
Abstract
CRISPR (clustered regularly interspaced short palindromic repeats) genome editing holds promise in the treatment of genetic diseases that currently lack effective long-term therapies. Patients with alpha-1 antitrypsin (AAT) deficiency develop progressive lung disease due to the loss of AAT's antiprotease function and liver disease due to a toxic gain of function of the common mutant allele. However, it remains unknown whether CRISPR-mediated AAT correction in the liver, where AAT is primarily expressed, can correct either or both defects. Here we show that AAV delivery of CRISPR can effectively correct Z-AAT mutation in the liver of a transgenic mouse model. Specifically, we co-injected two AAVs: one expressing Cas9 and another encoding an AAT guide RNA and homology-directed repair template. In both neonatal and adult mice, this treatment partially restored M-AAT in the serum. Furthermore, deep sequencing confirmed both indel mutations and precise gene correction in the liver, permitting careful analysis of gene editing events in vivo. This study demonstrates a proof of concept for the application of CRISPR-Cas9 technology to correct AAT mutations in vivo and validates continued exploration of this approach for the treatment of patients with AAT deficiency.
Introduction
A
Gene therapy approaches for AATD are challenging because mutant AAT must be corrected to its wildtype form and be sufficiently secreted in order to fully rescue the lung disease. Previous studies have demonstrated proof-of-concept gene augmentation of AAT in animal models. 5,6 In addition, RNAi have been used to downregulate Z-AAT within hepatocytes to decrease the accumulated mutant protein. 7 In clinical trials, M-AAT cDNA has also been delivered via adeno-associated virus (AAV) from the muscle to restore M-AAT in the serum. 8 In comparison to AAV-based M-AAT augmentation and Z-AAT knockdown, in vivo genome editing is a most direct method to permanently correct Z-AAT mutation into M-AAT, which has the potential to provide long-term therapeutic benefit. Recently a targeted gene editing approach using a nuclease free targeted homologous recombination of an AAV expressing microRNA to silence mutant AAT along with a microRNA-resistant wildtype AAT cDNA into the albumin locus was described. 9 The authors showed a selective advantage for corrected hepatocytes achieving both serum AAT level correction and liver disease amelioration.
A transgenic mouse model for the study of AATD was generated in the 1980s and has been widely used for preclinical studies for gene therapy of AATD-related disease. 10,11 A ∼14 kb human Z-AAT transgene was introduced into the germline of mice, resulting in a mouse strain (PiZ mice) that expresses artificially high levels of human Z-AAT. These transgenic mice recapitulate multiple components of the human PiZ liver disease, including hepatic accumulation of Z-AAT and failed secretion of AAT into the blood stream. PiZ mice do not have AATD-related lung disease because endogenous mouse Serpina1 loci are still present—an important limitation of the model. Nonetheless, this system provides a genetic model for exploring gene correction of PiZ mutation, with potential toward human application. 12
CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 has emerged as a transforming genome editing tool with powerful therapeutic applicability. 13,14 Single guide RNA (sgRNA) guides the Cas9 endonuclease to complementary 20 nucleotide genomic sequences that must possess a downstream NGG protospacer-adjacent motif (PAM). Catalytic activity of Cas9 generates double-stranded DNA breaks at that locus, which are typically repaired by either nonhomologous end joining or the more precise homology-directed repair (HDR) pathway.
Many groups have demonstrated correction of genetic disease genes by CRISPR in cells or mouse models. 15 –18 We have recently developed CRISPR delivery for correcting a disease point mutation in the mouse liver. 19 –23 Although CRISPR/Cas9 has proven an attractive approach for therapeutic genome editing, correction of CRISPR-mediated Z-AAT correction in vivo has yet to be investigated.
In this study, we applied dual AAV vectors to deliver Cas9 and sgRNA/HDR template into PiZ transgenic mice to explore the effectiveness of Z-AAT genome editing and HDR correction in the liver. We observed that CRISPR treatment restored M-AAT to 45–71 μg/mL in the serum, suggesting that our dual AAV vector strategy has potential to be optimized for exploring the treatment of AATD.
Materials and Methods
CRISPR vectors
Single guide RNA sequences (Supplementary Table S1; Supplementary Data are available online at
Cell culture, transfection, and sgRNA evaluation
Cell culture conditions were as previously described.
24
Human 293T cells were cultured in Dulbecco's modified Eagle's medium +10% fetal bovine serum and transfected in 24-well plates with 300 ng
Animal experiments
All animal protocols were approved by the UMass Medical School Institutional Animal Care and Use Committee. AAV vector was constructed using Gibson assembly. AAV virus was prepared and purified by UMASS Medical School Viral Vector Core. Two hundred microliters of virus liquid mixture including AAV9-Cas9 (2 × 1011) and AAV8-HDR (1.2 × 1012) was delivered to ∼8-week-old PiZ mice by tail vein injection or 1-week-old PiZ mice by intraperitoneal injection. Mice were humanely sacrificed by CO2 for analysis.
Histology and immunohistochemistry
Livers were fixed in 4% (v/v) formalin overnight and embedded in paraffin. Four micrometer liver sections were stained with hematoxylin and eosin or with Myc tag antibodies (ab19234, Abcam, 1:800 dilution) using standard immunohistochemistry protocols. The percentage of Myc+ cells was measured at low magnification lens from at least three regions per liver (4 mice).
CRISPR-induced insertion/deletion detection and AAT correction
Genomic DNA was purified from mouse liver followed by the protocol of the High-Pure PCR Template Preparation Kit (Roche). sgRNA target sites were PCR amplified through two-step PCR with the primers shown in Supplementary Table S2 and subjected to deep sequencing on Illumina NextSeq500. BWA 0.7.5 and SAMtools 0.1.19 were used for mapping the identified reads to the reference genomic sequence. Insertions and deletions were identified and detected by VarScan2 (version 2.3). The HDR rate was based on the ratio of reads of corrected M-AAT over the total mapped reads.
Myc tagged M-AAT ELISA
High binding extra 96-well plates were coated with goat anti-Myc tag antibody (1:1,000; ab19234, Abcam) in Voller's buffer at 4°C overnight, and 5% bovine serum albumin was used to block the plates at 37 °C for 1 hour. The concentration of Myc tag-AAT in the serum was then quantified as previously described 7 using a Myc tag-AAT standard curve.
Statistics
Student's t-test and one-way ANOVA were used to determine p values.
Results
To determine whether CRISPR can edit the AAT locus in human cells, we designed two sgRNAs, sgAAT.1 and sgAAT.2, targeting the AAT coding sequence adjacent to the PiZ mutation (Supplementary Fig. S1). We transfected human 293T cells with pX330 vectors co-expressing Cas9 and sgAAT and observed CRISPR-induced indel bands of predicted molecular weights by surveyor nuclease assay (Supplementary Fig. S1). sgAAT.2 was chosen for in vivo study because it demonstrated higher editing efficiency than sgAAT.1. All sgRNAs with NGG PAM within the 60 bp region flanking the PiZ mutation is listed in Supplementary Table S3. Notably, sgAAT.2 does not have high score predicted off-target sites in the human genome (Supplementary Table S4). Together these results suggest that CRISPR genome editing was achieved in cells.
Due to size limitations two AAV vectors were utilized to deliver CRIPSR in vivo. One AAV vector harboring a U1a-Cas9 expression cassette 25 was packaged using an AAV9 serotype (termed AAV9-Cas9 hereafter), which has broad tropism, and can target hepatocytes. We chose AAV9 because of the potential to investigate the safety profile of systemic Cas9 delivery. The second AAV vector, containing a U6-sgRNA expression cassette and HDR template (Fig. 1A) was packaged using an AAV8 serotype (termed AAV8-HDR hereafter), which mainly targets hepatocytes. We designed an HDR template containing a 1.6-kb sequence homologous to the AAT genomic region to (1) correct the mutant “A” nucleotide to wild-type “G” in order to effect an amino acid change from mutant 342Lys(AAG) to 342Glu(GAG); (2) change the “GGG” PAM into “GGC” to prevent recleavage of the repaired chromatid following the initial HDR event; (3) insert a 30 nt Myc tag before STOP codon (Fig. 1A). The Myc tag facilitates M-AAT detection in tissue and serum. 7

In vivo delivery of Cas9 and adeno-associated virus–homology-directed repair (AAV-HDR) template in 1-week-old PiZ mice partially restores wildtype alpha-1 antitrypsin (M-AAT) in the serum.
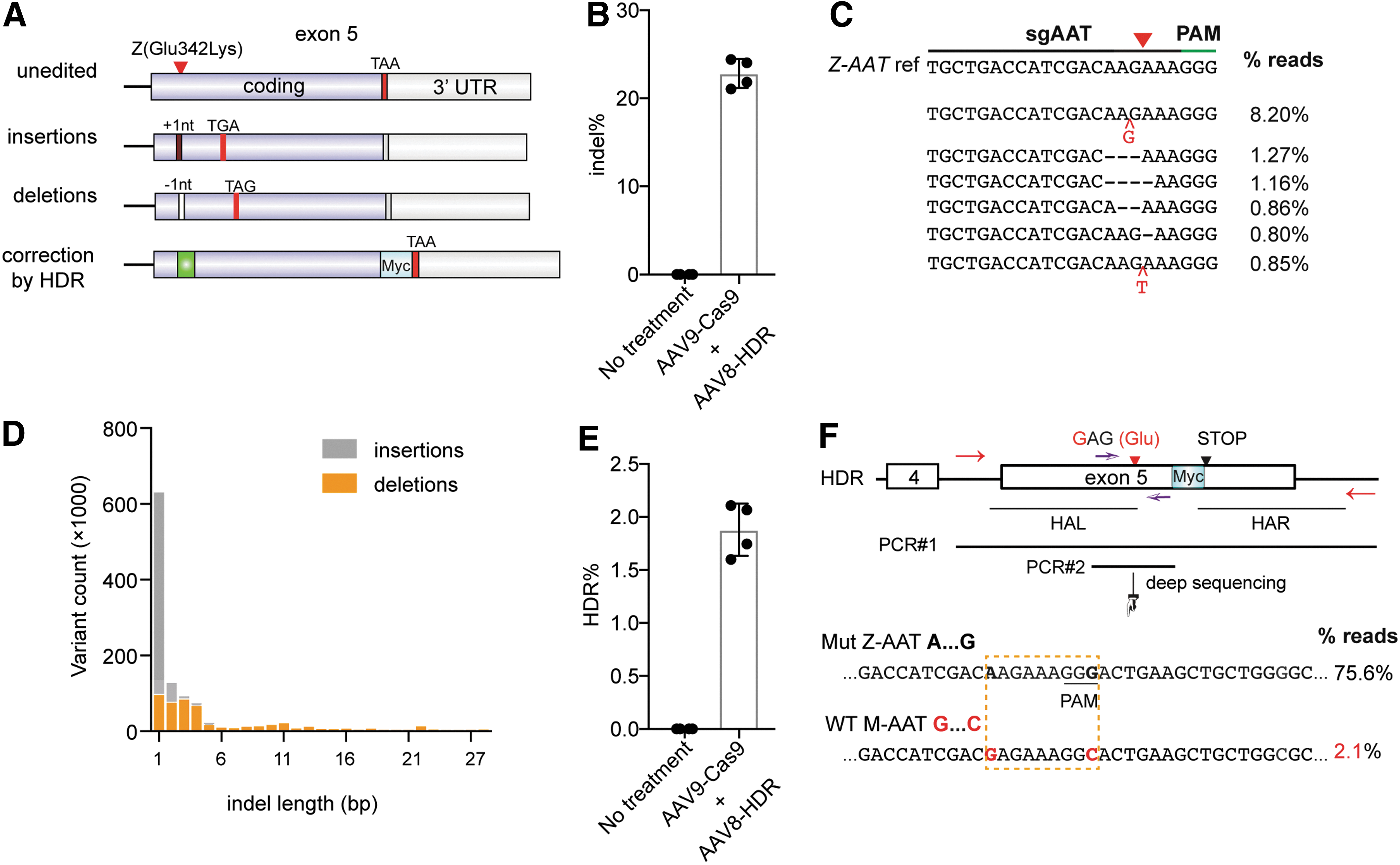
CRISPR treatment generates both indel mutations and precise gene correction in the liver.
To explore whether the dual-vector HDR approach can repair the Z-AAT mutation in vivo, we co-injected 1-week-old young PiZ mice (n = 4) with AAV8-HDR and AAV9-Cas9 (Fig. 1B), and the treated mice were euthanized for analysis after 3 weeks (Fig. 1B). Age-matched untreated mice were used as controls. To examine whether CRISPR generates M-AAT positive hepatocytes in vivo, we stained liver sections with a Myc tag–specific antibody by immunohistochemistry. Around 14% of hepatocytes stained Myc-tag positive in CRISPR-treated PiZ mice (Fig. 1C, D). To determine whether Myc tagged M-AAT can be efficiently secreted into serum, we performed ELISA on the serum of both treated and non-treated mice. Whereas the control mice had undetectable M-AAT levels, we detected that the amount of M-AAT in the serum was 45 ± 8 μg/mL in treated PiZ mice (Fig. 1E). Although it is difficult to compare the M-AAT levels between PiZ mice with multiple AAT gene copies and patient with two copies, this level of Myc tag M-AAT is comparable to approximately 8% of the 570 ug/mL M-AAT required to improve lung disease in AATD patients. 2
To examine the efficiency of genome editing at the human AAT locus in the liver, we performed the deep sequencing of the human Z-AAT gene locus in total PiZ liver genomic DNA. We observed an average of 22.3% small indels (insertions or deletions) at the predicted sgRNA target region (Fig. 2A–D, Supplementary Fig. S2, and Supplementary Table S5). Of note, a one nucleotide G insertion was the most abundant pattern observed in our analysis of genome editing outcomes, which may lead to truncated AAT (due to a shift in the reading frame and introduction of a premature “TGA” stop codon after the sgRNA PAM region). Because the truncated AAT may still be toxic to hepatocytes, we did not study the AAT liver phenotype. To minimize PCR bias, we designed a pair of primers (shown in Fig. 2F; red color) located outside the two homology arms, permitting specific amplification of the Z-AAT gene locus from the PiZ liver genome. A second primer pair (Fig. 2F; blue color) was chosen to generate a relatively unbiased genome-editing template without Myc tag. The analysis of deep sequencing data confirmed a corrected “G…C” pattern at the human AAT locus in 1.9 ± 0.2% of the total liver DNA (n = 4 mice) (Fig. 2E–F). These data indicate that CRISPR induced both indel mutations and precise gene correction in the PiZ mouse liver.
To perform an initial off-target analysis in mouse livers after long-term AAV expression of Cas9, we chose the top three predicted off-target (OT) sites in the mouse genome (Supplementary Table S6). The sgAAT.2 target and OT sites were PCR amplified and analyzed by Surveyor nuclease assay. On-target site showed indel bands (Supplementary Fig. S2). All three off-target sites OT1, OT2, and OT3 did not have detectable indel bands, indicating that the off-target effect is below the detection limit of this method. Future studies are required to explore sgAAT's off-target effects using deep sequencing methods in mouse tissue and human cells.
To further explore whether AAV-mediated HDR is also feasible in adult PiZ mice, we co-injected the same amount of AAV9-Cas9 and AAV8-HDR into 2-month-old adult PiZ mice (Fig. 3A). Two months later, we detected ∼18% Myc-tag positive hepatocytes (Fig. 3B–C) and 71 ± 14 μg/mL M-AAT in the serum (∼12% of the 570 μg/mL level) (Fig. 3D). To measure the genome editing efficiency, we used the same primers as in Fig. 2F to amplify the genomic DNA and performed Tracking of Indels by DEcomposition analysis to measure indel rate. 26 ∼15% indels were observed in the treated mice (Fig. 3E). Collectively, these data suggest that CRISPR treatment can partially restore M-AAT in adult PiZ mice.
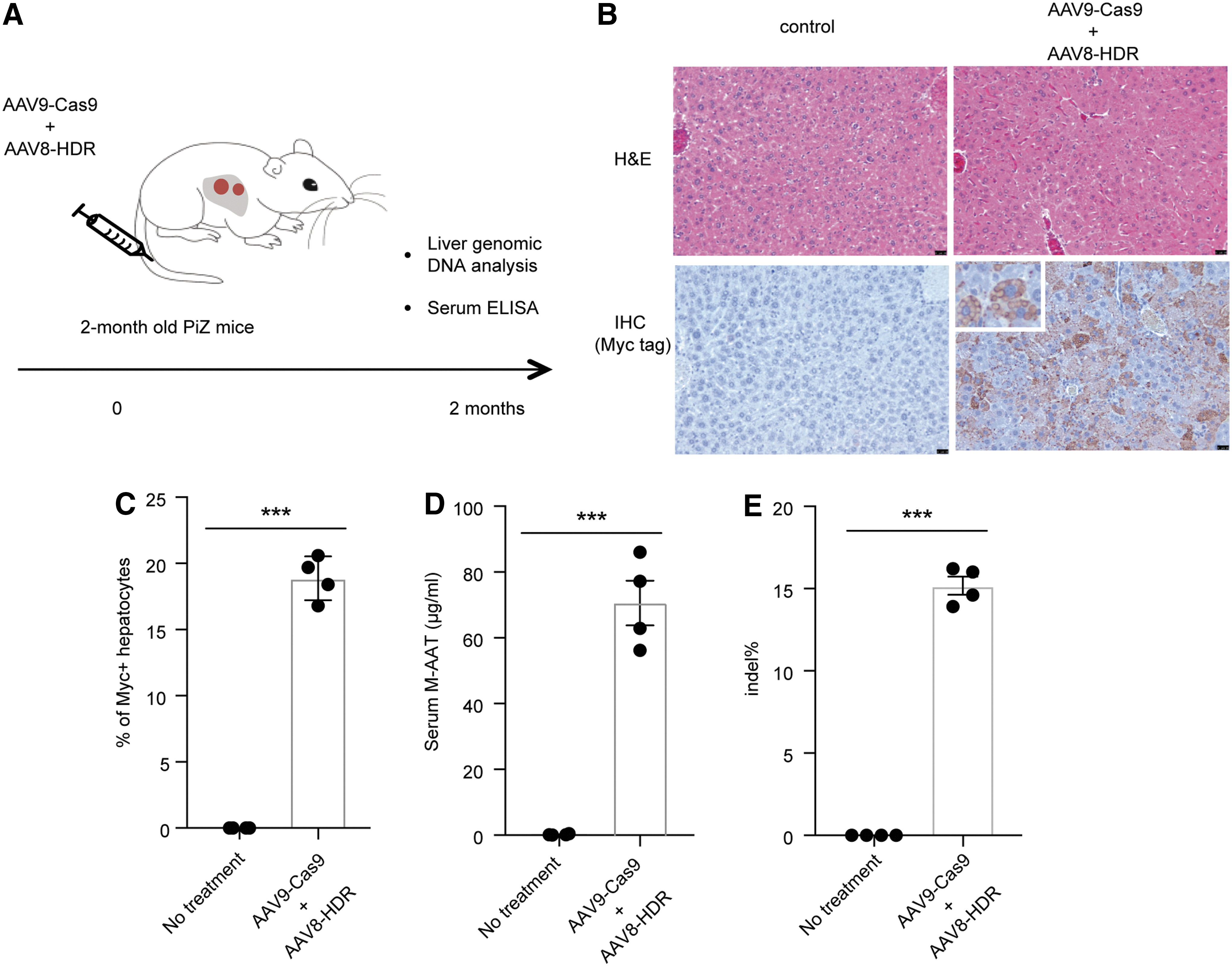
In vivo delivery of Cas9 and AAV-HDR template in adult PiZ mice partially restore M-AAT in the serum.
Discussion
Therapeutic editing has broad potential to treat a range of diseases through permanent correction of genetic mutations. We demonstrated that Cas9 delivered in tandem with sgRNA/HDR template using AAV vectors can correct the common Z-AAT mutation and partially restore M-AAT in the serum in both day-7 pups and 2-month-old adult PiZ mice. HDR was confirmed by deep-sequencing analysis. In line with a previous work demonstrating the application of CRISPR/Cas to correct the PiZ mutation in human cells, 27 our study is a proof of concept for CRISPR-mediated correction of a human-derived AAT mutation in a mouse model.
While serum levels of M-AAT failed to reach the suggested therapeutic threshold, our CRISPR AAV vectors offer a platform that may be further optimized to increase the efficiency of gene correction. A 30-nt Myc tag in the HDR template enabled the careful quantification of serum M-AAT levels by ELISA to assess successful M-AAT correction. Up to 85 μg/mL M-AAT in the serum can be detected after only a single round of treatment. Although the M-AAT level in treated mice is only 8–12% of the 570 μg/mL threshold, further improvement of HDR efficiency may be achieved by leveraging strategies to optimize HDR template design and nonhomologous end joining pathway inhibition. 28 The partial restoration of M-AAT levels in the blood serum of CRISPR-treated mice suggests that this approach may be effective in the treatment of AATD-related lung disease. We have not formally excluded MYC tag expression from nonintegrated or randomly integrated rAAV vector sequences mediated by the minimal inverted terminal repeats (ITR) promoter activity. The MYC tag levels we observed far surpass either the levels of ITR-mediated expression observed in the past in the liver or the levels of random-site integration of rAAV vectors previously reported. 29 Notably, the sgAAT targeting exon 5 may generate truncated AAT instead of depleting PiZ, preventing the investigation of liver phenotype in this study (Fig. 2A). Shen et al. 30 (co-submitted) showed that sgAAT targeting exon 2 can effectively deplete Z-AAT in PiZ mice, suggesting that CRISPR targeting exon 2 also improves Z-AAT liver disease. More effective sgRNA may help increase the gene editing and correction rate.
Previous studies have reported that the ratio of hepatocytes expressing gene-corrected protein can be higher than the ratio of HDR DNA level. 22 Because PiZ mice have ∼16 copies of Z-AAT transgenes, 9 only one corrected allele per dipoid or polyploid hepatocyte cell is sufficient to yield an M-AAT positive cell. It is also possible that a fraction of M-AAT was secreted to neighboring hepatocytes and was detected by immunohistochemistry. Future work is required to determine whether multiple copies of the PiZ transgenes help to increase HDR rate in this model compared with an endogenous gene locus.
It would seem likely that a higher efficiency of gene editing would be required to reverse or prevent AAT liver disease than to treat lung disease, since PiZ AAT acts within the cells that secrete it and could trigger inflammation in a subset of uncorrected cells, even if the production of PiM AAT from the corrected cells were sufficient to protect the lungs from protease-mediated injury. Offsetting this consideration is the fact that the introduction of indels capable of inactivating the PiZ allele could prevent expression of PiZ AAT and thus protect hepatocytes from injury even if HDR does not occur.
In addition to optimizing the CRISPR system, future studies should address the safety profiles of AAV and CRISPR. For example, CRISPR can induce off-target indels, and Cas9 has potential immune response in vivo. 31,32 The livers of AATD patients with PiZ alleles are overburdened by PiZ accumulation and consequently more prone to inflammation and to the development of hepatocellular carcinoma. 33 Since AAV integration has also been linked to an increased risk of hepatocellular carcinoma in some rodent models, 34 any increase in risk due to CRISPR-mediated mutagenesis also bears careful examination. Thus, the safety of AAV and CRISPR must be determined especially carefully in order to further evaluate the translational applicability of this platform.
Because AAV is a clinically relevant delivery vehicle, this study provides proof of principle for correction of AATD mutations in a mammalian system. The sgRNA for human AAT and related rAAV vectors will be valuable for future studies. Because AAV serotypes can target a wide range of tissues besides liver, our approach has broad basic research and clinical applications for other genetic diseases. In this study, we applied the combination of AAV8 and AAV9, future studies are required to test other AAV serotype combinations. In summary, our delivery vehicles and genome editing tools will provide a blueprint to guide future development of safe and effective CRISPR-mediated AAT gene therapy.
Footnotes
Acknowledgments
C.Q.S., G.G., C.M., T.R.F., and W.X. designed the study. C.Q.S., D.W., T.J., K.O.C., Q.T., and H.Y. performed experiments and analyzed data. C.M. helped design gene editing constructs, experimental design, data interpretation, manuscript writing and reagents. C.Q.S., K.O.C., T.R.F., and W.X. wrote the manuscript with comments from all authors.
We thank C. Mello, P. Zamore, A. Keeler, and E. Sontheimer for their insightful comments. We thank Y. Liu and E. Kittler in the UMass Medical School Morphology and Deep Sequencing Cores for support. This work is supported by grants from the National Institutes of Health: DP2HL137167 (to W.X.); P01HL131471 (to T.R.F.,W.X.,G.G. and C.M.); R01 DK098252 (to T.R.F and C.M); and R24OD018259 (to C.M). W.X. was supported by the Lung Cancer Research Foundation, American Cancer Society (129056-RSG-16-093), Hyundai Hope on Wheels, and ALS Association. K. O'Connor was supported on grant T32 GM107000 from the National Institute of General Medicine, National Institutes of Health awarded to the UMMS Medical Scientist Training Program.
Author Disclosure
G.G. is a founder of Voyager Therapeutics and holds equity in the company. G.G. is an inventor on patents with potential royalties licensed to Voyager Therapeutics and other biopharmaceutical companies. T.R.F. is a paid consultant for Beam Therapeutics and a scientific founder of AGTC and ApicBIO, and previously has been a consultant to Editas Medicine, all of which may have interest in therapies for AAT deficiency. C.M. is CSO and scientific founder of Apic Bio and SAB member of Gemini Therapeutics. W.X. is a consultant for the Cystic Fibrosis Foundation Therapeutics Lab.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.