
Abstract
Alpha-1 antitrypsin deficiency (AATD) is a hereditary liver disease caused by mutations in the SERPINA1 serine protease inhibitor gene. Most severe patients are homozygous for PiZ alleles (PiZZ; amino acid E324K), which lead to protein aggregates in hepatocytes and reduced circulating levels of AAT. The liver aggregates typically lead to fibrosis, cirrhosis, and hepatocellular carcinoma, and the reduced circulating AAT levels can lead to emphysema and chronic obstructive pulmonary diseases. In this study, two CRISPR/Cas9 gene editing approaches were used to decrease liver aggregates and increase systemic AAT-M levels in the PiZ transgenic mouse. In the first approach, AAT expression in hepatocytes was reduced more than 98% following the systemic delivery of AAV8-CRISPR targeting exon 2 of hSERPINA1, leading to reduced aggregates in hepatocytes. In the second approach, a second adeno-associated virus, which provided the donor template to correct the Z mutation, was also administered. These treated mice had reduced AAT expression (> 98%) and a low level (5%) of wildtype AAT-M mRNA. Taken together, this study shows that CRISPR gene editing can efficiently reduce liver expression of AAT-Z and restore modest levels of wildtype AAT-M in a mouse model of AATD, raising the possibility of CRISPR gene editing therapeutic for AATD.
Introduction
A
Transgenic mouse models expressing the human SERPINA1-Z loci were developed 10,11 to aid disease pathophysiology investigation 6,10 –12 and evaluate novel therapeutics. 13,14 In these mice, the complete 14.4 kb of hSERPINA1 locus was isolated from a PiMZ (heterozygous of the wildtype allele PiM and mutant allele PiZ) individual to generate a few transgenic founders, one of which is PiZ-11.03 (the PiZ mice). 10,11 This PiZ transgenic mouse model harbors 16 copies of the human SERPINA1 (hSERPINA1) transgene and recapitulates the progression and characteristics of AATD liver diseases, including AAT aggregation in rER of hepatocytes, fibrosis, cirrhosis, and consequential HCC. 10 Young PiZ mice have high circulating levels of human AAT-Z and abundant retention of misfolded AAT-Z proteins in hepatocytes. 13 Heterozygous PiZ mice carry eight copies of hSERPINA1 genes and the concentration of circulating human AAT is similar to that in PiZZ patients. Older mice accumulate AAT-Z protein in hepatocytes, thereby stimulating ER stress and apoptosis. Hepatocytes with less AAT-Z burden and damage are predisposed to more regenerative stimuli to compensate for injured cells, increasing the risk of HCC over time. 12 Because the endogenous mouse Serpina1 loci are present in PiZ mice, they do not manifest any lung abnormalities characteristic of AATD in human.
Therapeutic options are limited for AATD patients. A small fraction of newborn patients and patients at the advanced stage of liver disease undergo liver transplant. Although there is no reliable clinical prediction of lung diseases, natural history studies revealed that AATD patients with liver symptoms are at high risks of developing lung emphysema in their late 50s or 60s. Currently, AAT protein augmentation therapy is the standard of care for AATD lung emphysema in the developed world. 1,2,15 This treatment involves weekly infusion of AAT proteins purified from healthy blood donors. Novel therapeutics, including weekly-dosing of small interfering RNA (siRNA) or antisense oligonucleotides (ASOs), have demonstrated promising preclinical and clinical trial results in reducing the AAT-Z load in hepatocytes. 13 More recently, gene augmentation therapy using adeno-associated virus (AAV) vectors have shown sustained AAT expression at 2.0–2.5% of the target level after intramuscular injections of recombinant viral vectors in the absence of severe adverse effects in patients. 16,17
In this study, two CRISPR/Cas9 gene editing approaches were used to decrease liver aggregates and increase systemic AAT levels in the PiZ transgenic mouse. In both approaches, Staphylococcus aureus (saCas9) was used because the reduced coding sequence compared with Streptococcus pyogenes Cas9 (spCas9) allows packaging of Cas9 and single-guide RNA (sgRNA) expression cassettes into a single AAV8 vector. In the first approach, AAT expression in hepatocytes was reduced more than 98% following the systemic delivery of AAV8-CRISPR targeting exon 2 of SERPINA1, leading to reduced aggregates. In the second approach to correct the Z mutation (E342K) in vivo, saCas9 is expressed in the first AAV vector and a mutant-allele specific sgRNA was packaged together with a donor template targeting the Z mutation in the second AAV. These treated mice had reduced AAT expression (>98%) and a low level (5%) of wildtype AAT-M mRNA. Our results demonstrated the potential of different gene editing strategies in treating genetic diseases in the liver.
Materials and Methods
Construction of AAV vectors
Plasmids containing the thyroxine binding globulin (TBG) promoter, EFS promoter (short form of elongation factor-1α promoter), U6-driven sgRNA, saCas9, nuclear localization signals (NLSs), a triple flag tag, and homology arms were synthesized. These components were cloning into the pTR-UF-11 plasmid (Catalog No. MBA-331; ATCC) to obtain the all-in-one AAV8-CRISPR or AAV-homology directed repair (HDR) vectors flanked by inverted terminal repeats (ITRs) from AAV2. Multiple cloning sites at the junction of each components allow for subcloning of different elements in this AAV8-CRISPR vector system. All AAV vectors were designed and cloned to be within 4.7 kb packaging limit including both ITRs. The proviral vectors were packaged in to capsids from AAV8 using the “triple-transfection” method at Vector Core, University of Massachusetts Medical School. Recombinant AAV8 vectors were purified by two rounds of CsCl-gradient ultracentrifugation and titered by quantitative PCR and digital droplet PCR using primers and probes specific to the transgenes.
Animal experiments
PiZ mice were bred and maintained in an AAALAC-accredited facility at Saint Louis University School of Medicine as described previously, 10 and ALL procedures were in accordance with the animal protocol approved by the Institutional Animal Care and Use Committee. Mice homozygous for the human PiZ alleles were bred with C57B6 males (Jackson Laboratories) to obtain heterozygous offspring. All studies were conducted with 6- to 16- week-old heterozygous mice. Dosing groups were designed to include at least one PBS control animal in each litter of animals.
Quantification of editing efficiencies
Genomic DNA was extracted from transfected cells or liver pulverized by a Geno/Grinder (SPEX SamplePrep) using DNAdvance Kits (Agencourt) on a liquid-handling robot per manufacturer's instructions. To quantify indel percentage at target genomic loci, T7 Endonuclease I Assays (M0302, New England BioLabs) were conducted as described previously 1 using the PCR primers listed in Supplementary Table S1. Next-generation sequencing was carried out on a MiSeq System (Illumina) and analyzed to assess the specific DNA disruptions in detail.
Quantitative real-time PCR and RNA sequencing
Total RNA was extracted from pulverized liver samples using RNeasy Mini Kits (Qiagen). RNA concentration and integrity were assayed using RNA 600 Nano Kits (Agilent) on a bioanalyzer (Agilent) to determine an RNA integrity number (RIN) greater than 6.5. RNA was treated Turbo DNA-free Kit (Ambion) and reverse-transcribed using Superscript 3 RT (Invitrogen). Quantitative real-time PCR (qRT-PCR) was performed on a BioRad CFX384 with SYBR Green I Master (Roche) and primers as listed in Supplementary Table S1. Human SERPINA1 transcription was normalized to mouse beta-2-microglobulin (B2M) transcription levels and then to the corresponding value from the PBS-treated littermate.
RNA-sequence (RNA-seq) library preparations and HiSeq runs were conducted at Genewiz. Ribosomal RNA depletion was performed prior to stranded RNA library preparation. To be able to map RNA-seq reads to the mouse genome and to the human SERPINA1, we constructed a new reference genome by adding the sequence of human chromosome 14, containing hSERPINA1, to the mouse mm10/GRCm38 reference assembly. Likewise, we added the hSERPINA1 annotations in Gencode Release 24 to the mouse annotations in Gencode Release M11. We used the STAR aligner (PMID: 23104886) to generate genome indexes and align the reads. Default options were used, except for –outFilterMismatchNmax 100, to allow alignment of reads with indels and mismatches to the reference. We used pysamstats, v 0.24.2; pysam, v 0.8.4; and samtools, v 1.3 (PMID: 19505943) and custom python code to extract the rates of insertions, deletions, and HDR events around the cut sites. To count mapping reads to the different genes we used the Bioconductor package GenomicAlignments (PMID: 23950696), using the union mode in the summarizeOverlaps function. We calculated fold changes in gene expression using the regularized logarithm transformation in DESeq2 (PMID: 25516281).
Liver histology, PAS-D staining, and immunohistochemistry
Liver tissues were collected, fixed in 10% buffered formalin, embedded in paraffin, and sectioned into 4 um slices. Sections were stained with periodic acid-Schiff-diastase (PAS-D) to visualize AAT aggregation in hepatocytes, followed by quantitation of globule counts and total globule area using ImageJ (W.S. Rasband, National Institutes of Health). Three random areas were imaged and analyzed for each liver sample. Slides of hematoxylin and eosin staining were also imaged to visualize general morphology and AAT aggregates.
Plasma chemistry analysis by enzyme-linked immunosorbent assay
Blood samples were collected by tail nicking at interval time points and by cardiac puncture at terminal time points. Blood was further processed into serum and kept frozen at −80°C. Human AAT ELISA Kit (ab108799; Abcam) was used to measure the absolute concentrations of human AAT in serum samples from the PiZ transgenic mice. Briefly, serum samples were thawed on ice and diluted in supplied assay buffer containing 0.01% naïve C57BL/6 mouse serum. A standard curve was prepared by reconstituting the supplied recombinant A1AT as per the supplied protocol to 100 ng/mL and serial dilutions to 0.137 ng/mL. After antibody incubation and exposure steps, the plate was read on an Envision (Perkin Elmer, Santa Clara, CA) at the absorbance of 450 nm. A standard curve was calculated fitted to a four-parameter sigmoidal curve (GraphPad Prism, La Jolla, CA) using the background corrected absorbance values for each standard. For each serum sample, the dilution(s) lying in the linear range of the standard curve were extrapolated to generate raw values. The raw values were adjusted by multiplying by the dilution to give the actual value in serum in ng/mL. If there were two values in the linear range, both values were calculated and then averaged.
Protein expression analysis by Western blotting
Soluble and insoluble fractions of AAT was isolated as previously described. 18 Briefly, liver tissues were homogenized with chilled Dounce homogenizer and sheared by passing through a 28-gauge needle. Total protein content was determined to aliquot 5 μg of total liver lysate from each sample. After centrifugation at 10,000 g for 30 minutes at 4°C, supernatant was immediately separated from the insoluble layer with care. Both fractions were resuspended and denatured prior to being loaded on SDS-PAGE gels. Human AAT protein was probed using a goat anti-human alpha-1 antitrypsin antibody (DiaSorin, Saluggia, Italy) and a horseradish peroxidase conjugated rabbit anti-goat immunoglobulin G antibody (Agilent). Mouse GAPDH was probed as loading control. Scanned blots were analyzed using ImageJ (W.S. Rasband, National Institutes of Health).
Statistics
Two-tailed Student t-tests were performed using Excel and GraphPad Prism 7 (GraphPad). Statistical significance was assumed with a p-value ≤0.05 (*), ≤0.01 (**), ≤0.001 (***), and ≤0.0001 (****). Error bars represent s.e.m. unless noted otherwise. Group size (n) represents biological sample size.
Results
Identification of AAV8-CRISPR vectors that disrupt hSERPINA1 loci in PiZ transgenic mice after systemic delivery
In order to deliver the CRISPR/Cas9 system in an all-in-one AAV vector, saCas9 was used in the current study due to the more compact coding sequence relative to that of spCas9. 19 SaCas9 sgRNAs targeting hSERPINA1 exons were first identified in silico using a proprietary sgRNA design program with protospacer adjacent motif (PAM) sequences of NNGRRT or NNGRRV. To disrupt the transcription of hSERPINA1 transgenes in the PiZ mice, sgRNA prediction was carried out using the coding sequence of exon 2 and 25 bp flanking regions on either end as region of interest. Candidate sgRNA target sequences were then ranked based on predicted off-target sites in the human genome and their locations relative to the start codon of hSERPINA1. During in silico off-target prediction, genome assembly GRCh38/hg38 and GRCh38/mm10 were used for human and mouse genome reference, respectively. Twenty-three sgRNAs were identified with targeting sequences of 22-nucleotide (nt) in length that lacked predicted off-target sites containing zero, one, or two mismatches. These twenty-three hits were tested for their relative efficiency at cutting hSERPINA1 DNA sequences in two human cell lines, HEK293 and Hep3B (Supplementary Fig. S1). SgRNA-333 with an NNGRRV PAM was selected for subsequent in vivo experiments based on cutting efficiency and distance from the start codon on exon 2 (Fig. 1a).
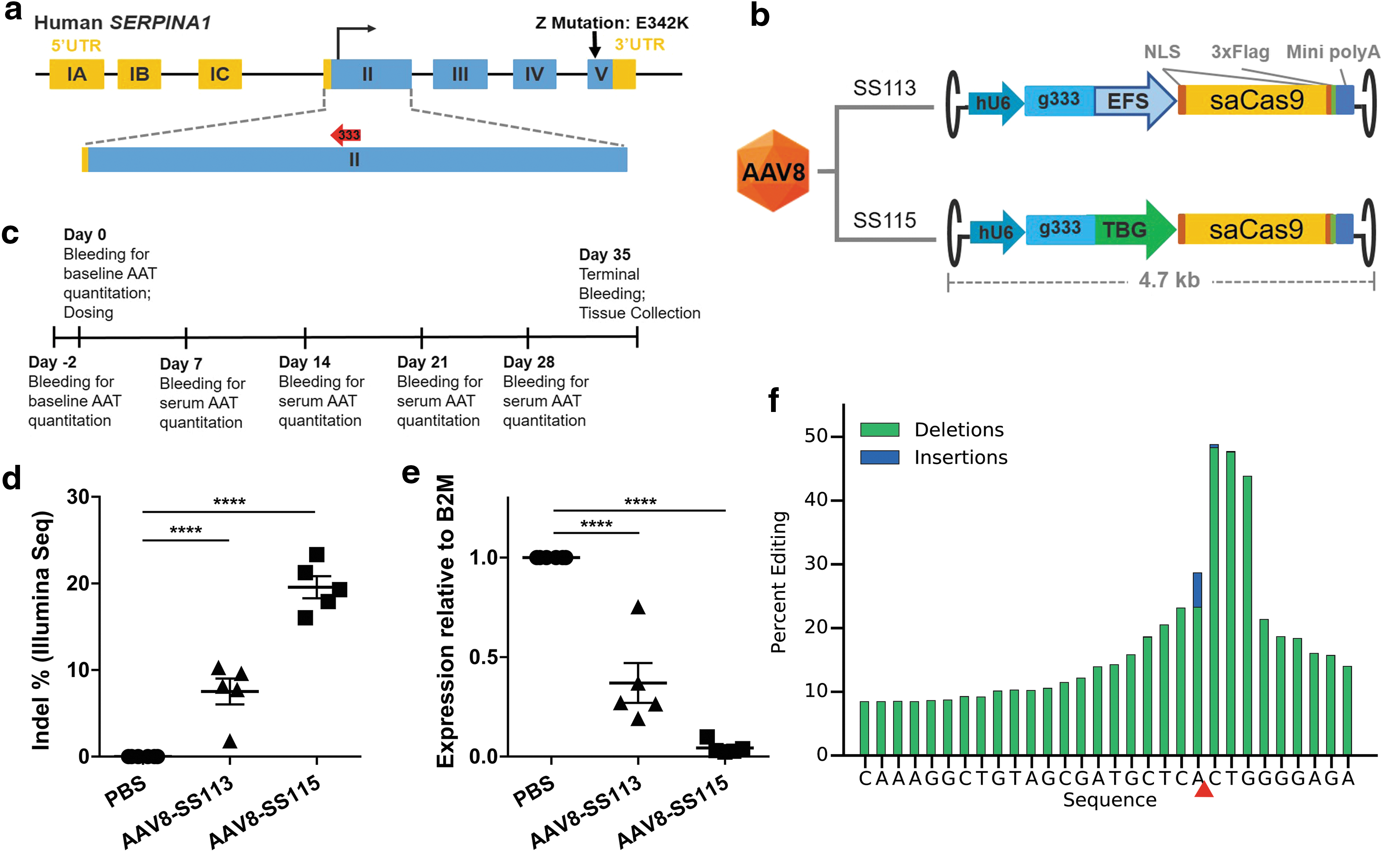
Gene disruption of human serine protease inhibitor hSERPINA1 loci by AAV8-CRISPR in adult PiZ transgenic mice.
AAV8 is a potent naturally occurring AAV serotype capable of transducing liver in multiple animal species, from mice to dogs to nonhuman primates. 20 –24 To construct a CRISPR experimental medicine based on AAV8, the complete transgenes of saCas9 and sgRNA expression cassettes are flanked by ITRs from wildtype AAV2 genome and pseudotyped into AAV8 capsids (Fig. 1b). Two AAV8-CRISPR vectors were constructed. In both vectors, the sgRNA is expressed from a U6 promoter, and the coding sequence of saCas9 is flanked by two NLSs and a C-terminal triple Flag tag. In AAV8-SS113, saCas9 coding sequence is driven by a constitutive EFS promoter, whereas in AAV8-SS115, saCas9 is expressed off a hepatocyte-specific TBG promoter. Both AAV8-CRISPR vectors are within the 4.7 kb packaging limit of recombinant AAV vectors.
Homozygous PiZ transgenic mice were bred with wildtype C57B6 mice to obtain heterozygous offspring with 8 copies of hSERPINA1-Z in each diploid genome. Mixed-gender PiZ mice at the age of 5–14 weeks were first bled on day −2 and 30 minutes prior to dosing to obtain the baseline values of serum human AAT (hAAT) concentrations prior to any intervention. Next, the animals were administered with PBS, 5 × 1013 vg/kg of AAV8-SS113, or 5 × 1013 vg/kg of AAV8-SS115 through bolus tail vein injections. Mice were bled weekly throughout the in-life portion of the experiments. On day 35 post injection, animals were euthanized, and liver samples were processed for molecular biology and histology analysis. The experimental procedure is outlined in Fig. 1c.
We first analyzed genome modifications in treated mice at day 35 post injection. Genomic DNA extracted from the homogenized livers were analyzed using next generation sequencing (NGS). AAV8-SS113 (EFS) and AAV8-SS115 (TBG) caused indel rates of 8% and 20%, respectively, near the saCas9 cut site corresponding to sgRNA-333 (Fig. 1d). To determine how these editing rates affected AAT-Z expression, hSERPINA1 transcription level in total RNA extracted from these PiZ mouse liver lysates was determined by qRT-PCR. The primer and probe set amplifies the junctions between exons 3 and 4 of hSERPINA1, downstream from the saCas9 cut site. The threshold cycle (C t ) value of hSERPINA1 were first normalized to the corresponding C t value of mouse B2M from the same animal to obtain ΔC t . Then the ΔC t value from each AAV8-CRISPR-treated mouse was normalized to the ΔC t value from the corresponding littermate treated by PBS to obtain ΔΔC t . Ten to the order of ΔΔC t represents fold changes of hSERPINA1 expression. It is believed that indels introduced by CRISPR/Cas9 system yield frame-shifts and downstream premature stop codons, which then leads to nonsense mediated decay of mRNA transcripts. These qRT-PCR results showed that AAV8-SS113 (EFS) and AAV8-SS115 (TBG) reduced hSERPINA1 transcription levels by 63% and 96%, respectively, compared to control animals. Transcription results were further supported by whole-transcription RNA-seq analysis on representative samples. By comparing the fraction of total RNA reads mapped to hSERPINA1 transcript, AAV8-SS113 (EFS) and AAV8-SS115 (TBG) reduced hSERPINA1 transcription levels by 58% and 93%, respectively, compared to control animals (Supplementary Table S2).
Analysis of the surviving RNA-seq reads around the cut site revealed high levels of indels (Fig. 1f and Supplementary Table S2). For instance, in one animal treated with AAV5-SS115, 10% of remaining hSERPINA1 transcripts had an indel rate of ∼50%. These molecular analyses suggest that AAV8-CRISPR targeting the early exon of hSERPINA1 knocks down AAT-Z protein expression by disrupting the coding sequences.
AAV8-CRISPR treatment reduced AAT-Z proteins in circulation and hepatocytes of adult PiZ transgenic mice
The effect of AAV8-SS113 (EFS) and AAV8-SS115 (TBG) on AAT expression in mice was determined. We first measured circulating levels of hAAT-Z protein as an indicator of hAAT-Z synthesis in the hepatocytes at several times predose and postdose. An ELISA assay with a human-specific AAT antibody was performed to eliminate interference of mouse AAT in the PiZ mice. By day 7 post dose, mice treated with AAV8-SS115 had reduced circulating AAT (69%) that was further reduced by day 14 (95%) and remained stably reduced (>98%) throughout the 35 days of the experiment (Fig. 2a). Mice treated with AAV8-SS113 showed a similar temporal reduction in AAT that reached a maximal reduction of 44% at the end of study on day 35 (Fig. 2a).
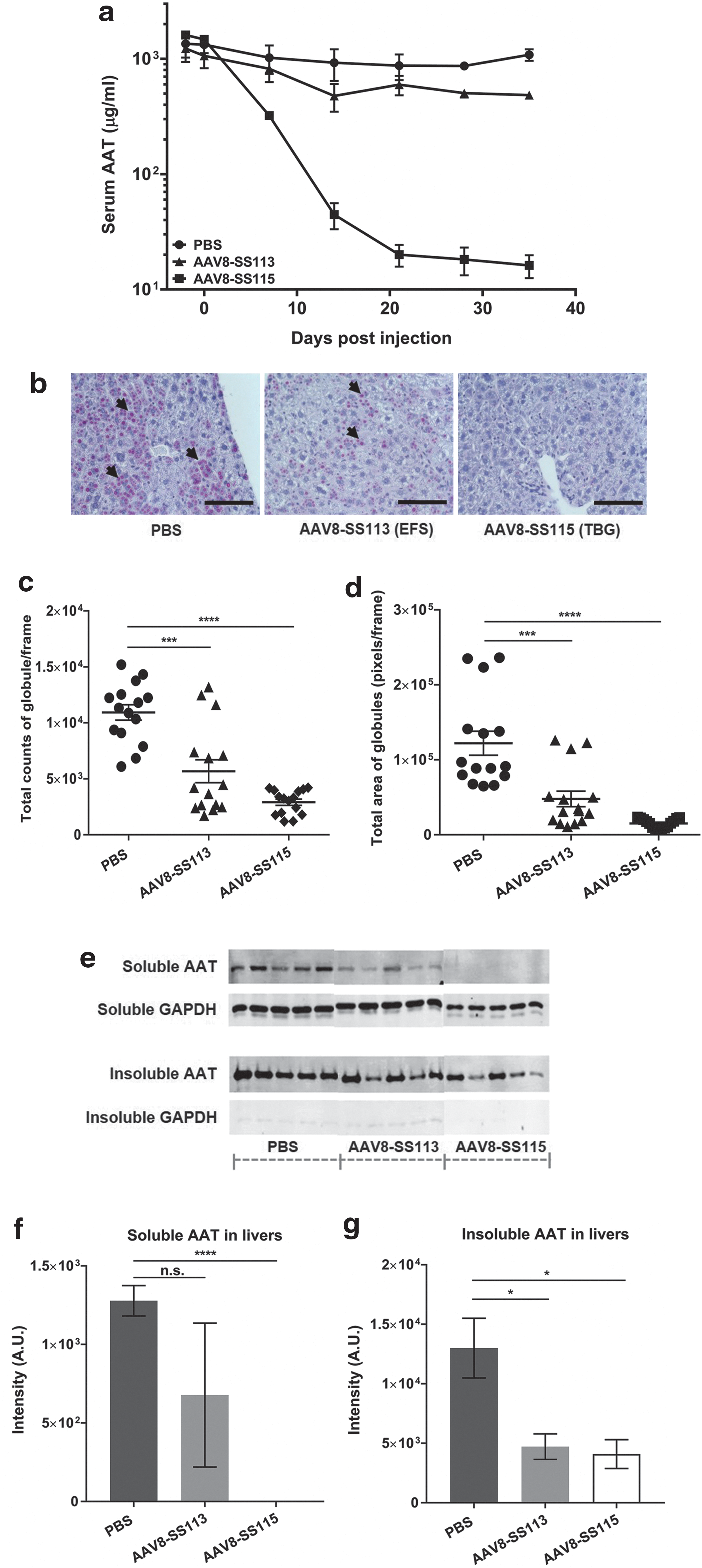
Knocking down hSERPINA1 transcription with AAV8-CRISPR ameliorates the liver phenotypes in adult PiZ transgenic mice. (
To further investigate the effects of our experimental therapeutics, histology analysis of AAT-Z in hepatocytes was conducted. AAT proteins are highly glycosylated and, therefore, can be visualized using PAS-D staining that reacts with the hydroxyl groups on glycans. Positive PAS-D staining in PBS-treated mice are identified as punctuated purple dots under white-field microscope (Fig. 2b and Supplementary Fig. S2). In PiZ transgenic mice, AAT globules form in the livers after birth and grow in size and quantity over time. By 4 weeks of age, clusters of hepatocytes could be detected by PAS-D staining. In the adult mice we treated, the terminal collection was performed around age of 10–19 weeks. Three representative images are taken from the PAS-D staining of one liver section for each animal to quantify the number and sizes of PAS-D positive areas. Compared to the PBS group, the treatment groups showed dramatic changes in the total number and sizes of AAT globules as analyzed by ImageJ (Fig. 2c and d). Specifically, AAV8-SS113 (EFS) and AAV8-SS115 (TBG) treated mice had a 52.2% and 73.4%, respectively, reduction in total counts of positive globules and 39.1% and 87.6%, respectively, reduction in the total area of globules in each histology image. Encouragingly, no change was observed in hepatocyte morphology, aspartate aminotransferase levels, or circulating albumin levels after AAV8-CRISPR treatment, indicating minimal liver toxicity (Supplementary Fig. S3).
We then separated the soluble and insoluble fractions of the homogenized PiZ livers to differentiate secreted and aggregated forms of AAT-Z in the PiZ livers (Fig. 2e). As measured by Western blotting with a human AAT specific antibody, AAV8-SS113 (EFS) and AAV8-SS115 (TBG) treated mice had 47% reduction and complete abrogation of, respectively, soluble AAT (Fig. 2f), and 63.7% and 68.5% reduction of, respectively, insoluble AAT (Fig. 2g) in PiZ liver lysates. The differences in AAT protein reduction in the liver by AAV8-SS113 and AAV8-SS115 corporate with the reduction of AAT protein in circulation – 44% by AAV8-SS113 (EFS promoter) and >98% by AAV8-SS115 (TBG promoter). Taken together, these results show that the CRISPR/Cas9 system delivered by AAV8 vectors elicits early onset and prolonged hSERPINA1 inhibition in PiZ mice that significantly reduces the AAT protein in both circulation and hepatocytes.
A dual-AAV vector system introduces targeted gene correction of the Z mutation in exon 5 of hSERPINA1 in vivo
To address the unmet medical need from lung symptoms in addition to the liver symptoms in AATD patients, we designed a gene editing strategy where a HDR donor template to correct the Z mutation was co-delivered with the gene editing viral vectors that disrupt the SERPINA1 near the Z mutation.
We first designed in silico sgRNAs targeting a region covering 300 bp centered at the Z mutation on exon 5. Previous reports claimed that HDR correction frequencies peak near the DNA double-stranded breaks regardless of the forms of donor template. Therefore, candidate hits of targeting sequences were ranked by their proximity to the Z mutation and by in silico off-target predictions. Twenty candidate hits were further tested in Hep3B cell culture and sgRNA-1889 was selected to be packaged into AAV vectors for the in vivo studies based on its cutting efficiency and close proximity between its cut site and the Z mutation (Supplementary Fig. S4). Given the packaging limits of AAV, the experimental therapeutic consisted of two AAV viruses (Fig. 3a): the first vector, AAV8-Cas9, encoded TBG-driven saCas9 flanked by NLS signals and then followed by a triple Flag-tag and a full-length bovine growth hormone poly A tail; the second AAV vector delivered U6-driven mutant-allele-specific sgRNA-1889 and the HDR donor template targeting the Z-mutation. We tested two HDR template configurations since an unbalanced design of single-stranded oligonucleotide as donor template was previously shown to enhance HDR frequencies in cell culture. 25 However, no investigation in how asymmetricity of AAV-delivered HDR templates impacts HDR efficiency has been reported, especially in an in vivo setting. One configuration, AAV8-MES06, contained homology arms of 1.7 kb in length on either side of the PiZ mutation, whereas the other configuration, AAV8-MES08, shifted the balance of the homology arms to 60% of sequence on the 5′ end and 40% on the 3′ end relevant to the PiZ mutation (2.1 kb +1.4 kb) (Fig. 3a). In addition, the donor template contained 10 point mutations. One mutation reverts the A-to-G mutation to restore the Z mutation to the wildtype M nucleotide; two silent mutations on the PAM domain and seven on the target sequence prevent sgRNA recognition of the same genomic loci after HDR correction; and two of the seven silent mutations on the target sequence also introduces an MfeI site to enable restriction fragment length polymorphism assay.
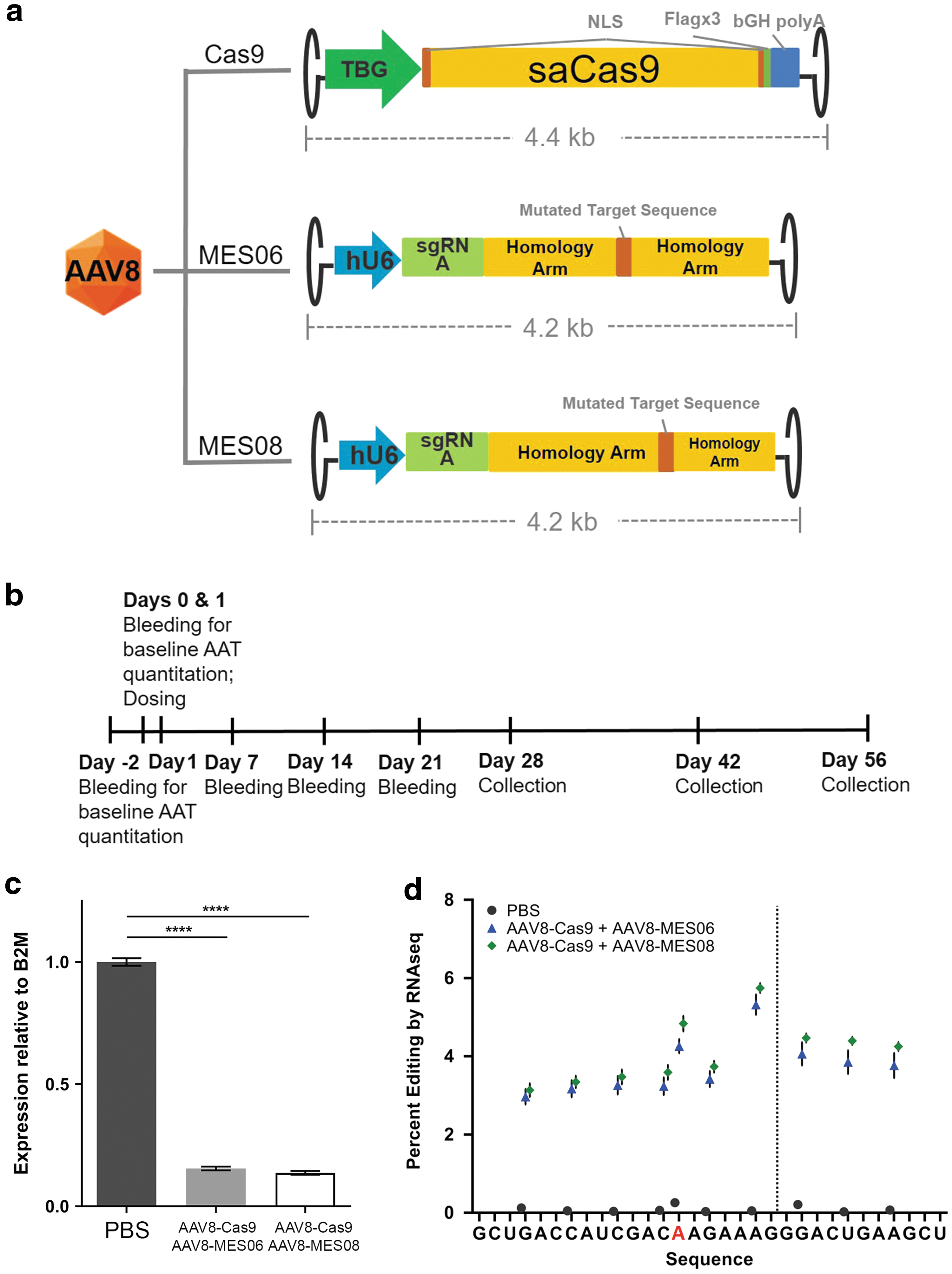
A dual-vector AAV8-CRISPR system corrects the Z-mutation in adult PiZ transgenic mice. (
To test these vectors and our hypothesis, mixed-gender, heterozygous PiZ mice at age of 5–11 weeks were dosed in four groups: (1) PBS control, (2) 5 × 1013 vg/kg of AAV8-Cas9 on Day 0 and 1.28 × 1014 vg/kg of AAV8-MES06 on Day 1, (3) 5 × 1013 vg/kg of AAV8-Cas9 on Day 0 and 1.28 × 1014 vg/kg of AAV8-MES08 on Day 1. The dose of AAV8-Cas9 vectors was the same as previous AAV8-Cas9 studies to ensure sufficient Cas9 expression in the liver. The dose of AAV8-MES06 or AAV8-MES08 was 2.56 × of AAV8-Cas9 to provide excess HDR donor templates. Animals were bled on days −2, 0 (3 hours prior to dosing), 7, 14, 21, 28, 35, 42, 49, and 56 to collect serum for AAT ELISA. Cohorts of mice from each treatment group were euthanized on Days 28, 42, and 56 to investigate the kinetics of therapeutic effects in hepatocytes by histology (Fig. 3b).
The 3.5 kb homology templates delivered by AAV8 imposed challenges on the analysis of gene loci near the Cas9 cut site by PCR amplification and subsequent sequencing assays. At a dose of 1.28 × 1014 vg/kg, it is estimated that there are more than 500 viral genome copies per quadruploid hepatocytes immediately after intravenous injections. For this reason, PCR amplification of 500 bp fragments for Illumina sequencing from genomic DNA will be difficult in the presence of >500 copies of highly homologous sequences. To overcome this problem, we depleted total DNA from RNA preparations and then conducted whole-transcriptome RNA-seq of the total RNA extracted from PiZ mouse livers at day 56 post injection. RNA-seq data revealed that the total hSERPINA1 expression is significantly reduced (by 85%) relative to mouse B2M in mice treated with the dual-AAV vector system. By contrast, hSERPINA1 expression decreased by 35% in the AAV8-MES06 control group. Next, we investigated the specific nucleotide changes on the targeting sequence and PAM in the hSERPINA1 mRNA. All 10 intended point mutations on the donor templates were detected from read alignments. Consistent with previous reports, the highest nucleotide substitution frequency occurs near the cut site at ∼6% of the total hSERPINA1 transcripts. The nucleotide responsible for Z-to-M transition (A > C substitution) showed 4.3% correction with AAV8-Cas9 + AAV8-MES06, and 4.8% correction with AAV8-Cas9 + AAV8-MES08 (Fig. 3d). The rest of eight mutations occur at frequencies of 3–4%, slightly decreasing for positions further away from the cut site. These results indicate that 4.3% to 4.8% of the remaining AAT transcripts from the PiZ mouse livers are edited to the more active M form of AAT at 56 days post the dual-vector injections in adult PiZ mice.
The dual-AAV vector system reduces AAT-Z expression in PiZ mice by disrupting exon 5 of hSERPINA1 in adult PiZ mice
Because of the expression of mouse AAT from the endogenous Serpina1 loci, these humanized transgenic PiZ mice do not manifest any lung emphysema symptoms. 10,11,13 Therefore, pulmonary function tests do not provide any insights in this current study. However, since majority of the gene modifications in exon 5 induced by the dual-AAV vector system are non-homologous end joining (NHEJ) events, we sought to investigate the pharmacological effects in these adult PiZ mice. First, a human-specific AAT ELISA was performed to understand the kinetics of effects in terms of circulating AAT. Consistent with previous observations, AAT levels were reduced by more than 99% in the serum mice treated with the combination of AAV8-Cas9 and AAV8-MES06 or AAV8-Cas9 and AAV8-MES08 (Fig. 4a). Due to lack of antibodies specific to AAT-M and the low abundance of the corrected AAT-M in circulation, we were not able to analyze what percentage of circulating AAT proteins were in either M or Z form.

The dual-vector AAV8-CRISPR system alleviates liver phenotypes in adult PiZ mice. (
PAS-D staining of liver sections harvested at day 56 demonstrated robust reduction in AAT globules in hepatocytes (Fig. 4b). Quantification of PAS-D staining images (Supplementary Fig. S4) revealed that both the globule counts and sizes are reduced by more than 80% on day 56 (Fig. 4c and d) in both dual-vector-treated groups, compared to either PBS group. Interestingly, the total globule counts and sizes increased from day 28 to 56 in both PBS and single-vector control groups, indicating accumulation of AAT globules in PiZ hepatocytes over time. However, this trend for globule accumulation was absent in the dual-vector treated group, suggesting long-lasting effects of gene modifications. Together, these molecular and pharmacological results suggest the potential of CRISPR/Cas9-mediated gene correction to simultaneously reduce the toxic protein load in livers and restores the expression of the functional M-AAT simultaneously.
Discussion
In this study, we demonstrated that two different gene editing approaches using the CRISPR/Cas9 system could correct the disease phenotypes in the humanized transgenic PiZ mice for AATD. The gene editing machineries, including saCas9 nuclease, sgRNA, and homology arms for DNA repair, can be delivered by recombinant AAV vectors to the hepatocytes in a tissue-specific manner. NHEJ events on either exon 2 or exon 5 significantly reduced hSERPINA1 transcription in the PiZ mouse livers, leading to robust and sustained decrease in circulating AAT-Z levels and AAT-Z aggregates sequestered in hepatocytes. In addition, we achieved modest level (4.8%) correction of the Z-mutation (E342K) by homologous recombination. This is the first report of in vivo correction of the human PiZ alleles by gene editing, and this study demonstrates the potential of developing a gene editing-based therapeutics to correct both the liver and lung disease symptoms associated with AATD simultaneously.
AATD patients with the PiZZ genotype represent the most prevalent subgroup of patients with the most severe disease manifestations and are predisposed to develop both liver diseases and early onset COPD at any age. Studies have shown a prevalence of PiZZ AATD as 1 in 1,500–5,000 individuals in the United States and Europe, estimating a total of 100,000 subjects. 26,27 Therefore, we used the PiZ genotype for our proof-of-concept study to treat AATD with the CRISPR/Cas9 technology. Results from this current study can potentially be adapted to other AATD genotypes with single-point mutations. In this report, both NHEJ and HDR approaches resulted in >98% of reduction of circulating AAT-Z protein, suggesting promising therapeutic potential for the liver diseases associated with AATD. In adult PiZ mice with globule-containing hepatocytes, significant reduction of mutant AAT-Z expression happened at 7 days post AAV-CRISPR delivery, and the repressive effect lasted throughout the duration of the in-life portion of studies. Interestingly, in the HDR approach, the majority of the gene modification outcomes is NHEJ repair at exon 5, and therefore, the AAT expression level also decreased dramatically. Although adult mice were only followed up to 10 weeks post AAV-CRISPR treatments in this study, long-term effects of AAT-Z down-regulation have been reported previously. Studies with autophagy enhancers, siRNA, and ASO technologies revealed that prolonged reduction of hAAT-Z proteins in the PiZ mice prohibits the development of hepatic fibrosis in adult mice and prevents accumulation of predisposition of liver diseases in neonatal mice. 1 Consequentially, less HCC nodules were observed in aged PiZ mice with lower hAAT-Z burden than the control groups. Similar levels of AAT knockdown were also observed in preclinical studies in nonhuman primates using RNAi (Alnylam Pharmaceuticals) and ASO (Ionis Pharmaceuticals). This phenomenon suggests that AAV8-CRISPR, capable of knocking down the mutant AAT by 100-fold, is likely to reduce the tendencies of liver fibrosis and tumorigenesis in long-term studies in transgenic PiZ mouse models.
In our attempt to develop a treatment for the AATD lung disease by correcting the Z-mutation in vivo, a smaller fraction (4.8%) of gene modification corrects the adenine to guanine, and consequentially, changes the Z allele to the wildtype M allele. The superior capability of AAV vectors in shuttling DNA into nuclei could in part explain the relatively high HDR rate when the donor templates are delivered by AAV vectors. 28 One recent publication reported 1.2% of mutation reversion in hepatocytes of adult mouse models of ornithine transcarbamylase with CRISPR/Cas9 delivery by AAV. The same study also reported a significantly higher HDR correction rate of 10% (6.7–20.1%) in the livers when AAV-CRISPR vectors were administered in neonates. 29 Active division of hepatocytes with the presence of abundant HDR donor templates might count for the exceptionally high HDR rate in this context. It would be interesting to attempt the PiZ-to-PiM correction in neonatal PiZ mice to obtain a higher gene correction rate that could potentially reach the therapeutic threshold of functional AAT-M in circulation. According to protein replacement therapy, the target serum level of AAT protein in asymptomatic PiSZ patients is 11 uM (570 μg/mL) of AAT-S and AAT-Z, which are less enzymatically active than AAT-M. Therefore, although 15–20% of gene correction might be sufficient to elevate the serum level of AAT to 11 uM in PiZZ patients, it is likely that less amount of wildtype AAT-M is necessary to functionally abrogate lung emphysema. In addition, the presence of HDR templates in the form of recombinant AAV genomes made it challenging to detect nucleotide correction at the chromosomal level. A direct measurement of nucleotide correction at the DNA level would shed light on the DNA repair pathways involved in the diseased hepatocytes at different developmental stages. Ultimately, a new transgenic mouse model with the mouse SerpinA1 genes deleted to generate a model of AATD lung disease in the correct genetic background is essential to evaluate the therapeutic effects of the HDR approach.
Another interesting observation in the study is the discrepancy between modest NHEJ rate (∼20%) at the Cas9 cut site and a high efficiency (>98%) of AAT-Z protein reduction in liver and circulation. One hypothesis is that this asymmetric gene editing might results from asymmetric transcription of hSERPINA1 loci in this mouse model. The eight copies of hSERPINA1 loci were incorporated at different chromosomal sites, some of which might be more accessible for transcription due to epigenetic modifications or chromosomal structures. It is likely that specific hSERPINA1 loci open for transcription are also prone to recognition by Cas9 ribonucleoprotein complexes, and therefore, a small fraction of editing could lead to dramatic reduction of protein expression. What makes this hypothesis more complicated is the polyploidy nature of mature hepatocytes. In both human and mice, hepatocytes are composed of cells with diploid, quadruploid, and even higher ploidy to compensate for genome toxicity. 30 The editing rates measured by DNA sequencing represent editing events on an ensemble of chromosomes from all the liver cells, but it is possible that only one hSERPINA1 alleles in each cell expresses the majority of AAT-Z protein. Therefore, a small editing rate might lead to a high reduction rate of AAT-Z in mice. In addition, the disparity of indel rate on the DNA level and the more significant protein down-regulation in serum and liver can also be attributed to AAV-CRISPR expression in a subpopulation of cells in the liver. It is plausible that only a subpopulation of AAT-producing hepatocytes are edited, especially around the central vein area. More preclinical studies with gene editing in the liver are critical in understanding the biology behind this disparity.
Preclinical and clinical development of gene augmentation therapy for AATD has accumulated promising results in animal models and patients 14,31 –33 in recent years. The small coding sequence of hSERPINA1 made gene augmentation therapy a plausible option for treating the lung diseases. On contrary, one major challenge of viral delivery of the CRISPR/Cas9 gene editing machinery was the limited size of viral vector transgenes, especially in AAV vectors (4.7–5.0 kb). The smaller coding sequence of saCas9 (3159 bp), compared to that of spCas9 (4104 bp), enables delivery of sgRNA and nuclease with their respective regulatory elements in a single AAV vector and allows for small tissue-specific promoters. The TBG promoter has been previously shown to restrict transgene expression in hepatocytes and its relative small size enables efficient AAV packaging. What is more interesting is that vectors with TBG-driven saCas9 could induce higher indel rates on target hSERPINA1 loci in vivo, compared to the ubiquitous EFS promoter. As expected, animals with higher gene editing frequency in the liver also showed more significant AAT reduction in both circulation and liver tissues.
One concern with systemic delivery of AAV8-CRISPR is the genotoxicity and immune responses induced by prolonged expression of Cas9 nucleases. A recent study comparing DNA repair profiles after spCas9 transfection in a plethora of cell lines demonstrated a nonrandom gene editing outcome distribution, leading to the conclusion that DNA repair outcomes are determined by the protospacer sequence and independent of Cas9 delivery method, cell line, or genomic context. 34 These results suggest that constitutive Cas9 expression does not alter off-target sites at a genomic level across model systems. In addition, scientists engineered molecular biology tools to prevent sustained exposure to Cas9 nucleases. For example, self-inactivating AAV-CRISPR constructs were designed to target sequences unique to the Cas9 coding sequence or AAV construct backbones in order to demolish the double-stranded episomal configurations of AAV vectors expressing Cas9. 35,36 Another study in the mouse model of Ornithine transcarbamylase deficiency took advantage of the growth and expansion of hepatocytes in the neonatal stage to diminish AAV-CRISPR vectors after correcting point mutations in diseased mice. 37 Nonviral delivery method is another mitigating approach to avoid sustained Cas9 expression in vivo. Delivery of either Cas9 ribonucleoprotein complexes or a combination of Cas9 mRNA and sgRNA allow for short exposure of DNA to gene editing machineries and, potentially, repeated dosing.
In summary, we developed two therapeutic approaches to treat diseases associated with AATD using the combination of CRISPR/Cas9 gene editing tool and AAV gene delivery vectors. In the transgenic mouse model harboring human PiZ alleles, we demonstrated >99% reduction of misfolded AAT protein in sera and hepatocytes without induction of liver toxicity. In addition, using a dual-vector system, we achieved 4-5% nucleotide correction at the Z-mutation and >99% reduction of the liver retention of AAT aggregates. Our results indicate that the gene knockdown and gene correction approaches could potentially benefit both AATD patient populations with liver or lung disease phenotypes.
Footnotes
Acknowledgments
The authors would like to thank Dr. Guangping Gao and Vector Core at University of Massachusetts Medical School for AAV vector production. We would also like to thank Tongyao Wang for her help with in silico prediction of off-target sites, Dawn Ciulla and Georgia Giannoukos for their assistance in NGS and RNAseq sample submission, Vic Myer for discussions, and Jenni A. Franey for tail vein injections. This work was supported by Editas Medicine.
Author Disclosure
S.S., M.E.S, C.J.Y., E.M., H.J., and C.F.A. are employees of Editas Medicine. E.M.C. and D.B. were employees at Editas Medicine during the course of this study. J.H.T. is a consultant to Editas Medicine.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.