
Abstract
Gene transfer targeting hematopoietic stem cells (HSC) in children has shown sustained therapeutic benefit in the treatment of genetic diseases affecting the immune system, most notably in severe combined immunodeficiencies affecting T-cell function. The HSC compartment has also been successfully targeted using gene transfer in children with genetic diseases affecting the central nervous system, such as metachromatic leukodystrophy and adrenoleukodystrophy. HSCs are also a target for genetic modification in strategies aiming to confer drug resistance to chemotherapy agents so as to reduce off-target toxicity, and to allow for chemotherapy dose escalation with the possibility of enhanced therapeutic benefit. In a trial of this strategy in adult glioma patients, significant engraftment of gene-modified HSCs expressing a mutant of the DNA repair protein O6-methyl-guanine-methyl-transferase (MGMT(P140K)) showed potential in conferring drug resistance against the combined effect of O6-benzylguanine (O6BG)/temozolomide (TMZ) chemotherapy. The aim was to test the safety and feasibility of this approach in children with poor prognosis brain tumors. In this Phase I trial, seven patients received gene-modified HSC following myelo-suppressive conditioning, but with only transient low-level engraftment of MGMT(P140K) gene-modified cells detectable in four patients. All patients received O6BG/TMZ chemotherapy following infusion of gene-modified cells, with five patients eligible for chemotherapy dose escalation, though in the absence of demonstrable transgene-mediated chemoprotection. Since all gene-modified cell products met the criteria for release and assays for engraftment potential met expected outcome measures, inadequate cell dose, conditioning chemotherapy, and/or underlying bone-marrow function may have contributed to the lack of sustained engraftment of gene-modified cells. We were able to demonstrate safe conduct of a technically complex Phase I study encompassing manufacture of the gene therapy vector, genetically modified cells, and a drug product specifically for the trial in compliance with both local and national regulatory requirements.
Introduction
I
HSC-targeted gene therapy strategies have also shown promising results for the treatment of non-hematopoietic genetic diseases such as X-linked adrenoleukodystrophy 13,14 and metachromatic leukodystrophy. 15 –17 In these diseases, for which allogeneic bone-marrow transplant (BMT) has shown therapeutic benefit, 16 successful engraftment of gene-corrected HSCs has resulted in the stabilization in progressive disease phenotypes 13 –15 and likely prevention of disease onset when undertaken at or before the emergence of symptoms in young children. 14,16
As the first institution in Australia to treat a child with SCID-X1 as part of a larger gene therapy trial, 18 and with a focus on developing the capacity to undertake gene therapy trials for cancer, this study sought to develop and initiate a cancer gene therapy trial targeting HSCs in pediatric patients as a means of conferring chemoprotection to the hematopoietic compartment against methylating chemotherapy drugs used for the treatment of brain tumors.
The strategy adopted relies on the expression of a mutated gene for O6-DNA-methyl-guanine-methly-transferase (MGMT), which, through substitution of a single amino acid at position 140, is insensitive to depletion by O6-benzylguanine (O6BG), a small molecule inhibitor of the endogenous protein. 19,20 O6BG, administered with the aim of depleting MGMT activity in the tumor, has been trialed in combination with the alkylating drug N,N′-bis(2-chloroethyl)-N-nitrosourea (BCNU) or the methylating drug temozolomide (TMZ) in patients being treated for brain tumours, 21 –25 where high MGMT activity within the tumor is associated with a poor prognosis. 26 However, the dose-limiting toxicities (DLTs) in the bone marrow that accompany the use of these drug combinations 21 –24 limit the extent to which improved therapeutic effect can be achieved. Gene transfer of MGMT(P140K) into the HSC compartment prior to delivery of O6BG/TMZ or O6BG/BCNU has been shown repeatedly to enable chemotherapy dose escalation under chemoprotection afforded to the gene-modified cells in both small- 27 –31 and large-animal models. 32,33 Preclinical data, generated using MGMT(P140K) transgenic donor bone marrow, demonstrated in vivo selection of MGMT(P140K) expressing HSCs in the bone marrow of allogeneic recipient mice following systemic administration of O6BG/BCNU. 31
Moreover, this strategy has shown promise for the treatment of adults with newly diagnosed glioma, as reported by Adair et al. in a trial treating seven patients, 34,35 and in which all patients showed engraftment of the gene-modified cells following conditioning chemotherapy using BCNU. In that trial, an increase in tolerance for repeated cycles of O6BG/TMZ chemotherapy administered post gene therapy was also demonstrated in addition to a delay in tumor growth in comparison to patients receiving TMZ in the absence of O6BG.
Using a similar approach, this study sought to test the safety and feasibility of gene modifying the HSC compartment of pediatric patients with recurrent brain tumors or newly diagnosed diffuse intrinsic pontine glioma (DIPG) with MGMT(P140K) in a Phase I study. The secondary aims included measures of the level of gene-modified cell engraftment, and the extent to which gene-modified cell populations may have facilitated dose escalation of O6BG/TMZ chemotherapy.
The trial results are reported for seven patients who received both gene-modified cells and post-infusion chemotherapy. Although the infusion of gene-modified cells was feasible and safe from a logistical standpoint, only transient and low-level engraftment of MGMT(P140K) gene-modified cells was detectable in four of the patients post infusion. Despite this, a total of 36 post-infusion chemotherapy cycles of the O6BG/TMZ combination were successfully administered (up to a maximum of 12 cycles for one patient), with the majority of patients eligible for dose escalation over the course of the study.
These escalations in chemotherapy were possible, however, due to individual patients' recovery from previous cycles in the absence of DLTs, rather than any contribution of transgene-mediated chemotherapy resistance. Although it was possible to manufacture and re-infuse gene-modified HSCs safely into this group of pediatric patients, it is concluded that conferring chemoprotection to the HSC compartment to allow for post-infusion chemotherapy dose escalation will require development of alternative protocols to those used in this study.
Materials and Methods
Pretrial preparation: vector construction and vector supernatant manufacture
The γ-retroviral construct pMFG-MGMT(P140K) was generated by polymerase chain reaction (PCR) amplification of the MGMT(P140K) sequence from pQEA-P140K (151) (A. Pegg, Pennsylvania State University, State College, PA), using primers that inserted a 5′ Nco-1 site and a 3′BamH-1 site to allow for insertion into the pMFG backbone plasmid (obtained from Richard Mulligan, Harvard University, Cambridge, MA). PA317 cells were transduced with pMFG-MGMT(P140K) and selected for stable transfection by exposure to O6BG and BCNU. Supernatant collected from selected PA317 cells was used to transduce PG13 cells, and individual clones were obtained by limiting dilution cloning. Supernatant collected from the clones was tested for vector titer on K562 cells and TF-1 cells. The stable producer cell line PG13#18 MGMT(P140K) was chosen for production of the Master Cell Bank, which was subjected to quality screening for mycoplasma, endotoxin, sterility, adventitious agents, and replication competent retrovirus (RCR). Two 10 L batches of serum-free retroviral vector supernatant were manufactured, frozen, and stored at −80°C within the Kid's Research Institute's (KRI) Gene and Cell Medicine Facility (GCMF) cleanrooms using X-VIVO™ 10 medium (Lonza, Sydney, Australia). Vector supernatant titer was measured at approximately 4 × 105 TU/mL (functional titer on HT1080 cells, determined by protein expression and antibody labelling of MGMT) with a molecular titer (by qPCR) of approximately 8 × 105/mL. Vector supernatant was quality tested for RCR, sterility, mycoplasma, and endotoxin by independent accredited laboratories.
Ethics and regulatory approvals
The study received approval from the Sydney Children's Hospitals Network Human Research Ethics Committee (HREC) following evaluation by the Therapeutics Goods Administration (TGA; Department of Health, Australian Government) under the Clinical Trial Exemption (CTX) Scheme. The TGA's evaluation included review of the clinical protocol and the manufacture of two investigational products: CD34+ MGMT(P140K)+ gene-modified cells and O6BG. HREC appraisal of the study proposal also included specific review of the gene therapy elements of the study by the Australian Government's National Health and Medical Research Council's (NH&MRC) Cellular Therapies Advisory Committee (CTAC). HREC approval was granted in February 2011, and the trial commenced in June 2012, and was conducted throughout at the Children's Hospital at Westmead (CHW), Sydney. The study is listed on the Australian New Zealand Clinical Trial Registry (ANZCTR) with identifier: 12612000535875.
O6BG manufacture
O6BG was manufactured for clinical use by Radpharm Scientific Pty Ltd. (a division of Global Medical Solutions Australia), under contract to the authors' institution. The drug was formulated as a lyophilized powder with accompanying buffer for reconstitution by pharmacy staff prior to administration.
Patient eligibility
Patients (aged between 1 and 21 years) with recurrent or progressive high-grade glioma, medulloblastoma, ependymoma, atypical teratoid/rhabdoid tumor, low-grade glioma, or recurrent or newly diagnosed DIPG, and who were assessed at high risk of dying from their tumor (>85%) were eligible for this study. In addition, patients were only eligible provided recurrence had followed standard therapy, and all alternative standard treatment options available had been exhausted, including surgery, chemotherapy, and radiotherapy, except when such therapy was considered unacceptably morbid due to young age or location of the tumor. Newly diagnosed DIPG patients were eligible for enrolment following completion of a standard radiotherapy regimen. Verification of the high-risk tumor was required either by histology or radiologic scans, with measurable disease at the time of study entry. For enrolment, patients were required to have a Karnofsky score ≥50% (patients >16 years of age) or Lansky score ≥50% (children ≤16 years of age), in addition to a predicted life expectancy of ≥8 weeks, and evidence of full recovery from the acute toxic effects of all prior treatment, with adequate bone marrow function using standard measures. Exclusion criteria included pregnant or breast-feeding patients, or patients with any severe uncontrolled infection. Patients were enrolled after obtaining informed consent from their parents following established CHW Oncology Department practice.
Gene therapy protocol
Peripheral blood stem cells (PBSCs) were harvested from patients following granulocyte-colony stimulating factor (G-CSF) mobilization according to institutional practice by the BMT team over one or two apheresis collections to reach a minimum target for CD34 selection of 5 × 106 CD34+ cells/kg patient weight. An un-manipulated backup PBSC product containing a minimum 1 × 106 CD34/kg CD34+ cells was cryopreserved for each patient. The remaining portion of the PBSC product was CD34 selected using the CliniMACS system (Miltenyi Biotec GmbH, Germany) by the Sydney Cellular Therapies Laboratory (SCTL; ICPMR, Westmead Hospital) prior to the purified CD34+ cell product entering the gene transfer culture process. CD34+ selected cells were set up in pre-stimulation culture in X-VIVO™ 10 serum-free medium (Lonza) and human recombinant cytokines interleukin 3 (IL-3; 20 ng/mL; CellGenix GmbH, Germany), thrombopoietin (TPO; 100 ng/mL; CellGenix GmbH, Germany), stem cell factor (300 ng/mL; Stemgen, Swedish Orphan Biovitrum AB, or Cellgenix GmbH, Germany), and FLt3-Ligand (FLT3-L; Cellgenix GmbH, Germany) for 40 h. Cells were then washed and transferred to fresh culture bags (Miltenyi Biotech, Australia) coated with Retronectin (RN; Takara Bio, Japan) for transduction culture in MGMT(P140K) vector supernatant containing the same cytokine cocktail for 24 h. The 24 h culture period in fresh vector supernatant was repeated for a second round of transduction, and a third period of 5–6 h transduction culture preceded final washing of the gene-modified CD34+ cell product with Albumex 4 (CSL, Australia) prior to re-suspension of the cells in Albumex 4 for re-infusion into the patient. The cell product was evaluated for cell count, viability, and CD34 and MGMT expression by flow cytometry (FACS) prior to infusion, with results for sterility, mycoplasma, endotoxin, and vector copy number by qPCR becoming available post infusion. Acceptance criteria for release of the product for infusion were approved by the TGA.
Conditioning chemotherapy
For patients 1 and 2, conditioning chemotherapy consisted of 120 mg/m2 of O6BG over 1 h on day −3 prior to infusion, with 472 mg/m2 of TMZ orally at the conclusion of the 1 h infusion, followed by 30 mg/m2/hour of O6BG for 48 h. Patient 3 did not receive conditioning chemotherapy due to withdrawal from the study prior to PBSC mobilization. Patients 4–7 were given busulphan (4 mg/kg intravenously [i.v.]) once daily for 2 days (on days −4 and −3 prior to cell infusion), and patient 8 was given BCNU (600 mg/m2) as an i.v. infusion over 1 hour given 48 h prior to cell re-infusion. For patients 1–7, Peg-G-CSF 100 μg/kg was given subcutaneously (s.c.) on the day following gene-modified cell re-infusion.
Post-infusion chemotherapy
All patients were eligible for post-infusion chemotherapy as soon as there was hematologic recovery (platelets >100,000/μL, neutrophils >1,000/μL), with resolution of non-hematologic toxicities, and at a minimum of 21 days from the time of infusion. The post-infusion chemotherapy was administered according to the regimen described by Warren et al. 21 Chemotherapy cycles 1 and 2 (dose level 0) consisted of O6BG (120 mg/m2/day i.v. over 60 min daily for 5 days), followed after 30 min of TMZ (75 mg/m2/day orally daily for 5 days). Subsequent cycles (2–13) commenced as soon as there was hematologic recovery and resolution of dose-limiting non-hematologic toxicities, and at a minimum of 21 days from the beginning of the previous cycle. Dose escalation of the TMZ dose was possible in individual patients provided no DLTs were observed in two successive cycles (according to the definitions above). Escalating post-infusion chemotherapy dose levels were: level 1 (cycles 3 and 4) TMZ given at 100 mg/m2/day for 5 days; level 2 (cycles 5 and 6) TMZ given at 133 mg/m2/day for 5 days; and level 3 (cycles 7 and 8) TMZ at 175 mg/m2/day for 5 days. The O6BG dose was not escalated, and Peg-G-CSF was given (100 μg/kg s.c.) at the end of each 5-day cycle of chemotherapy. Patients were no longer eligible to receive post-infusion chemotherapy if there was documented disease progression. However, safety and feasibility measures were scheduled to continue past this time point according to a program of trial-related testing.
Measures of safety and feasibility
The overall safety of the clinical trial protocol (in relation to both the infusion of gene-modified cells and delivery of chemotherapy) was assessed for each patient by documentation of toxicities graded according to the Common Terminology Criteria for Adverse Events v4. In relation to infusion of gene-modified cells on this experimental protocol, feasibility was assessed via documentation and review of parameters related to PBSC mobilization and harvest yield, deliverable dose of gene-modified cells for infusion, and measurement of engraftment of gene-modified cells.
Assessment of MGMT(P140K) expression and transgene integration
Both gene transfer efficiency measured in vitro at the end of the gene transfer protocol and engraftment of gene-modified cells were determined by protein expression and molecular assay. Expression of MGMT(P140K) transgene expression was measured using flow cytometric evaluation of fixed and permeabilized (Fix and Perm Kit; Thermo Fisher Scientific, Australia) gene-modified CD34+ cells prior to infusion, and peripheral blood (PB) subsets post infusion using monoclonal antibody labelling of human MGMT (Clone MT 3.1; Lab Vision; Thermo Fisher Scientific, Australia) and fluorochrome (FITC, PE, PE-CY5) conjugated antibodies against CD34, CD3, CD19, CD15, CD14, and CD33 (BD Biosciences, Australia). The transgene copy number was evaluated by qPCR using a Sybr-green based assay (QuantiFast; Qiagen, Australia) and a primer set designed to amplify across the vector backbone/transgene boundary. GAPDH was used as a standard housekeeping gene for normalization of genomic DNA (gDNA) concentration, and an HT1080 human fibrosarcoma cell line clone containing one copy of the transgene used to construct a standard for comparison with patient-derived gDNA. The sensitivity of this molecular assay was 1 vector copy/105 cells. The transgene copy number of the re-infused cell product was performed on an aliquot of the final product, which was cultured for 7 days in serum-free medium containing cytokines prior to gDNA extraction. Engraftment of gene-modified cells was assessed by analysis of PB at day 15 following re-infusion, and at the beginning of each cycle of post-infusion chemotherapy.
Evaluation of gene-modified cells for engraftment capacity
An aliquot of the final product for each patient was cryopreserved for testing using the HALO-96PQR assay (Preferred Cell Systems; previously known as HemoGenix) as a measure of engraftment capacity, using the manufacturer's instructions. Assay validation was carried out against a manufacturer supplied PBSC product and against an in-house CD34 selected pediatric PBSC collection that was subsequently used as a positive control for each assay.
Tumor evaluation
Magnetic resonance imaging (MRI) of each patient's tumor was undertaken at the time of enrolment onto the study, after every second cycle of post-infusion chemotherapy, and if clinically indicated as determined by the treating physician. An amendment to the clinical protocol for imaging studies was made following the change in conditioning chemotherapy from O6BG/TMZ to busulphan after treatment of patient 2, as busulphan was considered to be less brain tumor specific than the prior drug combination. This amendment added an MRI prior to administration of cycle 1 of post-infusion chemotherapy to monitor for tumor progression during the period between enrolment on the study and receiving the first post-infusion chemotherapy cycle.
Results
Study recruitment
Eight patients consented and were enrolled on the study, with seven patients receiving both gene-modified cells and post-infusion chemotherapy, and one patient being withdrawn during the period between enrolment and G-CSF mobilization for PBSC harvesting (Table 1).
Patient characteristics and outcome of PBSC harvests
Patient enrolled but withdrawn by parents prior to the patient receiving G-CSF mobilization for PBSC collection.
Radiotherapy directed at the brainstem.
Craniospinal radiotherapy.
Radiotherapy localized to tumor site.
Patient failed to mobilize using G-CSF alone.
PBSC, peripheral blood stem cells; DIPG, diffuse intrinsic pontine glioma; G-CSF, granulocyte-colony stimulating factor; ABMT, autologous bone marrow transplant.
PBSC mobilization and harvesting
After mobilization and PBSC collection, the protocol target CD34+ cell yield of 5 × 106/kg was achieved in all patients over one or two apheresis collections (Table 1). The lowest yields were obtained from patients 2 and 8, both of whom had received prior chemotherapy. Patient 8 received plerixafor + G-CSF on the second attempt to mobilize PBSC, after mobilization was unsuccessful using G-CSF alone. Patient 4 was mobilized with both G-CSF and plerixafor in the expectation that mobilization would be difficult due to the patient's previous exposure to myeloablative chemotherapy during two earlier autologous high-dose procedures. The proportion of the harvested CD34+ yield that entered the gene therapy process varied between 31% and 69% of the initial yield due to the cryopreservation of an un-manipulated backup product (minimum 1 × 106 CD34+/kg), and the expected losses accompanying CD34+ selection, carried out on pooled PBSC products on the day following collection.
Gene therapy protocol outcomes
All cell products met acceptance criteria for release, including CD34 positivity, viability, and negative Gram stain. Post-infusion testing confirmed sterility, absence of mycoplasma, and endotoxin levels below specification limits. Fold increases in CD34 cell number over the gene transfer culture period were highest (up to 2.5-fold) with cells harvested from chemotherapy naïve patients 1, 5, and 7, and these patients also received the highest doses of CD34+, MGMT(P140K)+ gene-modified cells (1.8 × 106/kg–3.7 × 106/kg; Table 2). For patient 8, there was an overall loss of CD34+ cells, possibly reflecting the poorer underlying bone-marrow function of this patient (demonstrated by failure to mobilize, and lower baseline hemoglobin, white cell count [WCC], and platelet counts) at the time of mobilization for PBSC harvesting. The proportion of cells expressing CD34 HSC marker in the final product (% purity) was maintained or increased over the culture period for all patients (range 75–97%), except for patient 8 for whom CD34 expression in the final product for infusion was lower than at the initiation of the gene transfer protocol (58% compared to 73% initially). Mean transduction efficiency, measuring the percentage of CD34+ MGMT(P140K)+ cells by FACS of the final product was 21.6% (range 14.65–32.7%), resulting in gene-modified cell doses ranging from 0.5 to 3.7 × 106 CD34+ MGMT(P140K)+ cells/kg (Table 2). A molecular measure of gene-modified cell dose, calculated using vector copy number, determined using qPCR of DNA extracted from the final product cell suspension after 7 days in culture, gave a mean transduction efficiency of 60% (range 45–70%) for patients 4–8 (cells from patients 1 and 2 were not tested). As expected, gene-modified cell doses determined using this measure were higher (range 2.2–6.6 × 106/kg) than those determined using flow cytometry, since the FACS based assay, performed immediately prior to infusion, is unlikely to detect cells transduced within the latter stages of the gene modification protocol that have not yet fully integrated the vector sequence.
Gene therapy protocol outcomes
FACS, fluorescence-activated cell sorting; qPCR, quantitative polymerase chain reaction; ND, not determined; O6BG, O6-benzylguanine; TMZ, temozolomide; BCNU, N,N′-bis(2-chloroethyl)-N-nitrosourea.
Recovery from conditioning chemotherapy and engraftment of gene-modified cells
Patients 1 and 2 received combination O6BG/TMZ chemotherapy that was intended to target their tumor, and provide sufficient myelosuppression to aid in the engraftment of gene-modified cells (Table 2). For patient 1 who received the highest dose of 3.7 × 106 gene-modified cells/kg, engraftment was detected in <1% of PB cells on day 15 post infusion. Gene-modified cells were not detectable in the PB at 21 days post infusion. Gene-modified cells were not detected in patient 2 at day 15 following infusion or at any subsequent time point, with this patient receiving the lowest dose of gene-modified cells. A modification to the protocol to allow for busulphan (4 mg/kg/day for 2 days) as non-myeloablative conditioning chemotherapy 36 was made after failure to detect substantial engraftment of gene-modified cells in these two patients, based on the reported successful engraftment of gene-modified cells in trials treating ADA-SCID and WAS. 5 –8 Patients 4–7 subsequently received busulphan conditioning chemotherapy, with engraftment of gene-modified cells evident for patients 5 and 6 (7.4% and 2.1%, respectively), and at very low levels in patient 7 (0.3%) following infusion (day 15 for patients 5 and 6; day 28 for patient 7). In patient 7, testing for engraftment was delayed until day 28 following infusion due to slow WCC recovery (>30 days) attributed to side effects associated with antibiotic induced renal complications. For patient 4 in whom gene-modified cells were undetectable following infusion, recovery of a WCC (to >0.5 × 109/L) was also slow and considered to be related to poor underlying bone-marrow function due to two previous autologous HSC high-dose procedures. Gene-modified cells were, however, not detectable immediately prior to cycle 1 of post-infusion chemotherapy in any of these patients. Following the use of busulphan in these four patients, with a lack of sustained engraftment and failure to see any increase in gene-modified cell emergence following repeated cycles of O6BG/TMZ chemotherapy, an additional modification to the study protocol was made, to use BCNU as conditioning chemotherapy, as used in the adult glioma chemoprotection study. 35 Patient 8 was the first and only patient enrolled who subsequently received this conditioning chemotherapy, and engraftment of gene-modified cells was not detected post infusion at any time point. Although a substantial level of transduction was achieved for this heavily pretreated patient's CD34+ cells (40% transduction efficiency using qPCR), indicating that some expansion of this population had occurred during the culture period, the final product showed a decline in purity (% CD34 cells) in contrast to all other patients. Patient 8 received one of the lower doses of gene-modified cells, and engraftment of gene-modified cells was not detected at any time point post infusion.
Assays for capacity of MGMT(P140K) gene-modified cells to engraft
The capacity of the gene-modified cell populations of all patients was tested post infusion using the Hemogenix HALO assay, which compared the CD34+ gene-modified cell final product performance against a third-party positive control non-gene-modified CD34 selected pediatric PBSC product. Results of these assays (Fig. 1) indicated that for patients other than patient 8, the final product cells had equivalent or greater proliferative capacity than the pediatric CD34 selected and un-manipulated positive control cell population. For patient 8, this assay returned a failed result with respect to proliferative capacity of the more primitive lympho-hematopoietic stem cells (SC-HPP) cell population, indicated by the lack of an increase in the production of intracellular (adenosine triphosphate [ATP]) with increasing cell numbers over the standard 5-day culture period in medium designed to support expansion of these populations. Also for patient 8, cellular ATP production under the conditions for determining the quality of mature, hematopoietic stem cells (SC-GEMM) did not meet that determined for the positive control cells (data combined over four independent assays for the same sample) or the other six patient samples. These results once again were considered to reflect the poor underlying HSC function of this patient attributed to prior chemotherapy.
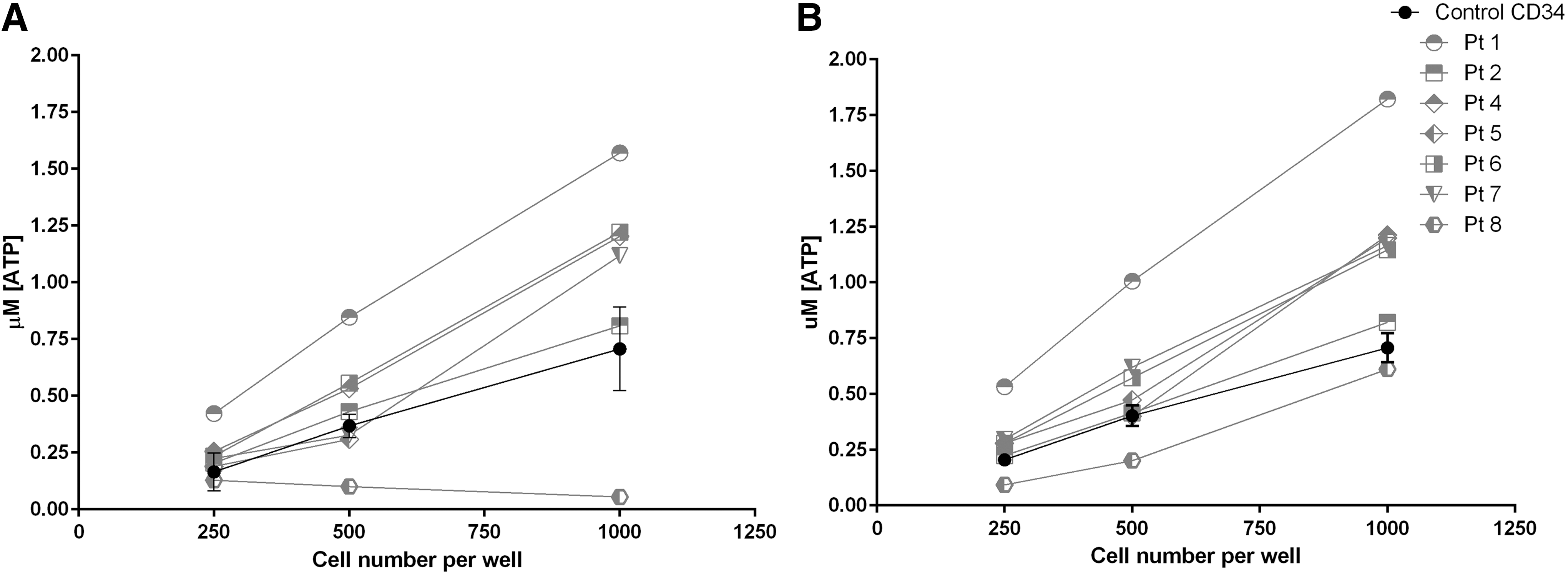
Outcome of HALO assay for gene-modified cells.
Post-infusion chemotherapy courses and patient outcome
All seven patients received post-infusion O6BG/TMZ combination chemotherapy (range 2–12 cycles; Table 3). Four patients (patients 2, 5, 6, and 7) were treated with TMZ dose escalation, and three patients did not receive escalated doses. Of those, one was eligible (no DLTs in prior two cycles) but did not receive chemotherapy due to tumor progression, and two patients were not eligible due to inadequate marrow function (patients 4 and 8). The only non-hematological DLTs seen in any patient was elevated liver enzymes (patient 2), which resolved with a de-escalated dose of chemotherapy. Although a minority of patients had stable disease noted for a period while on the study (for between 4 and 10 cycles of chemotherapy), all patients died of progressive disease between 1 and 13 months following cessation of trial protocol post-infusion chemotherapy.
Post-infusion chemotherapy courses and patient outcome
DLT, dose-limiting toxicities; ALT, alanine transaminase; HREC, Human Research Ethics Committee.
Toxicities and safety
For all three conditioning chemotherapy protocols used, patients experienced myelosuppression, recorded as grade 4 neutropenia for between 3 and 25 days (Table 4). High lactate levels were seen transiently in patients 1 and 2 following O6BG/TMZ chemotherapy, and patient 8 had an allergic reaction to the administration of BCNU. Recovery of neutrophil counts to >0.5 × 109/L following gene-modified cell infusion ranged between 14 and 33 days, with patients 4, 7, and 8 taking >28 days. In patients 4 and 8, this was attributed to underlying poor bone-marrow function related to previous treatment, and for patient 7, it was considered to be due to renal complications that accompanied antibiotic administration. There were no acute toxicities associated with the infusion of gene-modified cells, with all patients being discharged on the day following infusion. All patients except patient 8 received Peg-G-SCF prior to discharge. The protocol was modified in the interval between patient 7 and patient 8 to allow for BCNU conditioning chemotherapy, and the administration of Peg-G-SCF on the day following cell infusion was removed from the protocol due to concerns regarding a potential effect of this drug on engraftment of gene-modified cells. Patient 8 subsequently received Peg-G-SCF on day 28 post infusion due to slow neutrophil recovery. Post-infusion chemotherapy toxicities associated with O6BG/TMZ were as expected for this patient population, and included grade 4 hematological toxicities that were managed using standard supportive care measures. There were 29 serious adverse events (SAE) leading to unscheduled hospital admissions: 15 were related to tumor progression, and 14 were related to the study procedures, including fever/neutropenia (n = 10), tachypnea (n = 2), and single episodes of respiratory infection, drug reaction to BCNU, and mucositis. All SAEs were reported to the HREC and were reviewed periodically by the trial Data Safety Monitoring Committee.
Outcomes of conditioning chemotherapy and gene-modified cell infusion
ANC, absolute neutrophil count.
Discussion
The primary outcome tested in this Phase I study was the safety and feasibility of infusing gene-modified HSCs in pediatric patients being treated for high-risk brain tumors. Although sustained engraftment of gene-modified cells was not achieved, CD34+ cells were gene modified with efficiencies equivalent to those reported in similar studies, 2 –4,7 and the cell product met predefined quality standards, with no adverse events or toxicities attributable to the cell product infusion. Due to the lack of sustained engraftment, however, secondary outcome measures, such as evidence for in vivo selection of drug-resistant HSC populations following O6BG/TMZ combination chemotherapy and the extent to which dose escalation could be achieved under chemoprotection, could not be evaluated. This outcome also precluded any analysis of the longer-term safety of the use of the γ-retroviral vector in relation to the risk of insertional mutagenesis, or emergence of clonal dominance due to insertion site location under a chemotherapy-driven in vivo selection strategy.
Explanations for the lack of robust or sustained engraftment of gene-modified cells may be related to elements of the clinical protocol and/or the gene modification protocol and reagents used in the study. The clinical protocol dictated cell yield targets for PBSC collection that were met for all patients, although with quarantine of a portion of the harvest for an un-manipulated backup and losses due to CD34 selection, cell inputs for gene modification were reduced below this target for the majority of patients. Final doses of gene-modified CD34+ cells, were on the lower end of the range reported in other studies in children, both targeting hematological genetic diseases 1 –11 and non-hematological genetic disease. 12 –15.Lowest doses were received by patients who had been exposed to pre-enrolment chemotherapy as a cumulative result of lower initial harvest yield, lower CD34+ purity of the PBSC product after selection, and lower fold expansion of the CD34 population during the culture period. Cell doses achieved in the MGMT(P140K) chemoprotection trial in adult glioma patients 34 with robust engraftment and persistence of gene-modified cells were also higher than those obtained under the present protocol (Table 5). Other differences between the study conducted by Adair et al. and the present study (Table 5) include patient age, pre-enrolment therapy, conditioning chemotherapy regimen (except for patient 8 in the present study), post-infusion chemotherapy TMZ dose, and administration of Peg-G-CSF to the majority of pediatric patients following infusion of gene-modified cells. In addition, there were technical differences between the vectors used and transduction culture conditions in the gene transfer protocol.
Comparison of protocols for MGMT(P140K) gene transfer into patient HSCs
HSCs, hematopoietic stem cells; GAL-V, gibbon ape leukemia virus; IL, interleukin; SCF, stem cell factor; Flt3-L, Flt3-Ligand; TPO, thrombopoietin; VS, vector supernatant; RN, retronectin; SFEM, serum-free expansion medium.
In relation to conditioning chemotherapy, the combination of O6BG/TMZ initially used was chosen due to its application in other brain tumor targeted protocols not requiring stem-cell support, since the feasibility of the gene therapy protocol at the authors' institution was untested. With the lack of engraftment seen in these patients, a modification to the protocol to use busulphan was tested in the subsequent four patients, resulting in initial engraftment at low levels in three of these patients, but not sustained engraftment of gene-modified cells. The one patient receiving the most myelosuppressive conditioning, and that used in adults, had been heavily pretreated in preceding months, experienced poor mobilization with sub-standard stem-cell quality, and did not achieve gene-modified cell engraftment.
Engraftment of gene-modified cells in this pediatric population may have been compromised by, at alternate ends of the scale, poor marrow function in patients previously exposed to chemotherapy or healthy marrow that had a high endogenous capacity for recovery from non-myeloablative conditioning in chemotherapy-naïve patients. Notably, MGMT(P140K) expression in HSCs would not be predicted to provide a survival advantage to infused gene-modified cells during the early engraftment period and prior to any post-infusion chemotherapy. This is in contrast to the cellular survival advantage afforded to gene-corrected HSCs in the treatment of SCID 37 or WAS 9 from the time of infusion.
The extent to which the dose and timing of post-infusion chemotherapy may have been able to mediate in vivo expansion of gene-modified cells was not assessable in the study. Although patient 1 initially had a low level of engraftment and received two cycles of O6BG/TMZ chemotherapy, the possibility that gene-modified cells may have emerged with further cycles of chemotherapy could not be evaluated, as early progressive disease was documented after cycle 2, making the patient ineligible for further chemotherapy (Table 3). In patient 5, gene-modified cells were detected prior to cycle 1 of post-infusion chemotherapy (day 15) but were undetectable by the time the chemotherapy was given (day 25). Gene-modified cells were then not detected following subsequent rounds of O6BG/TMZ chemotherapy, which in this patient, and others, was not myelosuppressive (neutrophil count <1.5 × 109/L) until after dose escalation, some months after infusion.
Alternatively, or in combination with clinical protocol elements, technical aspects of the gene modification protocol differing from those used in adults (Table 5) may have had an impact on the engraftment of gene-modified cells. However, the culture conditions used both to generate vector supernatant and to gene modify CD34+ HSCs were successful in maintaining CD34 expression throughout the culture period, and were similar or the same as those used in other pediatric gene therapy protocols. 1 –5.The major difference between the current protocol and others used for pediatric patients was the use of PBSC-derived HSCs, rather than bone marrow, as in protocols for ADA-SCID 4 –8 and SCID-X1. 1 –3 Adair et al. 35 reported that in a subset of patients, an effect of the serum-free medium used for the gene transfer protocol led to a loss of gene-modified cells following engraftment that was correlated to a particular batch of serum-free medium. In this study, engraftment overall was low, making it difficult to discern any batch-to-batch variation in outcome following infusion, but similar problems could have influenced attempts to attain sustained engraftment. For all but one patient, gene-modified cells passed testing for HSC quality, equivalent to an un-manipulated control in a validated assay. All gene-modified cell products met predetermined and regulator-approved release criteria for infusion, including CD34 expression, transgene expression, and cell viability.
The conduct of this Phase I clinical trial in a population of pediatric patients was associated with many challenges—most notably, the need to weigh risks associated with the experimental protocol, such as the infusion of a gene-modified cell product, against the possibility of gaining scientific insight to enable further development or refinement of the strategy. Although adjustments to conditioning chemotherapy in the clinical protocol were made during the study with the aim of improving engraftment, the change most likely to enhance engraftment, and used successfully in the adult setting, remained untested in any chemotherapy naïve patients when the trial closed, not allowing factors that may contribute to robust engraftment of gene-modified cells expressing MGMT(P140K) in this patient population to be more fully understood.
This element of the protocol remained untested, as the trial closed due to the intermittent and unpredictable nature of enrolments, in addition to depletion of vector stocks. Although it was feasible and safe to undertake the study from a technical standpoint, the efficacy of this strategy in protecting the HSC compartment against combined O6BG/TMZ chemotherapy was not established in pediatric patients with brain tumors in the authors' institution.
In summary, it is concluded that a number of important changes to the clinical protocol, including higher initial PBSC harvesting targets and the narrowing of eligibility to patients with good marrow function, would be required to make improvements to the outcome of the strategy in children. Such changes, alongside technical improvements to the gene therapy protocol (improved vector transduction efficiencies, use of chemically defined culture medium) could result in higher cell doses and more robust engraftment as long as sufficient myelosuppression is provided by the conditioning chemotherapy. However, without certainty regarding predictable and timely recruitment, the testing of this strategy in this patient group would still remain a complex undertaking.
Footnotes
Acknowledgments
B.K, R.S., J.W., R.D., A.R., S.M., and YW.C. were funded by The Kid's Cancer Project (TKCP), which also generously supported the conduct of the preclinical work, initiation, and running of the trial. Funding was also provided by the Kids Cancer Alliance (KCA) and the Sporting Chance Foundation. Funding for O6BG Manufacture was provided by both Radpharm Scientific Pty. Ltd. and TKCP. The authors wish to acknowledge the work of clinical and scientific colleagues who enabled the conduct of the trial. A summary of findings was presented at the Joint 10th Australasian Gene and Cell Therapy Society and Australasian Society for Stem Cell Research Scientific Meeting, held in Sydney in May 2017.
Author Disclosure
All authors declare no conflicts of interest or competing financial interests in the conduct or reporting of this clinical trial.