demonstrated dramatically enhanced NK cell expansion levels after 14 days, which ranged from 169- to 300-fold using PBMCs as autologous or allogeneic FCs separated from corresponding healthy individuals or tumor patients, respectively. Therefore, resting NK cells were separated by an immunomagnetic two-step (CD3 depletion, CD56 enrichment) manufacturing process, whereas the negative fractions lacking NK cells were used as FC-PBMCs after 25 Gy irradiation. 9
In further steps, it was postulated that irradiated FCs derived from healthy individuals could efficiently induce autologous naïve NK cells in response to initial IL-21 and multiple doses of IL-2. This enhanced NK cell expansion may represent a useful strategy to overcome the hurdle of limited effector cell doses for adoptive NK cell-based therapies. Several publications describe that IL-21 is produced by activated CD4+ T cells and can affect resting NK cells. 33 Different cytokine compositions were used, including IL-2 or IL-15, respectively, to integrate IL-21 into some cultivation protocols for expansion and stimulation of NK cell activity. 34,35 Therefore, these specific cytokine compositions were analyzed for the NK cell culture batches that required NK MACS medium containing 25 Gy-irradiated FCs (20:1 FC-to-NK cell ratio). Highly purified NK cells from the immunomagnetic, one-step manufacturing process (Prodigy) were expanded with IL-21 and irradiated autologous FCs at the initiation of the ex vivo NK cell expansion followed by repeated (every 2–3 days) IL-2 (1,000 IU/mL) stimulation over 2 weeks. Under these culture conditions, the NK cell expansion rate was increased significantly in the range of 66- to 92-fold compared to NK cell expansion rates of 39- to 58-fold in the absence of initial IL-21 application. Similar results were also shown by Granzin et al., who used the irradiated clinical-grade Epstein–Barr virus-transformed lymphoblastoid cell line (EBV-LCL) as FCs for cultivation approaches. Over 1 week, this resulted in a 53-fold increase of NK cell expansion by IL-21 and IL-2 compared to 22-fold proliferation rate of NK cells in medium containing IL-2 alone and irradiated EBV-LCLs. In addition, these activated NK cells in the presence of initial IL-21 revealed increased TRAIL, NKG2D, and DNAM-1 surface expression levels, IFN-γ, and TNF-α secretions and an improved in vitro and in vivo cytotoxic response against several malignant cells (12). Compared to the cultivation experiments in the presence of initial IL-21, irradiated autologous FCs, and IL-2, the expanded NK cells exhibited significantly enhanced expression levels of surface NKG2D, NKp30, NKp44, CD69, CD137, CD178, and CD253 but not NKp46. In addition, other pro-inflammatory molecules that play an essential role in the induction of apoptosis followed by cancer cell killing, such as TNF-α, IFN-γ, and granzymes A and B, showed higher secretion levels after 14 days of expansion in relation to expanded NK cells activated with IL-2 alone and resting primary NK cells. Moreover, these alterations in phenotype and cytokine/apoptotic marker secretions in FCs/IL-21/IL-2-activated NK cells led to enhanced killing levels against K562 cells compared to naive NK cells, as previously described in several NK cell expansion studies. 1,28,31 However, the secretion levels of perforin did not change during cytotoxic reactions against K562, which is controversially discussed in the literature. On one hand, an increased perforin production including high expression levels of TNF-α and granzymes A, B, and K were detected after IL-2-expansion in the presence of EBV-LCL FCs for >1 week. 31 However, on the other hand, no changes of perforin levels combined with slightly increased intracellular expression of granzymes A and B were observed under similar culture conditions with a clinical-grade EBV-LCL FC line (TM-LCL, 100 Gy-irradiated, 20:1 FC-to-NK cell ratio) in X-VIVO 20 containing 500 IU/mL of IL-2 for a total of 28 expansion days. 1 This shows that the duration of cultivation is particularly important for cytotoxic and functional aspects of expanded effector cells in regard to production of respective surface receptors and degranulation markers, especially NKp46 and perforin. However, this might be in contrast to GMP-compliant protocols for large-scale ex vivo NK cell expansion based on long-term cultivation periods for multiple NK cell infusions. These differences could be overcome by combining IL-15 and IL-2 associated with autologous FCs. For this purpose, NK cells were immunomagnetically separated by a two-step manufacturing process (CD3 depletion, CD56 enrichment) and subsequently expanded with irradiated FCs at a 10:1 ratio (FCs:NK) in the presence of IL-2, IL-15, and anti-CD3 mAB (OKT3) for 19 days, which resulted in NK expansion rates of 117 ± 20-fold combined with cytotoxic potency against primary AML blasts in vitro and transplanted human AML in NOD/SCID mice in vivo. 30
As mentioned above, in addition to autologous FCs, allogeneic FCs are also suitable for proliferation and stimulation of NK cells. 36 Compared to allogeneic FCs, autologous PBMCs used as FCs revealed a lower potential and decreased potency to affect naïve NK cells efficiently. 9 Another reason to use allogeneic FCs is the availability of autologous FCs, which could be limited by clinical treatment of the patients. Therefore, established cell lines as allogeneic FCs might be a better source. FC lines, especially K562, RPMI 1866, Daudi, MM-170, HFWT, KL-1, and several EBV-LCLs, can be easily expanded to clinically required cell amounts to induce NK cell expansion. 18,37 –39 Granzin et al. clearly demonstrated that the cultivation of primary NK cells was feasible with the fully automated Prodigy device using EBV-LCLs and IL-2, which allowed NK cell expansion of 850- to 1,000-fold. 31
However, viewed from the regulatory guidelines, application of FC lines within a GMP-compliant expansion protocol revealed serious shortcomings because it must be clearly demonstrated that these lines are qualified as safe for application in patients. Accordingly, the characterization and subsequent validation of those cell lines as FCs, such as manipulated K562, includes costly viral testing followed by validated analyses for detection and exclusion of bacterial and mycoplasma contaminations. 40
The increasing importance of gene-modified NK cells, in particular CAR NK cells, involves the continuous optimization and improvement of the transduction and cultivation methods of those stable expanded effector cells to overcome TIEMs in a immunosuppressive tumor environment. Therefore, the transduction efficiency depends on various parameters such as the cell culture conditions, the pseudotype, the applied vector system, the critical time period for transduction during or after NK cell expansion, and transduction parameters as well as agents. 41 Using methods established in earlier studies that aimed to increase the transduction efficiency of primary donor NK cells after IL-2-cultivation over 2 weeks, 41 this study was able to transduce FCs/IL-21/IL-2-expanded NK cells successfully via alpha-retroviral vectors encoding anti-CD123 CAR (CD123CAR) or EGFP (EGFP) constructs, respectively, immediately after 14 days of expansion. It clearly demonstrated that gene transfer into activated NK cells is feasible, safe, and efficient, with transduction rates ranging from approximately 18% to 26% among all cultured NK cells. Importantly, gene modification had no negative influence on cytotoxic NK cell receptor expression, CD107a degranulation, or cytotoxicity against K562. However, the expression rates of the transgene seemed to decrease 9 days after transduction, which suggests that the turnover and degradation rate of these artificial CAR constructs on the effector cell surface may be high, as previously described for the activation of EGFP-CARs as a complex translocation process from the cytoplasm into the nucleus, followed by several stimulatory steps within the nucleus. 42 Nevertheless, in vitro experiments using KG1a cells or primary leukemia blasts as target cells showed enhanced secretion levels for CD107a, TNF-α, IFN-γ, and granzymes A and B and increased killing activities of NK cells modified with anti-CD123-CAR compared to EGFP CARs without anti-CD123 constructs and blocking control samples. While the anti-CD123-CAR utilized in the functional potency assays increased NK cell killing activity against CD123-expressing KG1a cells and native leukaemia blasts, different co-stimulatory domains should be compared to one another to develop the most efficient NK cell CAR design. For example, incorporation of sequences derived from the key accessory proteins DNAX activation protein 10 or 12, respectively, may improve CAR NK cell-redirected cancer cell elimination. 41,43 In accordance with the cytotoxic data, transmission/fluorescent microscopic imaging performed by Olympus scanR acquisition analysis showed significantly increased effector/target contacts in the presence of CD123CAR NK cells compared to time-limited and non-specific/confused cell cluster formations by EGFP-modified NK cells against CD123-expressing KG1a cells.
Regarding CD123-positive AMLs, in addition to CD33 antigen as recently described by Pizzitola et al., cytokine-induced killer cells (CIKs) modified with anti-CD123 CARs confirmed the high cytotoxic potency against several AML cell lines and native leukemia blasts in vitro. 22,44 High efficacy and cytolytic effects of T cells expressing CD123-specific CARs against human AML were also detected in vivo. 45,46 Recently, in the context of AML immunotherapies, clinical Phase I/II studies targeting CD123 by specific mABs and other immunotoxins (Clinical Trial.gov ID NCT 00397579 and NCT 004401739) revealed only moderate clinical benefit. Therefore, the development of novel therapeutic approaches with suitable vector systems and activated effector cells may generate future improvements. 47
Conclusion
The combination of alpha-retroviral vector technology for efficient NK cell transduction together with optimized NK cell expansion protocols containing suitable cytokine compositions and irradiated autologous FCs might be an essential advancement to clinical development of adoptive NK cell immunotherapies. The results demonstrate that activated NK cells transduced by CAR constructs, such as CD123CAR, might be an innovative strategy for efficient redirected elimination of resistant AML blasts. Gene-modified donor NK cells could be utilized to support current therapeutic protocols, especially combined with haploidentical stem-cell transplantation and other retargeted therapies.
Footnotes
Acknowledgments
This project was funded by grants of the Integrated Research and Treatment Center Transplantation (IFB-Tx, Ref. No. 01EO0802 and 01EO1302) and the German Federal Ministry of Education and Research (Collaborative Research Center 738, CRC738): “Optimierung konventioneller und innovativer Transplantate,” Cluster of Excellence REBIRTH (EXC 62/1) and the European Union (FP7 projects PERSIST, CELLPID, and Horizon 2020 project SCIDNET).
Author Disclosure
The authors declare no financial or commercial conflicts of interest, except that A.S. is co-inventor on a patent application describing alpha-retroviral SIN vectors.