
Abstract
Ovarian cancer represents the most lethal gynecological cancer. Although cytoreductive chemotherapy and surgery lead to complete macroscopic tumor removal, most of the patients in advanced stages suffer from recurrent disease and subsequently die. This may be explained by the activity of cancer stem cells (CSC), which are a subpopulation of cells with an elevated chemoresistance and an increased capacity for self-renewal and metastatic spread. Specifically targeting these cells by adoptive immunotherapy represents a promising strategy to reduce the risk for recurrent disease. This study selected the widely accepted CSC marker CD133 as a target for a chimeric antigen receptor (CAR)-based immunotherapeutic approach to treat ovarian cancer. A lentiviral vector was generated encoding a third-generation anti-CD133-CAR, and clinically used NK92 cells were transduced. These engineered natural killer (NK) cells showed specific killing against CD133-positive ovarian cancer cell lines and primary ovarian cancer cells cultured from sequential ascites harvests. Additionally, specific activation of these engineered NK cells was demonstrated via interferon-gamma secretion assays. To improve clinical efficacy of ovarian cancer treatment, the effect of the chemotherapeutic agent cisplatin was evaluated together with CAR-transduced NK cell treatment. It was demonstrated that NK cells remain cytotoxic and active under cisplatin treatment and, importantly, that sequential treatment with cisplatin followed by CAR-NK cells led to the strongest killing effect. The specific eradication of ovarian CSCs by anti-CD133-CAR expressing NK92 cells represents a promising strategy and, when confirmed in vivo, shall be the basis of future clinical studies with the aim to prevent recurrent disease.
Introduction
O
The concept of chemoresistant cancer stem cells (CSC) might provide a model to explain this observation. 3 –5 CSCs represent a subpopulation within the tumor and are equipped with a high tumorigenic potential and the ability to divide asymmetrically into distinct tumor subpopulations. 5 –8 These cells exhibit increased chemoresistance and clonal proliferation. 4,9 Several CSC-associated surface markers were identified after functional isolation from ovarian tumors. The most frequently reported markers are CD133, CD44, CD24, CD117, or the epithelial cell adhesion molecule (EpCAM). 5 –8 The observation that ovarian CSCs exhibit variable levels of surrogate marker penetrance, with some markers often only partially expressed, may result from the different methods used to isolate these cells. 5,6,8
One very promising CSC marker, CD133, was found on a variety of CSC entities, and the corresponding CD133-positive cancer cells exhibited enhanced tumorigenic potential and chemoresistance in preclinical studies. 6 Notably, elevated CD133 expression levels were detected in tumor samples of recurrent, platinum-resistant ovarian cancer. Furthermore, CD133-positive ovarian CSCs contributed to tumor evolution by enhancement of the metastatic potential of CD133-negative cells via paracrine stimulation. 10,11 Thus, the specific targeting and elimination of these cells represents a promising therapeutic approach in cancers harboring CD133-positive CSCs. 7,12,13
Chimeric antigen receptors (CAR) are a powerful strategy in current cancer research and clinical studies, as CAR-modified cells make antigen-specific cellular immunotherapy possible. CARs are fusion proteins that consist of a single-chain variable fragment (scFv) from an antibody and the intracellular signaling domain of a T cell receptor. 14,15 Genetic modification of immune effector cells such as T or natural killer (NK) cells with CAR expression cassettes endowed these cells with specific cytotoxic activity against the selected target cells. CAR-engineered T or NK cells showed specific cytotoxic effects against several tumors in vitro and in vivo, further underlining the promising clinical perspective of this treatment strategy. 16 –18
This study used NK92 cells, a clinically applied human NK cell line, engineered with a completely codon optimized anti-CD133-CAR to target CD133-positive cells specifically in ovarian cancer cell lines and primary cells. Furthermore, increased elimination of ovarian cancer cells was demonstrated upon combination of cisplatin chemotherapy with CAR-NK92 immunotherapy.
Methods
Cell lines
Lentiviral vector production was performed in HEK-293T (ATCC CRL-3216) cells. Human cell lines used in cytotoxic assays include the established human ovarian cancer cell lines SKOV3, A2780, Ovcar3, and the human NK cell line NK92. 19 Primary ovarian cancer cells were cultured from sequential ascites harvests using a protocol described by Shepherd et al. 20 and following informed consent in accordance to the Declaration of Helsinki and internal ethic committee approval. Primary cells were primarily cultured in low-attachment flasks (Corning, Wiesbaden, Germany). For cytotoxic assays, primary cells were cultured in standard-attachment flasks (Sarstedt, Nümbrecht, Germany). Ovarian cancer cell lines (SKOV3, A2780, and Ovcar3) cells were modified to express green fluorescent protein (GFP) for later analyses. All cell lines were maintained in RPMI-1640 medium (PAN Biotech, Aidenbach, Germany), except HEK-293T (Dulbecco's modified Eagle's medium; Biochrom, Berlin, Germany), supplemented with 10% heat inactivated fetal bovine serum, 100 IU/mL of penicillin, 100 mg/mL of streptomycin sulfate, and 10 mmol/L of HEPES. The CD133/1 (AC133) antibody conjugated to APC was purchased from Miltenyi Biotec (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). The Human IFN-gamma DuoSet enzyme-linked immunosorbent assay (ELISA; R&D Systems, Minneapolis, MN) was used to measure interferon (IFN)-γ concentrations in the supernatants, according to the manufacturer's instructions.
Cloning of vectors and virus production
The sequence of the CD133 scFv was adopted from a novel monoclonal antibody recently described by Swaminathan et al. 13 To increase the efficiency of transcriptional processing and protein expression, this sequence and all CAR sequences were codon optimized, the GC content was improved, and cryptic splice sites were removed before DNA synthesis (GeneArt; Thermo Fisher, Regensburg, Germany). CAR constructs were generated within a lentiviral vector backbone and adapted from a third-generation anti-CD19-CAR. 21 The protein sequence of this CAR was kindly provided by Richard Morgan (bluebird bio). The anti-CD19 CAR consisted of an anti-CD19 scFv derived from a mouse hybridoma FMC63 and expression units for CD28, CD137 (4-1BB), and CD3ζ. 22 The scFv fragment was inserted between AgeI and NotI sites. Thereafter, an internal ribosomal entry site–driven dTomato expression cassette was inserted between SalI sites.
293T cells (5 × 106) were seeded in 10 cm dishes and cultured overnight. The following plasmids for each dish were mixed, diluted in water with the desired volume, and transfected using the calcium phosphate method in the presence of chloroquine: 12 μg of the vector plasmid, 12 μg of pcDNA3.GP.4×CTE (gag/pol), 5 μg of pRSV-Rev, and 2 μg of RD114/TR envelope plasmids. 23 The lentiviral packaging plasmids were produced and purified by PlasmidFactory (Bielefeld, Germany). Lentiviral supernatants were harvested 36 h after transfection, filtered through Millex-GP 0.22 μm filters (Millipore, Schwalbach, Germany), concentrated via ultracentrifugation, and stored at −80°C until use.
Transduction of NK cells
NK92 cells were transduced using Retronectin to assist co-localization of NK92 and lentiviral vectors as well as transduction. In brief, 48-well plates were coated with Retronectin (Takara, Shiga, Japan; 210 μL of 24 mg/mL in phosphate-buffered saline [PBS] per well) overnight at 4°C or 2 h at room temperature. The wells were blocked with sterile-filtered PBS containing 2% bovine serum albumin for 30 min at room temperature. After washing with HBSS/HEPES, viral vector supernatants were added to the Retronectin-precoated plates and centrifuged for 30 min at 400 g and 4°C. Afterwards, 5 × 104 NK92 cells were added and incubated for 24 h. Determination of transduction efficiency was performed 72 h after transduction.
Cytotoxicity assays
Fluoroskan Ascent™ FL
Ovarian cancer cells were seeded in flat bottom 96-well plates (Sarstedt) at appropriate densities (A2780, 2 × 104 cells/well; SKOV3 and Ovcar3 1.5 × 104 cells/well). The next day, NK92 and CAR-NK92 cells were added at the designated effector/target (E/T) ratios. Before measuring, culture medium was completely removed. A total of 200 μL of 5% (w/v) SDS was added into each well. Then the fluorescence intensities of GFP in the cell homogenate, which corresponds to the cell numbers, was measured at excitation 485 nm/emission 520 nm using the microplate fluorometer Fluoroskan Ascent™ FL (Thermo Fisher Scientific, Waltham, MA).
xCELLigence
The xCELLigence system—a real-time, label-free monitoring device to determine cell health and behavior—was used for ovarian cancer cell index (CI) determination in the presence of NK cells via impedance measurements according to the manufacturer's instructions (Hoffmann La-Roche, Basel, Switzerland). Before seeding the target cells, 100 μL of cell culture media was added to each of the 96 wells in the E-Plate 96 (ACEA Biosciences, San Diego, CA). The plates were left in the cell culture hood for 30 min at room temperature to equilibrate the media and E-Plate surface. In step 1, the background impedance of cell culture media was measured. These data were then used as the reference impedance to calculate the confidence interval (CI). After step 1, the E-Plate 96 was removed from the incubator, and the desired cells were added in 50 μL of medium. The cell suspension was prepared for the appropriate cell concentrations previously determined by titration (A2780: 2 × 104; Ovcar3: 1.5 × 104; SKOV3 and primary ovarian cancer cells: 1 × 104). The plate was left in the culture hood for 30 min to allow the cells to settle to the bottom of the wells. Then the E-Plate 96 was reinserted. The next day, when the cells reached the logarithmic growth phase, effector NK cells were added in 50 μL of medium at the desired effector-to-target (E/T) ratio. The experiments were manually stopped 48–72 h after addition of effector cells.
Chemotoxicity assays
A2780 (ovarian cancer) or primary ovarian cancer cells were seeded in 48-well plates in the previously determined cell number. The designated treatment was performed by adding the desired amount of cisplatin (Sigma–Aldrich, St. Louis, MO), representing the previously determined IC50 concentration and NK cells (E/T 2:1) in 200 μL of medium. Total incubation time was 96 h. NK treatment was performed for 24 h, and cisplatin treatment for 72 h. In the sequential treatment groups, cells were washed twice before changing conditions. Controls were analyzed at each step to reduce systematic bias. After treatment, all wells were washed twice and analyzed with the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, Fitchburg, WI) following the manufacturer's protocol.
Statistical analysis
Data from the experiments are expressed as means ± standard deviations. Student's t-test was used for comparison of differences between the indicated groups. A p-value <0.05 was considered significant.
Results
A new codon-optimized anti-CD133-CAR specifically kills CD133-positive ovarian cancer cells
After codon optimization for favored human codon usage and removal of cryptic splice sites, the newly designed anti-CD133-CAR (see modular architecture and CD19 control vector in Fig. 1) was efficiently transduced into human NK92 cells, which have been applied in clinical trials. 4,19,25 First, ovarian cancer cell lines A2780 and Ovcar3 were modified to express GFP and then analyzed in a Fluoroskan based killing assay. A2780 served as negative controls, since these cells were negative for CD133 expression by flow cytometry (Fig. 1A). As a positive control for cell lysis, 1% Triton was used. As shown in Fig. 1B, C, no evident killing effect was observed in the CD133-negative A2780 (p = 0.90) or in the slightly CD133-positive Ovcar3 (p = 0.16) cells using the Fluoroskan assay. Due to the very low expression level of CD133 in Ovcar3, a more sensitive flow cytometric–based assay was used to determine the CD133-positive cells after NK cell co-incubation. With this assay, a significant decrease in CD133-positive cells was observed upon co-incubation with CAR-NK92 cells in comparison to control NK92 cells (p < 0.01; Fig. 1D). Interestingly, an unspecific reduction of CD133-positive cells by untransduced NK92 cells was also observed, possibly due to the innate capacity of NK92 cells to kill cancer cells.

A new codon optimized anti-CD133 chimeric antigen receptor (CAR) specifically kills CD133-positive ovarian cancer cells.
Engineered NK92 cells specifically kill CD133-positive primary ovarian cancer cells
Primary ovarian cancer cells, which were cultured from three sequential ascites harvests from one patient (P1 before chemotherapy and P2 and P3 during chemotherapy), were analyzed in an xCELLigence device–based killing assay (Fig. 2A). This assay allows for an indirect analysis of the cell density and the cytotoxic effect by real-time impedance monitoring. All primary cell samples (P1, P2, and P3) showed different percentages of CD133-expressing cells, that is, 4.2% positive cells in P1, 42.9% in P2, and 78.4% in P3 (Fig. 2B). Co-incubation with CD133-specific CAR-NK92 cells caused a significant (p < 0.05) reduction in the cell index (CI) values, which indicates a reduction of the cell number that was also confirmed by microscopy. Microscopic images before and after killing of primary ovarian cancer cells by engineered NK cells are shown in Fig. 3A. There was a dose-dependent killing effect in P2 and P3 cells, which expressed the highest levels of CD133. Noteworthy, primary cells presented different susceptibilities to unspecific NK cells. For example, P3 cells showed a dose-dependent unspecific killing effect by control NK cells, whereas P2 cells were not affected. In CD133 very low expressing P1 cells (4.2%), no significant killing effect was observed with this assay.
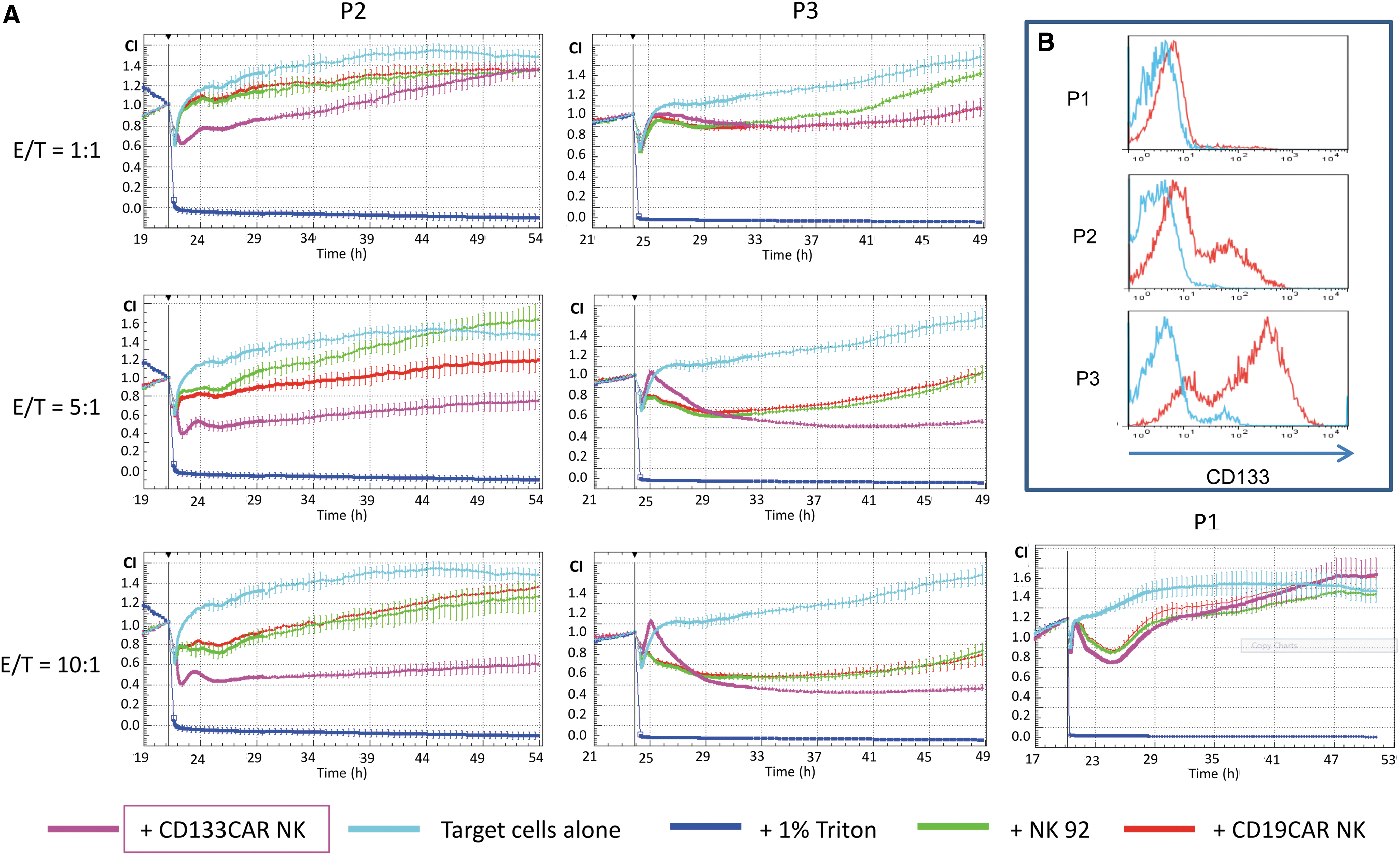
Engineered NK92 cells specifically kill CD133-positive primary ovarian cancer cells.
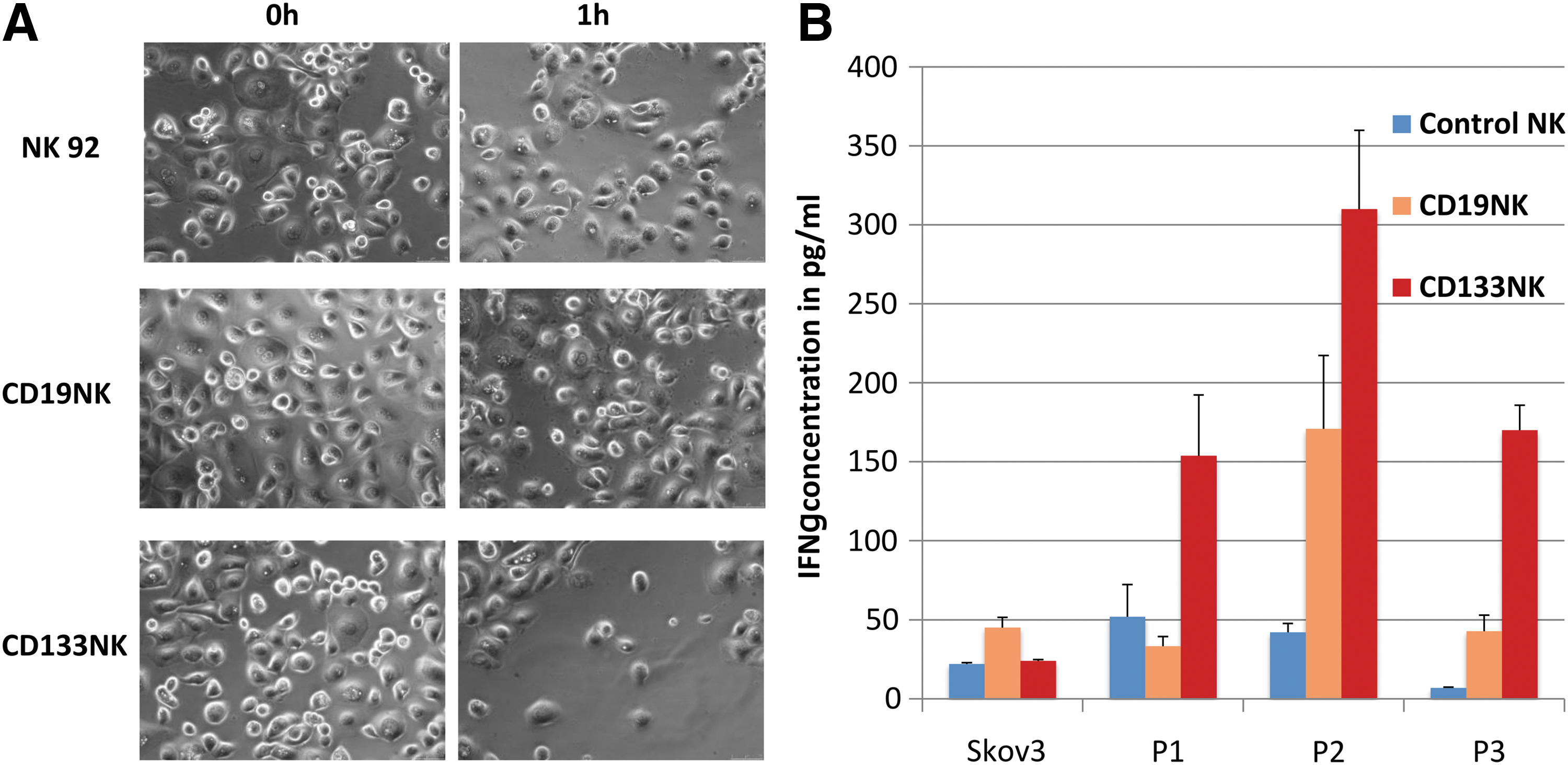
Engineered NK92 cells were activated upon co-incubation with CD133-expressing cancer cells.
Engineered NK cells were activated upon co-incubation with CD133-expressing cancer cells
To analyze the activation status of the NK92 cells, an ELISA was performed to quantify IFN-γ levels in the supernatant of the co-incubated cells (Fig. 3B). Of note, IFN-γ represents a typical cytokine that is secreted by NK cells upon activation. This assay clearly supported the previous results and showed strong activation of the CD133-CAR-expressing NK92 cells compared to untransduced and CD19-CAR control NK92 cells. Additionally, an unspecific elevated IFN-γ secretion by CD19-specific NK92 cells was observed when co-incubated with P2 or P3 cells. Interestingly, a significantly increased IFN-γ secretion was also observed compared to control cells after co-incubation of anti-CD133 NK92 cells with CD133 low expressing P1 cells (p < 0.05). SKOV3 served as CD133-negative control cells.
Cisplatin treatment enhances NK92 cell-mediated killing
Platinum-based chemotherapy in ovarian cancer is very effective in massively reducing the tumor load. However, CSCs, especially CD133-positive CSCs, exhibit increased chemoresistance and may survive cytoreductive therapy and subsequently lead to recurrent disease. To try to improve tumor control, the effects of combinatorial treatment strategies of cisplatin and CAR-expressing NK92 cells were explored. The previously determined IC50 dose of cisplatin was used for each specific cell line, and CAR-NK92 cells were added at an E/T ratio of 2:1. This low-dose treatment was used with the aim to better visualize possible additive effects. Co-incubation with NK92 cells was performed over 24 h, and incubation with cisplatin followed for 72 h. Figure 4A and B show the results of the different sequential treatment strategies tested. Changes in viability were calculated by dividing the results obtained for CD133-specific NK92 cells by the results of unspecific NK92 cells in the designated conditions. Interestingly, as shown in Fig. 4B, NK92 cell-mediated killing is still detectable under cisplatin treatment. Additive effects of cisplatin and NK92 cell-mediated toxicity were only observed in experimental conditions using CD133-specific NK92 cells to treat CD133-positive primary ovarian cancer cells (p = 0.03). Notably, this effect was not observed in CD133-negative A2780 cells (p = 0.82).
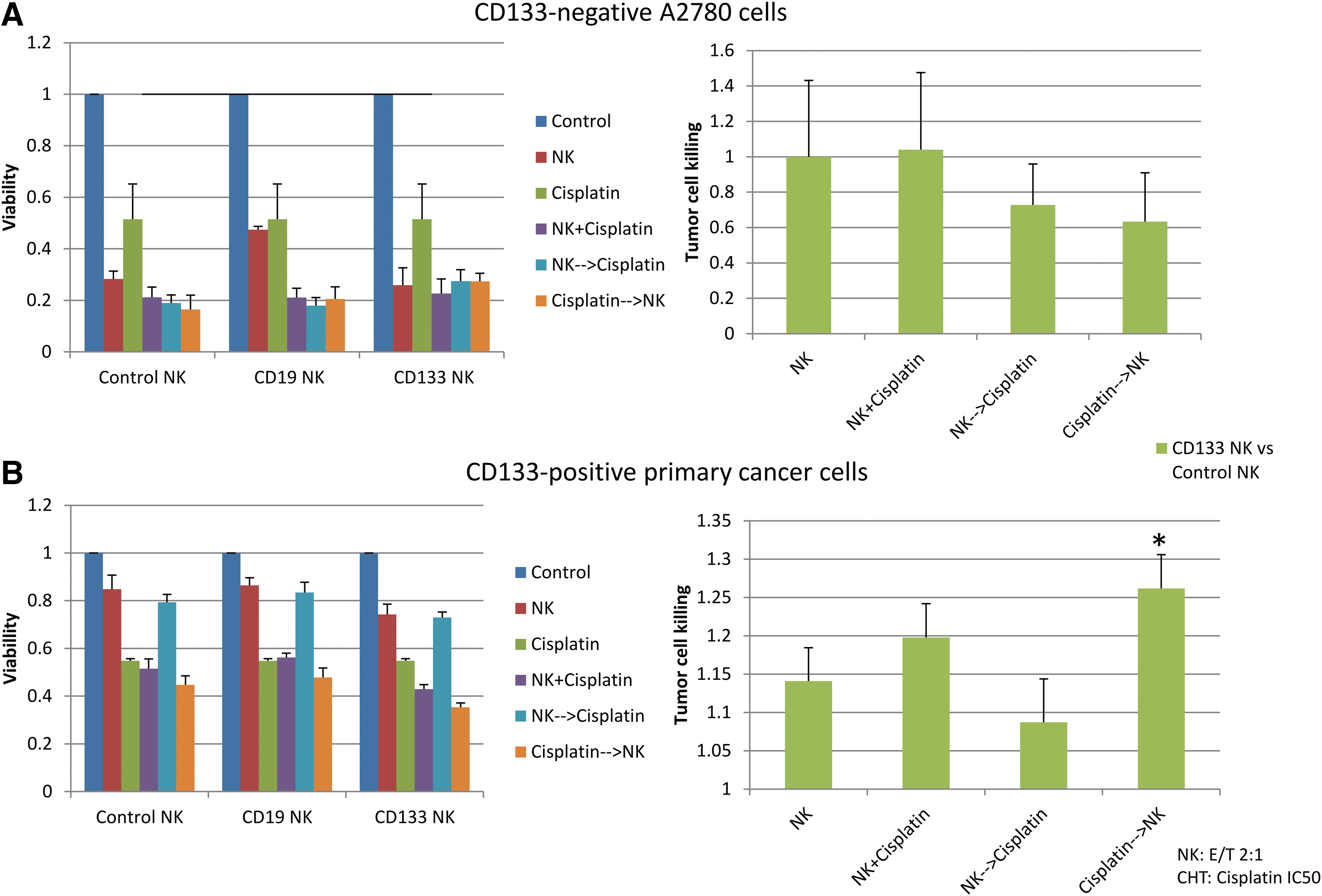
Cisplatin treatment enhances NK92 cell-mediated killing. Results of an MTS assay of surviving ovarian cancer cells under the indicated conditions. Co-incubation was performed for 4 days (cisplatin treatment for 72 h, NK92 treatment 24 h) with the previously determined IC50 dose of cisplatin and an E/T ratio of 2:1. Tumor cell loss was calculated by dividing the results of CD133-specific NK92 cells by the results of unspecific NK92 cells for the designated conditions.
In addition, to further confirm these results and to rule out possible unspecific effects, the relative killing effect of the CD133-specific NK92 cells was evaluated in relation to the unspecific NK92 cells in the designated conditions. The simultaneous treatment with cisplatin and specific NK92 cells resulted in a tendency for increased killing of CD133-positive primary cells, which was not significant (p = 0.46). However, using a sequential treatment of cisplatin followed by NK92 cells resulted in a significantly improved killing effect (p = 0.03) of the CD133-specific NK92 cells compared to treatment with control NK92 cells. Again, this effect was not detected in CD133-negative A2780 cells (p = 0.23).
Discussion
To the authors' knowledge, this is the first report that analyzes a CSC-specific CAR against ovarian cancer cells and importantly combines this novel CAR-based immunotherapy with platin-based chemotherapy. For this, a novel CAR directed against the promising CSC-antigen CD133 was generated. It was shown that NK92 cells engineered with this CAR were activated upon CD133 antigen presentation and mediated specific tumor cell killing. First, the combinatorial treatment of engineered NK92 cells and cisplatin on ovarian cancer cell lines and primary ovarian cancer cells was analyzed, and it was possible to show an additional killing effect after sequential treatment with engineered NK92 cells after cisplatin.
Although medical technology has advanced considerably, chemotherapy and surgery have only moderately extended the survival expectancy for ovarian cancer patients in the last decades. 26 Recurrent disease after cytoreductive surgery and chemotherapy represents the main drawback in current ovarian cancer therapy. 1,2 The model of CSCs may represent a reasonable explanation for recurrent disease, since these cells are characterized by increased chemoresistance and tumorigenicity. 4,9 The exact surface markers to characterize ovarian CSCs still remain controversially discussed, but CD133, CD44, and CD24 are most widely described as ovarian CSC markers in current studies. 5,6 Immunotherapies that directly target these CSCs might represent a promising approach against recurrent disease. In this regard, CAR-based cell therapies belong to the most promising strategies. The first Phase I study tested an anti-FRα-CAR in ovarian cancer patients. 27
In contrast to a recently published CD133 (AC133)-specific CAR that recognized glycosylated epitopes, the CD133 scFv sequence adopted in this project recognizes a more common non-glycosylated epitope of CD133. 12,13,28 Based on this anti-CD133 scFv sequence, antibody-toxin conjugates (dCD133KDEL) and CD133-targeted nanoparticles loaded with paclitaxel were developed and successfully tested on breast carcinoma cells, ovarian cancer, or head and neck cancer in preclinical studies. 7,29,30
In addition to CSC properties, CD133-positive ovarian cancer cells release several metastasis-related chemokines and cytokines, 11 which were shown to influence epithelial-to-mesenchymal transition (EMT) of human carcinoma cells. 10,31 On account of this, targeting CD133-positive ovarian cancer cells by anti-CD133 NK cells might reduce tumor progression by direct cytotoxic effects, as well as indirect effects on EMT and paracrine stimulation.
In contrast to most published approaches, a decision was made to use CAR-engineered NK-cells instead of T cells. 16 –18,24,32 The feasibility of CAR-engineered NK cells as effector cells was demonstrated. 33 However, the efficacy and clinical expansion of primary NK cells remain variable due to individual differences of patients or donors. A human NK cell line is a promising alternative to overcome this problem. NK92 is the most commonly studied NK cell line, and has been used in clinical trials. 19,24,25 Compared to patient-derived NK cells that might be functionally impaired, NK92 provide a more homogeneous and well-defined cell population. NK92 cells have been shown to be generally safe, with only moderate and transient toxicities. 24,25 Importantly, NK92 cells express a relatively large number of activating receptors and high levels of cytolytic pathway molecules, such as perforin and granzyme B. 34 Additionally, they express only few inhibitory receptors, and they lack most of the killer inhibitory receptors that are expressed on normal NK cells. 34 This confers NK92 cells with a superior cytotoxicity against a broad spectrum of tumor targets. Furthermore, CAR-expressing NK92 cells can be provided in a standardized quality and can be used for many different patients, increasing the practicality of these cells in clinical trials. However, similar to other established NK cell lines, NK92 cells do not express CD16. Thus, they require transfection of CD16 in order to perform antibody dependent cellular cytotoxicity. Also, NK92 cells were established from a patient with lymphoma, and they were shown to carry multiple cytogenetic abnormalities. 35 However, non-irradiated NK92 cells did not engraft after repeated infusions and administration of interleukin-2 in mouse models, 35,36 and Phase I/II trials did not reveal oncogenic effects, which suggests safe application in future studies. 36
In the current experiments, the cytotoxic activity of NK92 cells engineered with a newly designed anti-CD133-CAR on ovarian cancer cell lines and primary ovarian cancer cells is demonstrated. Especially on CD133 high expressing primary ascites cells (P2, P3), cytotoxicity was easily measured with the xCELLigence device. Although no cytotoxic effect was directly detectable on CD133 low expressing cancer cells (P1) with respect to the total cell count, NK92 activation as measured by IFN-γ secretion showed specific activation of engineered CAR cells upon antigen presentation. The main reason for the lack of significant cell number reduction after NK92 treatment might be caused by the very low proportion of CD133-positive cells. However, the possibility to eradicate this small cell population might be clinically meaningful in combination with chemotherapy due to the potential role for chemoresistance and recurrence of these CD133-positive cells. A combinatorial treatment of strongly cytoreductive chemotherapy and specific eradication of CD133-positive chemoresistant tumor cells might be a promising approach to prevent recurrent disease. On account of this, combinatorial treatment strategies of engineered NK cells and cisplatin on the effect of ovarian cancer cells were evaluated.
Interestingly, in these experiments, a significant additive killing effect was revealed when anti-CD133 NK92 cells were administered after previous cisplatin treatment. This sequential treatment strategy resulted in a 26% stronger cell reduction after 4 days compared to control NK cells. An even stronger effect can be assumed if a higher E/T ratio or a higher amount or repetitive doses of cisplatin were administered. A decision was made to use a low E/T ratio of 2:1 and only the IC50 concentration of cisplatin to clearly display combinatorial effects. The increased killing effect of the sequential treatment with engineered NK cells after chemotherapy can be explained by the enrichment of CD133-positive cancer cells after chemotherapy, as already described. 37,38 Interestingly, there was also an additive killing effect by simultaneous treatment with NK cells and cisplatin. This indicates a persistent cytotoxic activity of NK92 cells, even during cisplatin treatment, as already described for carboplatin. 39 It remains to be shown whether this effect is consistent at even higher cisplatin concentrations. To the authors' knowledge, these combinatorial treatments of chemotherapy and CAR-engineered NK cells have not been described so far in ovarian cancer. However, these results, if confirmed in appropriate animal studies, could be the basis for future treatment strategies in ovarian cancer. In contrast to most approaches, in which CAR therapy is applied in recurrent ovarian cancer, a primary treatment directly after or in parallel to cytoreductive surgery and chemotherapy might be a promising therapeutic strategy to prevent recurrent disease by specifically eradicating chemoresistant CSCs.
Of note, several studies showed comparable cytotoxic effects of CAR effector cells in mice and in vitro. 16,17 However, cytotoxic effects in immunocompetent humans are lacking. For example, an anti-FRα-CAR against ovarian cancer cells was successfully tested in vitro and in mice but lacked efficiency in humans. 27 One explanation might be that the first generation design of the CAR only included CD3ζ as an effector domain. In the present study, a third-generation CAR equipped with CD28 for enhanced proliferation and 41BB for improved survival of the cells was used in order to promote activity and persistence in humans. 14,15,40 The incorporation of these additional cofactors in CAR design enhanced survival and proliferation of engineered T cells. 14,40 Another factor that influences cytotoxicity in vivo is the persistence of CAR effector cells in the immunosuppressive tumor microenvironment. 41 In these settings, NK cells might be advantageous over T cells due to their native antitumor activity, which will be activated after specific localization in antigen expressing tumors. 32 Thereby, NK cells are also able to eradicate antigen-negative cancer cells. Engineering NK cells to produce immune-modifying cytokines after activation could also improve NK cell survival in an autocrine manner. 41,42 Synergistic administration of CAR-NK cells and antibodies, which block immune inhibitors expressed on tumor cells, could also be an effective treatment to enhance efficacy of adoptive cell therapy.
NK92 cells engineered with a CD19 control CAR also showed elevated IFN-γ secretion when co-incubated with one of our primary ovarian cancer cell samples. This might be explained by an unspecific stimulation of the native NK cell-mediated antitumor activity by overexpression of stimulating co-domains in transduced cells or could be due to increased levels of CD19+ lymphocytes in this ascites sample. 43 However, a significantly increased killing of primary cancer cells by CD19-CAR-engineered NK92 was not observed in these experiments. CD133 is also expressed in normal tissues, especially in hematopoietic stem cells, endothelial progenitor cells, as well as adult kidney, mammary glands, and some other cell types, which may lead to “off-cancer, on-target (CD133)” side effects. 13,44,45 Interestingly, preclinical studies using an antibody conjugated toxin against CD133 did not reveal severe off-target effects on hematopoietic cells or other somatic cells. 30,46 This might be due to lower antigen expression levels of CD133 in hematopoietic stem cells or somatic cells compared to cancer cells or posttranslational protein modifications in different cell types. 46 Another explanation might be a variable role of CD133 as a stem cell marker in hematopoietic stem cells or CSCs. 47 Since CD133 also represents a very promising CSC marker for several other malignancies such as breast, colon, pancreatic, or lung cancer (reviewed in Schmohl and Vallera 48 ), this therapeutic approach might not only be restricted to ovarian cancer but might also represent a potential treatment strategy against other chemoresistant malignancies.
Due to their shorter life time compared to T cells, NK cells might be advantageous regarding off-target effects, since their activity can better be controlled. 32 To date, a number of studies reported strategies for CAR design, for example bispecific CARs, to improve the tumor cell specificity and limit the off-target side effects. 49,50 Two CSC markers (e.g., CD133 and CD44) can be combined as two activating signals in the bispecific CARs due to their co-localization in the same membrane microdomains. 51 Alternatively, combination of one CSC marker and an ovarian cancer associated antigen, such as CA125 or mesothelin, could also be a promising strategy in future studies.
In summary, a new third-generation CAR against the CSC epitope CD133 was developed. Specific CD133 targeted cell killing was shown by engineered anti-CD133 NK92 cells in vitro. In addition, a combinatorial treatment strategy of anti-CD133 NK cells and cisplatin therapy was analyzed, and it was shown that the engineered NK92 cells retained their cytotoxic potential during cisplatin incubation and that the strongest killing effect was achieved after sequential treatment of cisplatin followed by NK cells. In vivo studies are already planned to investigate this newly generated CAR and its interaction further in parallel to or in combination with sequential chemotherapy. These results will be the basis for upcoming clinical studies in primary and recurrent ovarian cancer.
Footnotes
Acknowledgments
This study was supported by HILF (Hochschulinterne Leistungsförderung, Hannover Medical School) and the Young Academy (Hannover Medical School; both granted to Dr. Rüdiger Klapdor) as well as DFG (SFB738, Cluster of Excellence REBIRTH and KFO 286). We thank the Claudia-von-Schilling-Stiftung for the opportunity to use the xCELLigence real-time impedance analyzer. We gratefully acknowledge the technical assistance of Britta Wieland and helpful advice of Dr. Kristine Bousset.
Author Disclosure
No competing financial interests exist.