
Abstract
Infusion of patient-derived CD19-specific chimeric antigen receptor (CAR) T cells engineered by viral vectors achieved complete remission and durable response in relapsed and refractory (r/r) B-lineage neoplasms. Here, we expand on those findings by providing a preclinical evaluation of allogeneic non-viral cytokine-induced killer (CIK) cells transfected with the Sleeping Beauty (SB) transposon CD19CAR (CARCIK-CD19). Specifically, thanks to a large-scale 18-day manufacturing process, it was possible to achieve stable CD19CAR expression (62.425 ± 6.399%) and efficient T-cell expansion (23.36 ± 3.00-fold). Frozen/thawed CARCIK-CD19 remained fully functional both in vitro and in an established patient-derived xenograft (PDX) of MLL–ENL rearranged acute lymphoblastic leukemia (ALL). CARCIK-CD19 showed a dose-dependent antitumor response and prolonged persistence in a PDX, bearing the feature of a Philadelphia-like ALL with PAX5/AUTS2 translocation, and in a survival model of lymphoma, achieving complete eradication of disseminated tumors. Finally, the infusion of CARCIK-CD19 proved to be safe and well tolerated in a biodistribution and toxicity model. The infused cells persisted in the hematopoietic and post-injection perfused organs until the end of the study and consisted of CD8+, CD56+, and CAR+ T cells. Overall, these findings provide important implications for non-viral technology and the proof-of-concept that donor-derived CARCIK-CD19 are indeed effective against relapsed ALL, a possibility that will be tested in Phase I/II clinical trials after allogeneic hematopoietic stem-cell transplantation.
Introduction
A
Adoptive immunotherapy based on lymphocytes engineered ex vivo with chimeric antigen receptors (CARs) constitutes a potentially curative treatment for relapsed cancers. Treatment with patient-derived CAR T cells transduced using viral vectors targeting CD19 results in complete remission (CR) rates as high as 90% in children and adults affected by relapsed/refractory (r/r) ALL. 4 –8 Several studies using distinct CAR designs have produced similar CR rates, with durable remissions observed in many patients without the need of additional therapy. However, gene modification by gamma-retroviral and lentiviral vectors is hampered by the costs and complexity of manufacturing clinical grade CAR T cells, 9 as well as managing the many regulatory aspects of gene therapy products, including safety requirements. 10
Sleeping Beauty (SB) is a non-viral vector derived from the Tc1/mariner superfamily of DNA transposon reconstructed from salmonid genome. 11,12 The system is composed of a transfer plasmid, including the mobilized sequences and a synthetic transposase, which catalyzes the integration, provided by a separate plasmid. 13,14 As for SB, the potential genotoxicity is strongly mitigated by integration occurring into TA dinucleotides, granting close-to-random distribution of integrations, in the absence of a preference for integation near or within transcriptional units, unlike other gene transfer systems. 15,16 Furthermore, SB has no significant immunogenicity per se, contrary to viral vectors. 15,16 In this regard, a recent study validated the clinical use of SB-engineered T cells for the treatment of minimal residual disease in advanced non-Hodgkin lymphoma and B-cell ALL (B-ALL) after HSCT. 17,18 In the reported trials, no toxicity, including the exacerbation of graft-versus-host disease (GvHD), was observed.
We recently published a novel platform for T-cell non-viral manipulation using SB, 19 which allows engineering of cytokine-induced killer (CIK) cells with CAR to target different antigens. 20 Unmodified CIK cells, a T-cell population characterized by the enrichment of highly cytotoxic CD3+CD56+ cells and reduced GvHD risk, have shown high safety and efficacy profiles in leukemic patients, including those affected by B-ALL. 21,22 Allogeneic CIK cells engineered with non-viral vectors appear to represent a safe platform for ALL patients who relapse after transplantation, who are often lymphodepleted, 23 and it is likely to reduce cost and complexity within CAR immunotherapy landscape. 24
Since both non-clinical pharmacology studies in patient-derived xenograft (PDX) and toxicology of SB-transfected T cells are lacking, the goal of this study was to perform a preclinical evaluation of donor-derived CARCIK-CD19. It is shown that large-scale CARCIK-CD19 exhibited a fully competent T-cell response in vitro and potent dose-dependent antitumor activity in vivo. Remarkably, CARCIK-CD19 persisted over time in the hematopoietic compartment and post-injection-perfused organs in the absence of detectable toxicity. Collectively, the findings strongly support the clinical application of CARCIK-CD19 in B-ALL patients who relapse after HSCT.
Methods
Plasmids
The SB vector, which includes the CD19CAR/pTMNDU3 (MNZDU3) and the pCMV-SB11 transposase (MNZSB11) plasmids encoding for the anti-CD19CD28OX40ζ CAR (CD19CAR) and transposase SB11, respectively, were obtained from anti-CD19/pTMNDU319 and pCMV-SB1113 by replacing the ampicillin resistance with the kanamycin resistance gene (kindly provided by Eugenio Montini, Ospedale San Raffaele). Synthetically generated plasmids were then made available to VGXI Inc. (The Woodlands, TX) as starting material for good manufacturing practices (GMP).
CARCIK-CD19 manufacture and in vitro functional analysis
The study was conducted in accordance with ethical standards and approved by the Institutional Review Board of the Ethical Committee. Informed consent was obtained from patients or their guardians in compliance with institutional guidelines and the Declaration of Helsinki. CARCIK-CD19 were generated, as previously described.
19
Functional analyses were performed on the bulk cell-product. Peripheral blood mononuclear cells (PBMCs; 107) were electroporated using 4D-NucleofectorTM (Lonza, Basel, Switzerland) system with P3 Primary Cell 4D-Nucleofector kit (Lonza) with 15 μg of MNZDU3 and 5 μg of MNZSB11. As recently described, autologous PBMCs irradiated with 60 Gy of 137Cs γ-rays were added after electroporation. Interferon-gamma (IFN-γ; 1,000 IU/mL; Boehringer Ingelheim, Ingelheim, Germany) was added on day 0 and interleukin (IL)-2 (300 IU/mL; Novartis, Basel, Switzerland) and OKT-3 (50 ng/mL; Takara, Kyoto, Japan) were added on day 1. Cells were then cultured for 18–28 days, and IL-2 was added weekly. Flow cytometry analysis is detailed in the Supplementary Methods (Supplementary Data are available online at
Cytotoxic assay
CARCIK-CD19 cytotoxicity was evaluated by 4 h short-term and 24 h long-term co-culture assays. Cell death and apoptosis were detected using GFP-Certified™ Apoptosis/Necrosis detection kit (Enzo Life Sciences, Farmingdale, NY), according to manufacturer's instructions. CIK cells were co-cultured for 4 h with CD19+ targets that were previously labeled with 5- (and 6-) carboxyfluorescein diacetate succinimidyl ester (CFSE; 1 μM; eBioscience, San Diego, CA), at the indicated effector:target (E:T) ratio. For the potency assay, CARCIK-CD19 were co-cultured for 4 or 24 h with CD19+ targets at different transfection percentages, obtained by serial dilutions of CARCIK-CD19 with non-transfected (NT) cells, starting from the 60% transfection at a 5:1 E:T ratio. In this way, the E:T ratio is equivalent in all assays, but both the transfection percentage and the ECARCIK-CD19:T ratio are different. The final percentage of killed cells was determined adding the percentage of Annexin V+Necrosis Detection Reagent− to that of Annexin V+Necrosis Detection Reagent+ in CFSE+ target cells co-cultured with the effectors compared to target cells alone. All samples were run in duplicate.
Proliferation
CIK cell proliferation was assessed through CFSE-based tracking. Cells were stained with 1 μM of CFSE and co-cultured at a 1:2 effector:stimulator (E:S) ratio with irradiated target cells for 96 h. Following stimulation, CIK cells were labeled with CD3 and CAR using anti-human IgG (H+L). Proliferation of CFSE-labeled cells was detected as the progressive halving of cellular fluorescence following cell division. CFSE dilution was analyzed by flow cytometry and calculated on CD3+ cells.
Mouse models
This study was approved by the Italian Ministry of Health (approval no. 102/2013-B, issued on the April 23, 2013). Procedures involving animal handling and care conformed to protocols approved by the Milano-Bicocca University in compliance with national and international law and policies. Human leukemia xenografts were generated in NOD.Cg-Prkdcscid Il2rgtm1Wj1/SzJ (NOD scid gamma [NSG]) mice, as previously described. 25,26 To establish the PDX model, male NSG mice were sublethally irradiated (200 cGy), and mice were intravenously (i.v.) injected with splenocytes and/or bone marrow (BM) cells from primary or secondary leukemic recipients. Each secondary or tertiary mouse received from 0.5 to 1 × 106 cells in a single injection. For the survival model, mice were i.v. injected with CD19+ Burkitt lymphoma cells, Daudi (ATCC, Manassas, VA). Upon leukemia engraftment, mice were i.v. injected with a single dose of fresh or frozen CARCIK-CD19 or control NT cells. The good laboratory practice (GLP) toxicity study was conducted at GLP SR-TIGET (Milan, Italy) using GMP-manufactured lots produced by the Cell Factory Laboratorio di Terapia Cellulare e Genica Stefano Verri (Monza, Italy), detailed in Supplementary Methods. Vector copy number (VCN) analysis was carried out by the Tettamanti Research Center. Test and control items were produced and characterized by the Cell Factory laboratorio di Terapia Cellulare e Genica Stefano Verri—ASST Monza, Ospedale San Gerardo, under GMP conditions (European Directive 2001/83/CE and Decreto Legislativo 24 aprile 2006 n. 219, European Regulation on Advanced Therapies 1394/2007, Volume 4 Eu GMP).
Detection of integrated copy number of CD19CAR by quantitative polymerase chain reaction
DNA samples (100 ng) were performed in duplicate or triplicate, and VCN was determined by normalizing to the endogenous reference gene RNase P, detailed in the Supplementary Methods.
Statistical analysis
Mean values are reported as the mean ± standard error of the mean. A paired t-test or two-way analysis of variance with Bonferroni multiple comparison was used to determine statistical significance of the measurements, with the exception of the in vivo experiments, which were analyzed by the Mann–Whitney U-test. Two-tailed paired analysis was performed, unless otherwise specified in the text. Statistical calculations were performed with GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA).
Results
In vitro non-clinical pharmacology studies of CARCIK-CD19 after large-scale production
The transfection optimization of the CIK cell manufacturing process has been previously reported. 19 To verify whether CARCIK-CD19 could be propagated to clinical-size numbers, peripheral blood (PB) samples (approximately 45 mL each) were processed from three healthy donors (HDs; HD #1–3). Five additional large-scale productions were performed starting from buffy coats, with 30–60 × 106 PBMCs (HD #4–8).
The CARCIK-CD19-cell culture revealed an exponential growth trend, with a 23.36 ± 3.00-fold T-cell expansion after 18–21 days. An average of 8.63 × 108 ± 1.82 CAR+ T cells was obtained (Fig. 1a). CD19CAR expression level reached a value of 62.425 ± 6.399% (Supplementary Fig. S1a). Immunophenotypic analysis of CARCIK-CD19 cells showed the typical enrichment of the CD3+/CD56+ proportion, while no changes were detectable in naïve and central memory CD8+ and CD4+ T cells (Fig. 1b and Supplementary Fig. S1b).
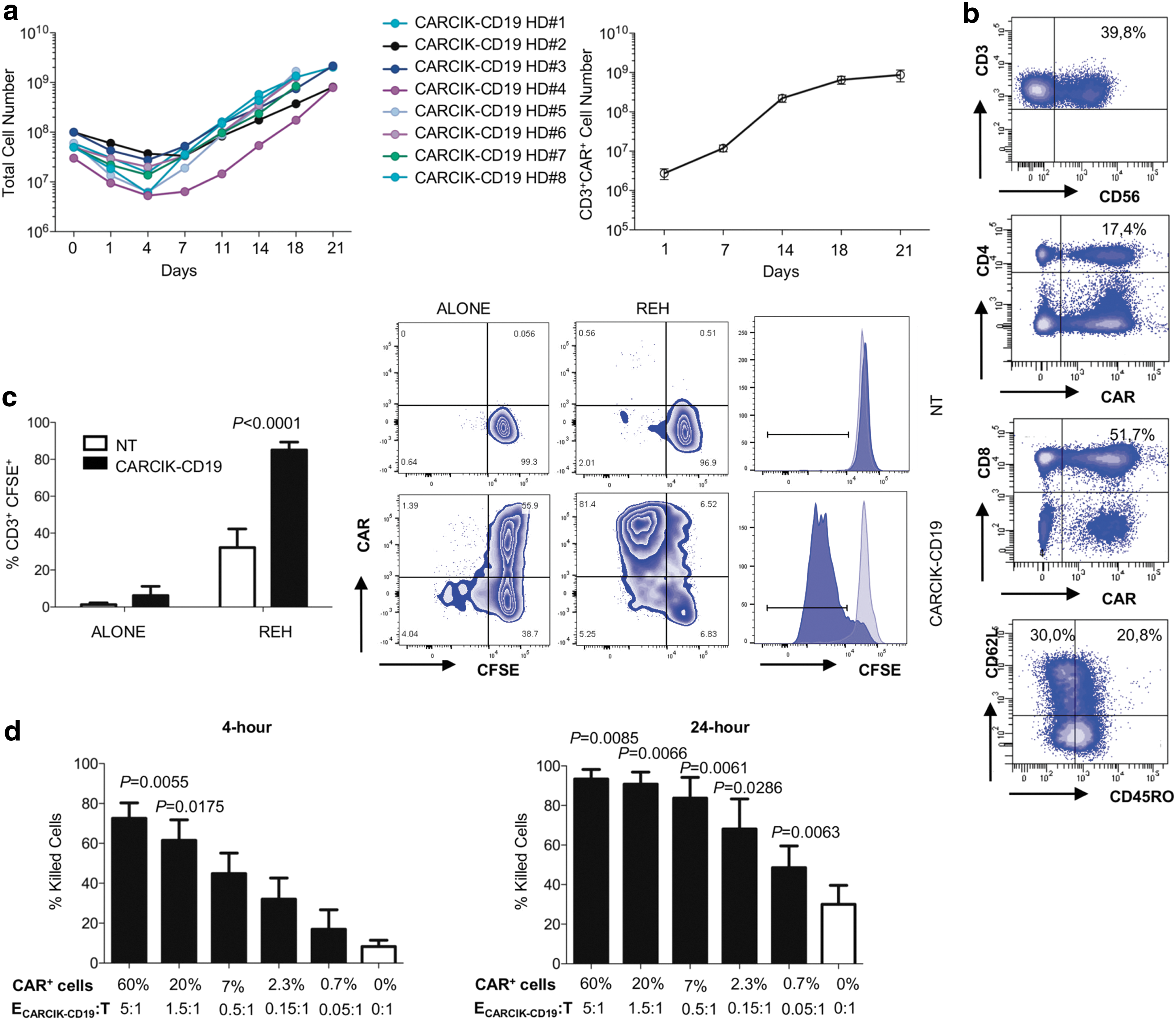
Expansion kinetics, chimeric antigen receptor (CAR) expression, and characterization of large-scale production.
Cytokine production was evaluated in response to CD19+ REH stimulating cells. The average of IL-2 and IFN-γ-producing T cells was 21.5 ± 2.1% and 38.28 ± 10.58%, respectively (n = 5). The production was restricted to the CAR+ population, while it was absent in NT cells (Supplementary Fig. 1c). CARCIK-CD19 showed a specific killing of REH at 4 h post stimulation, while NT cells did not show significant cell killing (Supplementary Fig. S1d). Moreover, the ability of CARCIK-CD19 to proliferate upon CAR-specific stimulation was confirmed (Fig. 1c).
The in vitro biological activity of CARCIK-CD19 associated to different levels of transgene expression was measured as drug substance potency. At least 20% of CAR expression was necessary to achieve a statistically significant higher cytotoxicity toward REH compared to NT at 4 h post stimulation (Fig. 1d). Furthermore, the percentage of killed cells correlated with the percentage of CAR+ T cells (Supplementary Fig. S1e). In a 24 hour assay, the potency of CARCIK-CD19 drug substance remained elevated up to 0.7% of CAR expression, as judged by the ability of the transfected cells to kill, even when outnumbered by target cells (Fig. 1d).
Development of formulation of CARCIK-CD19 from fresh to frozen cells
Cryopreservation is an essential step in translating cell therapies to the clinic. To evaluate whether cryopreservation affects cell-product functionality, both fresh and frozen/thawed CARCIK cells were characterized. After freezing/thawing, both CAR expression and CD3+/CD56+, CD3+/CD8+, and CD3+/CD4+ cell subsets were preserved (Supplementary Fig. S2a). With regards to the immunological memory, thawing determined a slight increase in the effector memory RA subset (Supplementary Fig. S2b). As shown in Fig. 2a, after cryopreservation, CARCIK-CD19 viability was not different from the fresh product.

Viability and functionality of CARCIK-CD19 after cryopreservation.
Next, the functional activity of frozen/thawed CARCIK-CD19 was evaluated in vitro and in vivo. CARCIK-CD19 exerted potent lytic activity, with an average of 63.6 ± 5.7% target-cell killing after thawing compared to 81.5 ± 9.5% of fresh cells (Fig. 2a). Upon stimulation with target cells, levels of cytokine production and proliferation of fresh and thawed CARCIK-CD19 were of comparable magnitude (Supplementary Fig. S2c and d and Fig. 2b). To establish a PDX model (Supplementary Fig. S2e), samples were derived from serial transplantations into recipient mice of human leukemic cells isolated from a 2-month-old patient (UPN1) diagnosed with pro-B infant ALL, carrying the t(4;11) translocation. Fresh and frozen/thawed CARCIK-CD19 showed comparable in vivo functionality (Fig. 2c and Supplementary Fig. S2f and g), resulting in significantly decreased engraftment levels in the spleen as well as diminished splenomegaly compared to mice treated with NT cells (Fig. 2c).
Dose-finding study in PDX model
To determine CARCIK-CD19 activity in a xenograft model of high-risk B-ALL and the correct dose for optimal activity, common B-cell precursor (BCP) ALL cells were isolated from a 1-year-old patient (UPN2) diagnosed with ALL, with a Philadelphia chromosome (Ph)-like genetic translocation involving the PAX5 and AUTS2 genes 27 (46, XX, der[7]t[7;11][q11.2;q12], der[9] t[7;9][q11.2;p13], −11, +mar[10]/46,XX[2]). BCP-ALL cells were used to establish leukemia in immunodeficient mice that were subsequently treated after 15 days with 5 × 106, 10 × 106, or 15 × 106 cells. The in vitro activity of CARCIK-CD19 toward UPN2 was confirmed (Fig. 3a). Given that the extent of CARCIK-CD19 cell cytotoxicity largely depended on the percentage of CAR expression, the optimal therapeutic dose was defined according to the number of CAR+ T cells. Leukemic burden was monitored in the PB two days after the treatment together with CIK-cell engraftment, which was consistent with the infused dose (Fig. 3b). As shown in Fig. 3c, the treatment was effective and dose dependent. At the time of sacrifice, the treatment determined a significant decrease in the engraftment of leukemic blasts in the spleen and BM of animals infused with 10 × 106 CARCIK-CD19, and in the PB, spleen, and BM of animals treated with 15 × 106 cells. In contrast, despite the presence of CIK cells in the PB, BM, and spleen (Fig. 3c), both mice treated with 5 × 106 CARCIK-CD19 and NT cells did not show any significant tumor reduction. As expected following infusion of a high number of cells, the infusion of 15 × 106 or 10 × 106 CARCIK-CD19 cells led to an initial body weight loss compared to untreated mice. By contrast, no adverse events were detected following the infusion of 5 × 106 cells (Fig. 3d). At sacrifice, 5/8 and 4/8 animals treated with 15 × 106 CARCIK-CD19 had detectable presence of CAR+ T cells in the BM and spleen, respectively (Fig. 3e).

Efficacy of CARCIK-CD19 cells in human ALL patient-derived xenograft model is associated with CAR expression in vivo.
CARCIK-CD19 cells eradicated leukemia in a survival model of CD19+ lymphoma
The ability of CARCIK-CD19 to improve survival was evaluated in a Burkitt lymphoma model by xeno-transplanting Daudi in NSG mice (Supplementary Fig. S3a). Leukemic burden and CARCIK-CD19 engraftment were monitored in PB after treatment. CD8+ T cells represented the predominant T-cell subset in vivo, whereas CD3+CD56+ CIK cells were found mostly at later time points (Fig. 4a).
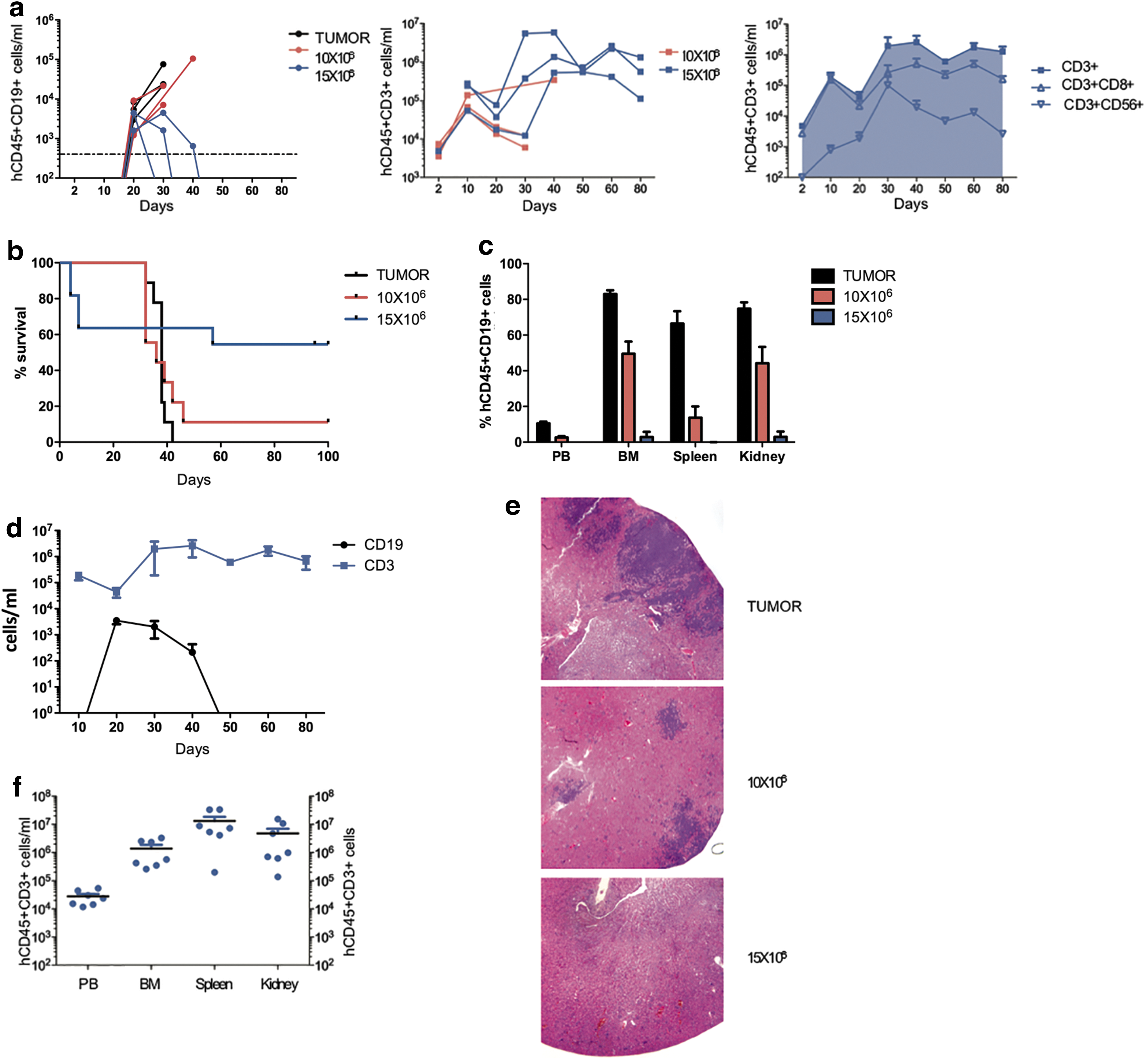
Leukemia eradication and prolonged survival by CARCIK-CD19 cells.
Untreated mice developed progressive highly disseminated leukemia and succumbed to the disease after 32–42 days (n = 9; median survival = 38 days; Fig. 4b). CARCIK-CD19 cells (10 × 106) had a significant inhibitory effect on tumor growth (Fig. 4c), which, however, did not impact on survival compared to untreated mice (n = 9; median survival = 36 days; p = 0.820, log-rank test; 95% confidence interval [CI] 0.433–4.63; hazard ratio [HR] = 1.417; Fig. 4b). This effect was enhanced in mice treated with 15 × 106 in which both leukemia and T cells were found in the blood in the early phase of treatment, with evident signs of tumor control (Fig. 4d). Following this coexistence state, CARCIK-CD19 completely eradicated the disease, inducing long-term tumor regression (Fig. 4c), along with a statistically significant survival prolongation compared to untreated mice (n = 11; p = 0.0495, log-rank test; 95% CI 1.003–11.50; HR = 3.396; Fig. 4b). Histopathological analysis revealed severe multicentric embolic and metastatic tumors in the kidneys of untreated mice, which was diminished in terms of pathological score in the 10 × 106 cell-treated group and was completely absent in the 15 × 106 cell-treated group (Fig. 4e). Early deaths were observed in 4/11 mice treated with 15 × 106 CARCIK-CD19. From day 10 to the end of study, all animals gained weight (Supplementary Fig. S3b). Interestingly, CARCIK-CD19 cells persisted in the PB and organs up to 3 months, well after tumor eradication (Fig. 4f and Supplementary Fig. S3c).
General toxicity, tissue damage, and biodistribution of CARCIK-CD19
Biodistribution and the general toxicity were evaluated in NSG mice infused with 15 × 106 CAR+ cells, corresponding to 22.5 × 106 cells of the manufactured CARCIK-CD19 cell product (CAR expression: 67%; Supplementary Fig. S4a and b). The number of human (h)CD45+CD3+ cells in the PB decreased in the first 20 days post infusion and then stabilized, decreasing from the initial levels of 16,714 ± 8,641 cells/mL (day +2) to 6,015 ± 5,661 cells/mL (day +90) in mice infused with 15 × 106 CARCIK-CD19 (Fig. 5a). Half of the group was sacrificed at day +30, while the remaining half was then sacrificed at day +90, showing engraftment of CARCIK-CD19 cells in organs (2/5 of mice infused). No premature deaths occurred in this study.
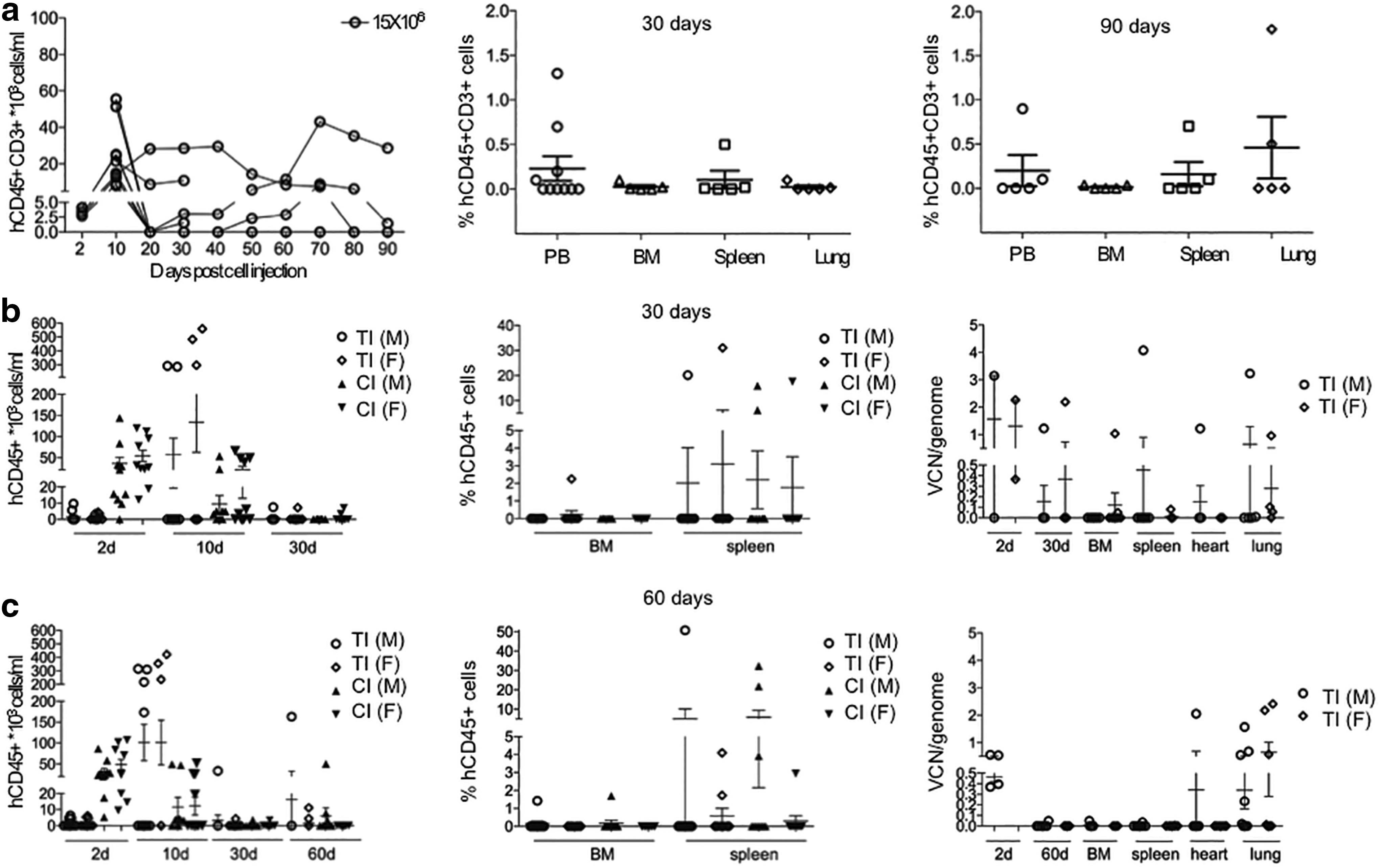
Biodistribution and toxicity studies of CARCIK-CD19.
Further, a GLP study was conducted by using 15 × 106 CARCIK-CD19 cells from two different GMP batches, corresponding to 23.55 × 106 total cells for batch 1 and to 32.27 × 106 total cells for batch 2, differentiated according to GMP-compliant protocol. NSG mice were divided in six experimental groups: group 1 (UT-30days) and 4 (UT-60days) were untreated, while groups 2 (TI-30days), 5 (TI-60days), 3(CI-30days), and 6 (CI-60days) received the test item (TI), namely CARCIK-CD19 cells, and the control item (CI), namely NT cells, respectively. Groups 1, 2, and 3 were sacrificed 30 days after the infusion and groups 4, 5, and 6 after 60 days. On day 2, groups 2 and 5 showed a lower number of hCD45+ cells compared to groups 3 and 6. As shown in Fig. 5b and c, on day 10, this trend was reversed, suggesting that CARCIK-CD19 had a delayed engraftment in PB compared to that of NT cells. At termination, no difference was observed in TI compared to the CI-treated groups. A fair number of animals (6/20 in group 2 and 9/20 in group 5) showed detectable levels of human transfected cells in both the PB and organs. In the engrafted animals, transfected cells persisted as single- or multi-organ engraftment, mainly with a constant VCN value. In a limited number of animals, hCD45+ cell engraftment reached high levels, mainly in relation to NT cells, showing a VCN reduction. The brains and gonads isolated from the mice in groups 2 and 5 were negative, with an exception where PB had a VCN/cell of 0.049 versus 0.013 in the testes, suggesting blood contamination of the testicular tissue. On histopathological examination, in a limited number of animals treated with either the TI or the CI cells, focal or multifocal aggregates of lymphoid-like cells were detected in the liver, gall bladder, lungs, spleen, kidneys, and stomach at both 30 and 60 days, in most cases as a multi-systemic organ involvement. Aggregates of lymphoid-like cells occurred in most cases with minimal or mild severity and did not cause loss of organ architecture or noteworthy parenchymal damage. Thus, these data clearly indicate that CARCIK-CD19 cell-treated mice do not exhibit any adverse events.
Discussion
Donor-derived non-viral CAR+CD3+ CIK cells could represent a valid therapeutic option for relapsed leukemic patients after HSCT. A number of reported clinical trials have used complex manufacturing processes exploiting apheresis- and viral modification-based protocols for manufacturing clinical-grade autologous lymphocyte. This study describes a large-scale production and preclinical evaluation of donor-derived CARCIK-CD19 developed for a pilot Phase I/II non-viral gene therapy trial to treat relapsed B-ALL. It shows that CARCIK-CD19 cells can be propagated to clinical-size numbers, achieving ∼60% CAR expression, with phenotypic and functional attributes indicative of therapeutic potency. Indeed, CARCIK-CD19 cells exerted potent antitumor activity in immunodeficient mouse models engrafted with tumor cells from patients with high-risk leukemia. Furthermore, they eradicated the disease in a survival study, thereby promoting prolonged leukemia-free survival. Infused CAR+ T cells were readily detectable in the BM and spleen and persisted in vivo for 3 months while proving to be safe and well tolerated in a biodistribution/toxicity study.
The functionality and persistence of non-viral SB cell products in vivo, especially in advanced diseases, still raises questions, and the differences into composition of CAR-T cell product profoundly impact the efficacy. Recently, the SB transposon has been validated in trials for ex vivo gene delivery of CD19CAR to T cells to treat low burden B-cell malignancies in both autologous and allogeneic setting, showing encouraging safety and feasibility results but reduced levels of persisting T cells. 17 In this case, the manufacturer used repetitive stimulations with artificial antigen presenting cells expressing the targeted antigen to select and expand CAR+ T cells in order to improve the efficacy of non-viral modification. 28 Our group has previously developed a protocol based on a single stimulation/expansion cycle coupled with a non-viral modification by using autologous gamma-irradiated PBMCs as feeder cells with soluble OKT3, reaching high transfection and T-cell expansion efficiency. This approach turned out to be improved in reducing the complexity and the number of manipulation steps with the advantage to minimize costs and prevent exhaustion of the T-cell product. 19 The present study shows that CARCIK-CD19 can be manufactured and propagated to clinical-size numbers, reaching a total of up to 109 nucleated cells in 18–21 days starting from 50 mL of PB, with important implications for non-viral technologies. The composition of CARCIK-CD19 and their effector functions indicate the acquisition of T-cell fitness, which is essential to provide optimal antitumor activity. 29 Using a developed potency assay, the ability of CAR+ cells to detach rapidly and recycle from one target to another was demonstrated. Other groups optimized SB-mediated engineering with proved in vivo efficacy. 30 The results obtained in this study demonstrate that engineered CARCIK-CD19 cells display long-term persistency and antitumor activity in PDX models of high-risk leukemia, observed despite the freezing/thawing process.
The use of a donor-derived CAR T cells represents an option to treat patients suffering from relapsing and progressing diseases following allo-HSCT. Donor cells have the advantages of outperforming autologous T cells, not being conditioned by the tumor or any previous treatment, and bypassing the restriction of individualized products, envisaging sources on basis of best HLA-matched 31 or TCR gene-edited T cells. 32 Even if there is still a small number of clinical examples, these approaches certainly represent a promising perspective likely to overcome many issues related to pharmaceutical distribution. Despite early-phase clinical trials having shown limited onset of GvHD using donor-derived CAR T cells in a post-transplant setting in the absence of lymphodepletion, 33,34 there is still the concern about eliciting GvHD with lymphocyte-depleting chemotherapy. A recent study supporting the hypothesis that donor CAR T cells have a reduced risk of GvHD demonstrated that when CD28z is used as costimulatory domain, alloreactive T cells undergo exhaustion and deletion upon cumulative TCR and CAR activation. 35 A decision was made to evaluate an alternative T-cell population, namely CIK over standard CAR T cells, which is known to be safe and well tolerated with minimal GvHD occurrence. 22,36,37 A recently concluded Phase II trial using donor-derived CIK cells in patients who relapsed after HSCT demonstrated a remarkably low incidence of GvHD, even for the highest dose administered, which was easily manageable with standard treatments. 37 In agreement with these observations, an exacerbation of xenogeneic GvHD reactivity was not seen in the biodistribution and toxicity study performed with high doses of non-CAR-purified cell products, despite CARCIK-CD19 being able to reach and form aggregates of lymphoid-like cells in hematopoietic and post-injection perfused organs, consistently with engraftment of the infused cells in multiple organs. Furthermore, detectable presence of CAR+ T cells in organs was not associated with hyperproliferative disorders and germline transmission.
Another remarkable observation emerging from this study is that the treatment with SB-modified CIK cells in vivo induces regression of malignancies and complete disease eradication. Results obtained using viral CAR T cells demonstrated that CAR T-cell expansion and long-term post-remission persistence correlate with subsequent remission and tumor surveillance in both animals 38,39 and patients. 8,40 Similarly, antitumor activity and disease eradication were observed, which were associated with CARCIK-CD19-cell engraftment and ongoing persistence for an adequate interval of time. The presence of human lymphocytes expressing the CAR transgene was detected in target organs. A dose-dependent effect of the single-dose administration was demonstrated. Indeed, hCD45+CD3+CAR+ occurrence in treated animals correlated with the dose infused, as well as the intensity of the pharmacological effect. Tumor eradication induced by high doses of CAR+ T cells impacted on survival prolongation and was associated with increased expansion and long-term persistence of both CD3+CD8+ T and CD3+CD56+ CIK cells, even following lymphoma clearance. The ability of SB transposon to mediate modification of nondividing cells, including the naïve T-cell subset, 19 could partially explain the rate of persistence achieved. Concerning the dose finding, 15 × 106 CARCIK-CD19 per animal represented the most effective and curative dose evaluated, while 10 × 106 was the lowest effective dose evaluated. Furthermore, 5 × 106 CARCIK-CD19 per animal represented the “no observed effect level” and the “no observed adverse event level.” A similar dose dependency was previously demonstrated in T cells engineered with lentiviral vectors with a CAR targeting the thymic stromal lymphopoietin receptor. 41 Clearly, new approaches to decrease CARCIK-CD19 effective dose are warranted and are being pursued through the optimization of a 10-day differentiation process to prevent T-cell exhaustion.
In conclusion, this study provides important information about the quality, safety, and efficacy of donor-derived SB-transfected CIK cells performed in a patient-derived xenograft, as well as in survival and biodistribution/toxicity models. This donor non-viral approach will ultimately benefit patients by broadening the gene transfer-based therapeutic options. Phase I/II clinical protocol based on the infusion of CARCIK-CD19, combined with lymphodepleting conditioning after HSCT, will evaluate the safety and activity of the proposed approach using a dose escalation scheme. 42
Footnotes
Acknowledgments
We thank Riccardo and Donatella Ruschi and the “Amici di Duccio” association, supporting C.F.M. fellowship, “Quelli che … con Luca” association, supporting S.T. fellowship and the “Comitato Maria Letizia Verga” and “Stefano Verri.” This work was supported by grants from the AIRC Foundation (AIRC Molecular Clinical Oncology 5 per mille, 9962, AIRC 2014, 15992, and AIRC 2015, 17248). C.F.M., S.T., A.B., and E.B. own a patent on the method used in this report.
Author Disclosure
Fondazione Tettamanti submitted a PCT application on Nov 6, 2015 (PCT/EP2015/075980), “Improved method for the generation of genetically modified cells”, Biondi, A, Biagi, E, Magnani, CF, Tettamanti, S. CARCIK-CD19 Development is partially supported by a Sponsored Research Agreement funded by Formula Pharmaceuticals, Inc. (Berwyn, PA). C.M., C.C., M.B., G.F., L.J.N.C., G.D., and G.C. have no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.