
Abstract
Acute myeloid leukemia (AML) is a kind of a malignant hematologic tumor caused by uncontrolled repopulation of myeloid hematopoietic stem cells (HSCs). Current therapeutic effects for AML patients are unsatisfactory. In particular, relapsed and refractory AML still have a poor prognosis. T cells modified by chimeric antigen receptor (CAR) was an immunotherapeutic strategy for malignancies, which has a broad developing prospect. Most AML cells overexpress the myeloid antigen CD33. Therefore, CD33-specific CAR-T cells with different co-stimulators (CD28, 4-1BB, or both, referred to as CD33 28z.CAR-T cells, CD33 BBz.CAR-T cells, or CD33 28BBz.CAR-T cells, respectively) were developed to evaluate their efficacy against AML. The effectiveness of three types of CD33 CAR-T cells against AML was verified by specific killing effect to AML cells and prolonged survival of a xenograft mouse model. In terms of CAR-T cell efficacy, especially when transfused into human bodies, the persistence of T cells is also an important index, as it is closely associated with the long-term effect of CAR-T cells. Therefore, the characteristics of three types of CD33 CAR-T cells related to the persistence of T cells were examined. It was found that during expansion, CD33 BBz.CAR-T cells had an increased central memory compartment, while CD33 28z.CAR-T cells were predominantly effector memory T cells. In addition, CD33 28z.CAR-T cells were more inclined to become exhausted. The study suggests that incorporation of 4-1BB in CARs may endow T cells with long-lasting survival ability, thus improving the long-term anti-leukemia effect of CAR-T cells, especially when transfused to the human body.
Introduction
A
The myeloid differentiation antigen CD33 is mainly expressed on myeloid progenitors, the CFU-GM, mature granulocytes, monocytes, and dendrite cells. 6 –8 It is reported that 85–98% of AML cells express CD33 molecules, 9,10 and the expression level of CD33 on AML blasts is significantly higher than that on normal cells. 11 Thus, CD33 was the first target antigen for immunotherapy of AML. In 2000, the Food and Drug Administration approved a humanized anti-CD33 monoclonal antibody, gemtuzumab ozogamicin (GO), to treat relapsed AML in the elderly. Clinicians found that the relapse and survival rates were improved with a combination of chemotherapy with GO at 6 years, although the drug was voluntarily withdrawn in 2010 due to the occurrence of adverse effect (veno-occlusive disease) in some patients. 12 It is suggested that CD33 is a potential target antigen of AML.
Chimeric antigen receptor (CAR) T-cell therapy is an inspiring immunotherapeutic strategy for tumor that has been developed recently. CAR is a kind of artificial T-cell receptor (TCR) consisting of an extracellular antigen binding domain, which is usually a single-chain variable fragment (scFv) from an antibody, a hinge, a transmembrane domain, and the intracellular signal transduction domain. 13,14 According to co-stimulators in the signal transduction domain, CARs can be divided into four generations. At present, second-generation CARs are commonly used. They contain one of the following co-stimulators: CD28, 4-1BB (CD137), OX40 (CD134), CD27, ICOS, and CD244, among others. Compared to first-generation CARs, second-generation CARs have more effective function and proliferative capacity. 15 –18 The efficacy of CAR-T cells may be influenced by the structure of CARs, particularly the co-stimulator component. Studies have shown that CAR-T cells containing the 4-1BB co-stimulator have superior anti-leukemic efficacy and improved persistence compared to those containing the CD28 co-stimulator. 16 However, how co-stimulators affect CAR-T cell function remains to be studied further.
In this study, three different CD33-CARs with co-stimulators CD28, 4-1BB, and both CD28 and 4-1BB (referred to as CD33 28z.CAR-T, CD33 BBz.CAR-T, and CD33 28BBz.CAR-T, respectively) were constructed, and the anti-leukemic functions of corresponding CD33 CAR-T cells were reported. In addition, the characteristics of CAR-T cells were further analyzed. It was demonstrated that CD33 CAR-T cells exhibit potent anti-leukemic efficacy, and CD33 BBz.CAR-T cells have an increased central memory compartment and show resistance to exhaustion. These results reveal the reason that CAR-T cells with 4-1BB perform better in anti-leukemic function, further illustrating that 4-1BB CAR might be superior to incorporate into T cells to obtain persistent antitumor efficacy, especially when transfused to patients.
Methods
Patients and samples
Healthy donors' peripheral blood (PB) for isolation of T cells and umbilical cord blood (UCB) were supplied by the Tianjin Blood Center. Bone marrow mononuclear cells (BMMNCs) or peripheral blood mononuclear cells were obtained from 13 AML patients who were enrolled at the Institute of Hematology and Blood Diseases Hospital. All subjects signed an informed consent in accordance with the Declaration of Helsinki. Studies using patient samples were approved by the ethical advisory board of the Institute of Hematology and Blood Diseases Hospital.
Construction of CD33 CAR lentiviral vectors and production of lentivirus
The murine anti-human CD33 scFv derived from mouse hybridoma cells (clone HIM3-4/HI33a, which was established at the Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College) was cloned into previously constructed pCDH-CAR plasmids 19 in order to obtain lentiviral vectors pCDH-CD33-28z-CAR, pCDH-CD33-BBz-CAR, and pCDH-CD33-28/BBz-CAR. The lentivirus was packaged and produced as previously described. 19 In brief, 293T/17 packaging cells were transfected with the three kinds of pCDH-CAR plasmids, together with the packaging plasmids and polyethylenimine (Polysciences). After incubation and culture medium replacement, the supernatants were harvested after 24 and 48 h, concentrated, aliquoted, and stored at −80°C until use.
Generation of CAR-T cells
T cells from donors' PB were isolated and enriched using RossetteSep T cell enrichment Cocktail (Stem Cell Technologies) and Ficoll solution (TBD Science). T cells were cultured in X-VIVO15 (Lonza) plus 5% fetal bovine serum (FBS; Gibco), and were incubated in 24-well plates at 1 × 106/mL. Dynabeads™ Human T-Activator CD3/CD28 (Gibco; 1 × 106/mL) and recombinant human interleukin (IL)-2 (R&D; 100–200 IU/mL) were added to the culture. After stimulation for 24 h, T cells were transduced with lentiviruses, centrifuged, and incubated for another 24 h. Then, the culture medium was replaced every other day, with IL-2 routinely added for up to 10–15 days. Cells were cryopreserved or used for the following experiments.
Cell lines
The leukemia cell lines U937, HL60, THP-1, Kasumi-1, KG-1, KG-1a, K562, and Nalm-6 were cultured in RPMI 1640 medium supplemented with 10% FBS. The 293T/17 cell line was cultured in Dulbecco's modified Eagle's medium with 10% FBS. Nalm-6 cells were transduced with lentivirus encoding firefly luciferase, followed by sorting of luciferase-positive clones with Aria III (BD Bioscience), and labeled as Nalm6-luci-RFP.
Flow cytometry analysis
Samples were tested with LSRII or FACS Canto II (BD Biosciences), and data were analyzed using FlowJo v7.6.1. CD33 CARs were detected with green fluorescent protein (GFP) and/or Alexa Fluor 647-conjugated goat anti-mouse immunoglobulin G, F(ab′)2 fragment specific antibody (Jackson ImmunoResearch). CD33 expression on leukemia cells was tested by anti-human CD33 Ab (clone WM53; Biolegend). The T-cell phenotype was assessed by antibodies from Biolegend (clone numbers in parentheses): CD3 (HIT3a), CD107a (H4A3), PD-1 (EH12.2H7), Tim-3 (cF38-2E2), and LAG-3 (11C3C65). To assess the phenotypes of CAR-T cells, data were analyzed based on gated CAR+/GFP+ cells. For non-transduced control T cells (NTD), whole cells were analyzed.
T-cell function assays
Cytotoxicity assays
CD33+ cell lines were used for cytotoxicity assays as target cells and CD33– cell lines as control target cells. T cells and target cells were co-cultured at the indicated effector/target (E:T) ratios for 0 to 72 h. Then, harvested cells were labeled with anti-human CD3 and CD33 antibody. The proportion of residual target cells was detected by flow cytometry to reflect the cytotoxic activity of the T cells.
Enzyme-linked immunosorbent assays to detect release of cytokines
T cells and target cells were incubated at an E:T ratio of 1:1 for 12 h or 24 h. Supernatants were collected and stored at −80°C. Detection of cytokines in the supernatants, including IL-2, interferon (IFN)-γ, and tumor necrosis factor (TNF)-α was performed following the enzyme-linked immunosorbent assay kit instructions (R&D).
T-cell degranulation assays
T cells and target cells were incubated at an E:T ratio of 1:1 or 1:20 in triplicate in 200 μL of medium containing IL-2 (50 IU/mL) and anti-CD107a antibody. One hour later, monensin was added (1 μM; Sigma–Aldrich). After another 4 h of incubation, cells were labeled by CD3 and CD33 antibody and assayed by flow cytometry. Analysis of CD107a expression was performed based on live CD3+ cells.
Colony-forming assay
CD34+ cells were isolated from human UCB using a CD34 MicroBead kit (Miltenyi Biotec), and then co-cultured with T cells at an E:T ratio of 1:1 or cultured alone for 24 h. The equivalent of 500 CD34+ cells was plated in methylcellulose medium (MethoCult H4434; Stemcell Technologies) and incubated at 37°C in a humidified atmosphere of 5% CO2. On day 14, colony-forming-units (CFU) were counted and photographed.
Quantitative reverse transcription polymerase chain reaction
Total RNA of T cells was extracted using RNAiso reagent (TaKaRa), and RNA was reverse transcribed into cDNA. All reactions were performed with SYBR green (TaKaRa) and 10 μM of forward and reverse primers using real-time polymerase chain reaction (PCR; StepOne; Applied Biosystems). The primers for Blimp-1 and T-bet were 5′-GTGTCAGAACGGGATGAAC-3′ (forward) and 5′-TGTTAGAACGGTAGAGGTCC-3′ (reverse), and 5′-GGGCGTCCAACAATGTGA-3′ (forward) and 5′-CGGCAATGAACTGGGTTT-3′ (reverse), respectively. Expression levels of the target genes were normalized to GAPDH. All amplifications were done in triplicate, and at least three different samples were performed.
Establishment of CD33+ leukemia murine model and treatment with CAR-T cells
To establish a human CD33+ leukemia mouse model, Nalm6-luci-RFP was transduced with lentivirus encoding human CD33 to generate Nalm6-luci-CD33-RFP (labeled as Nalm6-CD33). NOD/SCID mice aged 6–8 weeks were irradiated and inoculated with Nalm6-CD33 cells (5 × 105) intravenously. Mice then were randomized into groups for adoptive transfer of 107 CAR-T cells (with equivalent transduction efficiency), NTD cells, or phosphate-buffered saline (PBS) intravenously 7 days later. CD33+ leukemia cells in PB were detected by CD33 Ab with flow cytometry to assess disease development. Mice were weighed every week, and other symptoms such as diarrhea and paralysis were monitored. Once the mice were euthanized, the bone, liver, spleen, heart, kidney, and lung were removed for pathological examination, and flow cytometry analysis was carried out to confirm the occurrence of leukemia and the phenotype. The survival of the mice was measured from the date of inoculation to death. All animal experiments were approved by the Institutional Animal Care and Use Committee of Chinese Academy of Medical Sciences and Peking Union Medical College.
Statistical analysis
All graphs are reported as the mean ± standard error of the mean values of at least three biological replicates. Statistics were performed using GraphPad Prism v5 (GraphPad Software, Inc.). Unless otherwise stated, Student's t-test was used to determine the significance of the differences between groups, and a significant difference was accepted when p-values were <0.05.
Results
Construction of CD33 CAR and preparation of CAR-T cells
Second- (with CD28 or 4-1BB as co-stimulator) and third-generation (with both as co-stimulators) CD33 CARs were constructed (Fig. 1A). To obtain CAR-T cells, activated T cells were transduced with lentivirus. Then, CAR expression was detected on the surface of the T cells by flow cytometry (Fig. 1B). CAR-T cells could proliferate to about 800-fold in culture condition (Fig. 1C). This suggested that the system could successfully prepare CD33 CAR-T cells.
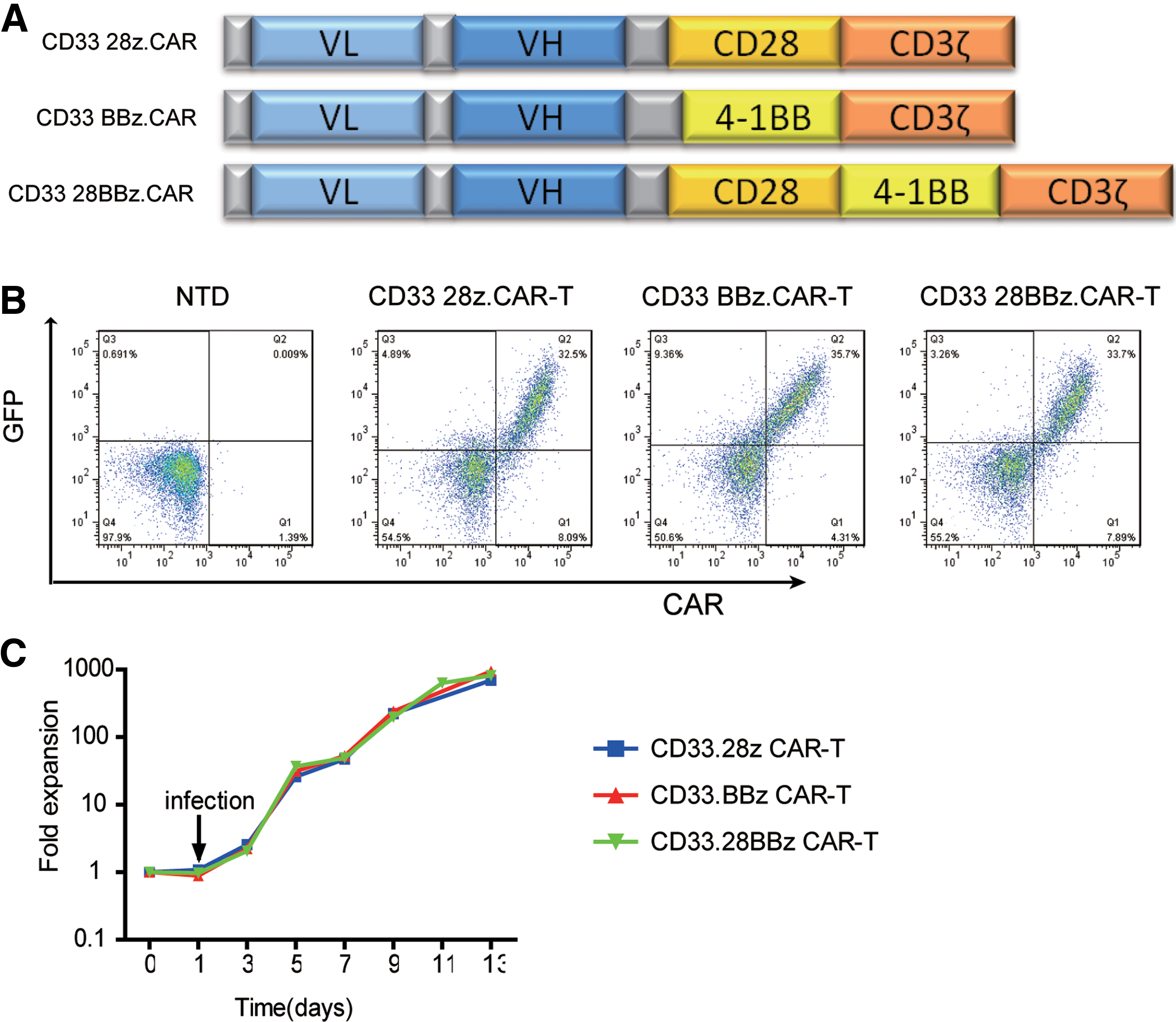
Construction of CD33 chimeric antigen receptors (CARs) and preparation of CAR-T cells.
CD33 CAR-T cells exhibited potent and specific cytotoxicity against AML cell lines
To assess the efficacy of constructed CD33 CARs, 28z.CAR-T, BBz.CAR-T and 28BBz.CAR-T cells were prepared and co-cultured with leukemia cell lines. First, the CD33 expressions on several leukemia cell lines were examined (Supplementary Fig. S1A and B; Supplementary Data are available online at
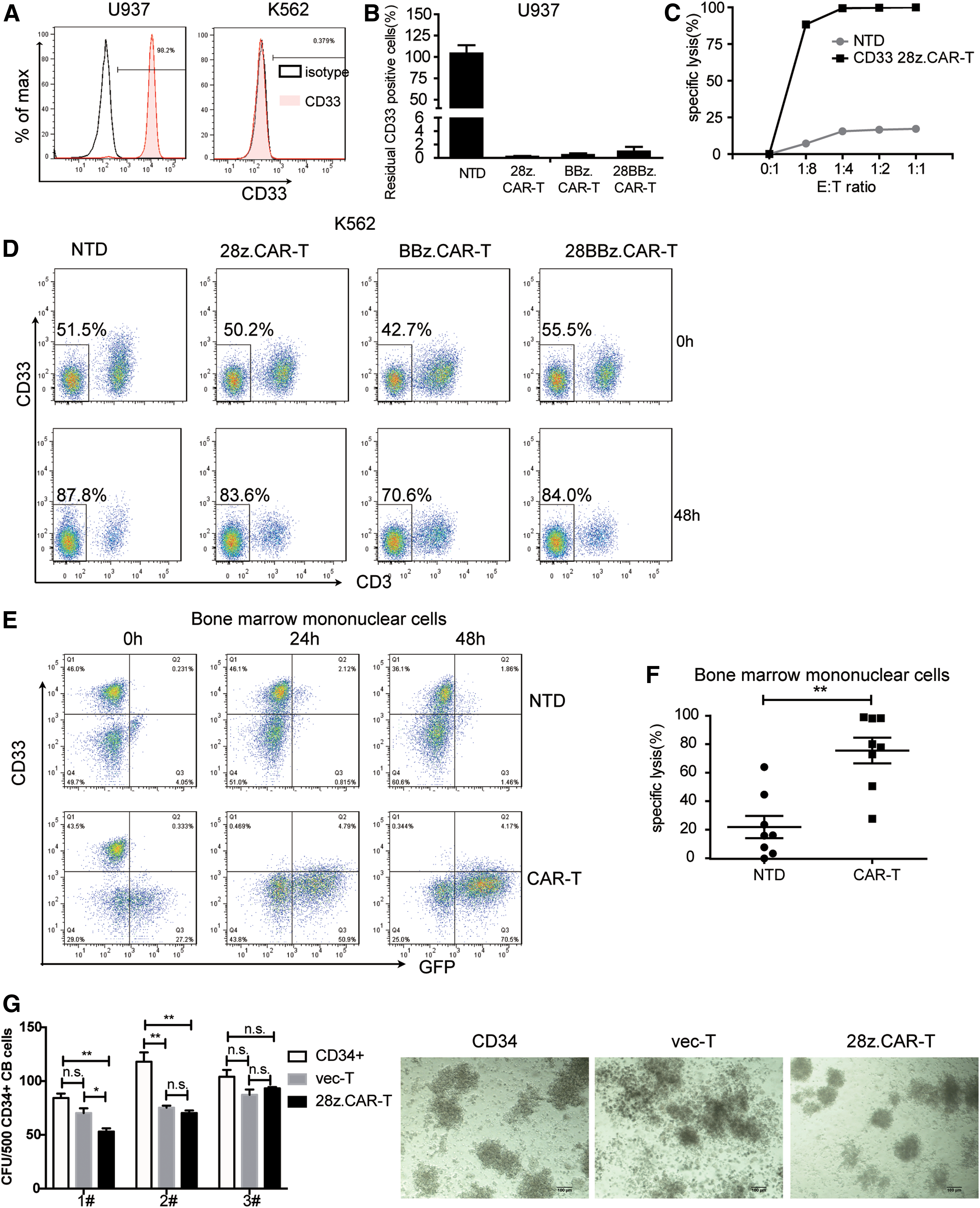
Killing effect of CD33 CAR-T cells.
CD33 CAR-T cells showed specific cytotoxicity against primary AML cells and less influence on CD34+ cord blood cells in vitro
The efficacy of CD33 CAR-T cells was further tested on AML patients' primary cells. Most AML cells overexpress the myeloid antigen CD33. So, CD33 expression of several AML samples was assayed by flow cytometry, and the results shown in Table 1. AML samples of P1–P8 were selected to validate the anti-AML potential of CD33 CAR-T cells. When co-cultured with CD33 28z.CAR-T cells at an E:T ratio of 1:1, primary CD33+ AML cells were almost eliminated within 24 h (Fig. 2E and F). These results further confirmed the potent efficiency of CD33 CAR-T cells against AML.
CD33 expression of acute myeloid leukemia cells from bone marrow or peripheral blood of 13 patients detected by flow cytometry
CD33+ cells were gated by isotype control antibody. MFI of CD33 expression was calculated by FlowJo v7.6.1.
BM, bone marrow; PB, peripheral blood; MFI, median fluorescence intensity.
Since CD33 is also expressed on myeloid progenitors, the potential on-target off-tumor effect was then assessed. CD34+ cells were sorted from human UCB, analyzed for CD33 expression (Supplementary Fig. S1D), and then co-cultured with 28z.CAR-T cells or vector T cells or cultured alone for 24 h. The cells were then plated in methylcellulose medium and cultured for 14 days. For samples 1 and 2, the number of CFU was significantly reduced after being pre-cultured with CAR-T cells compared to the untreated group. However, for sample 2, the number of CFU was comparable in the vector-T and 28z.CAR-T pre-cultured groups, implying that the reduced CFU colonies may be due to the incompatibility of donor T cells with sample 2, not the off-tumor effect of the CD33 CAR. For sample 3, the number of CFU in the three groups was similar (Fig. 2G). This suggests that the CD33 CAR-T cells had less of an influence on normal CD34+ cells.
CAR-T cells displayed significant activation during cytotoxicity
When incubated with CD33+ U937 leukemia cells, CAR-T cells were significantly activated for degranulation compared to NTD cells (p < 0.0001; Fig. 3A). When incubated with CD33– K562 leukemia cells, CAR-T cells showed no apparent increase in CD107a expression (Fig. 3A). To assess T-cell function, cytokines were detected in the co-culture supernatant. When co-cultured with CD33+ U937 cells, CAR-T cells displayed significant activation demonstrated by robust cytokines release. The levels of IL-2, IFN-γ, and TNF-α in the supernatant after 24 h of incubation showed a significant increase compared to those of the NTD control (Fig. 3B). The 28z.CAR-T cells and BBz.CAR-T cells released higher levels of cytokines than the 28BBz.CAR-T cells (Fig. 3B). However, when co-cultured with CD33– K562 cells, the levels of IFN-γ and TNF-α in the supernatant only had a slight increase, and IL-2 had no obvious change (Fig. 3C). When co-cultured with other CD33+ AML cell lines (e.g., 28z.CAR-T cells), CAR-T cells released a significantly high level of cytokines as well (Fig. 3D).

CAR-T cells were significantly activated during cytotoxicity.
CD33 CAR-T cells revealed a significant antitumor effect in vivo
Next, in order to estimate the in vivo antitumor effect, a CD33+ xenograft mouse model was established by intravenous inoculation of Nalm6-CD33 cells. First, the in vitro killing effect of CAR-T cells against Nalm6-CD33 cells was determined. Incubation of 28z.CAR-T cells with Nalm6-CD33 resulted in obvious degranulation (Fig. 4A) and potent cytotoxicity at E:T ratios of both 1:1 and 1:4 (Fig. 4B).
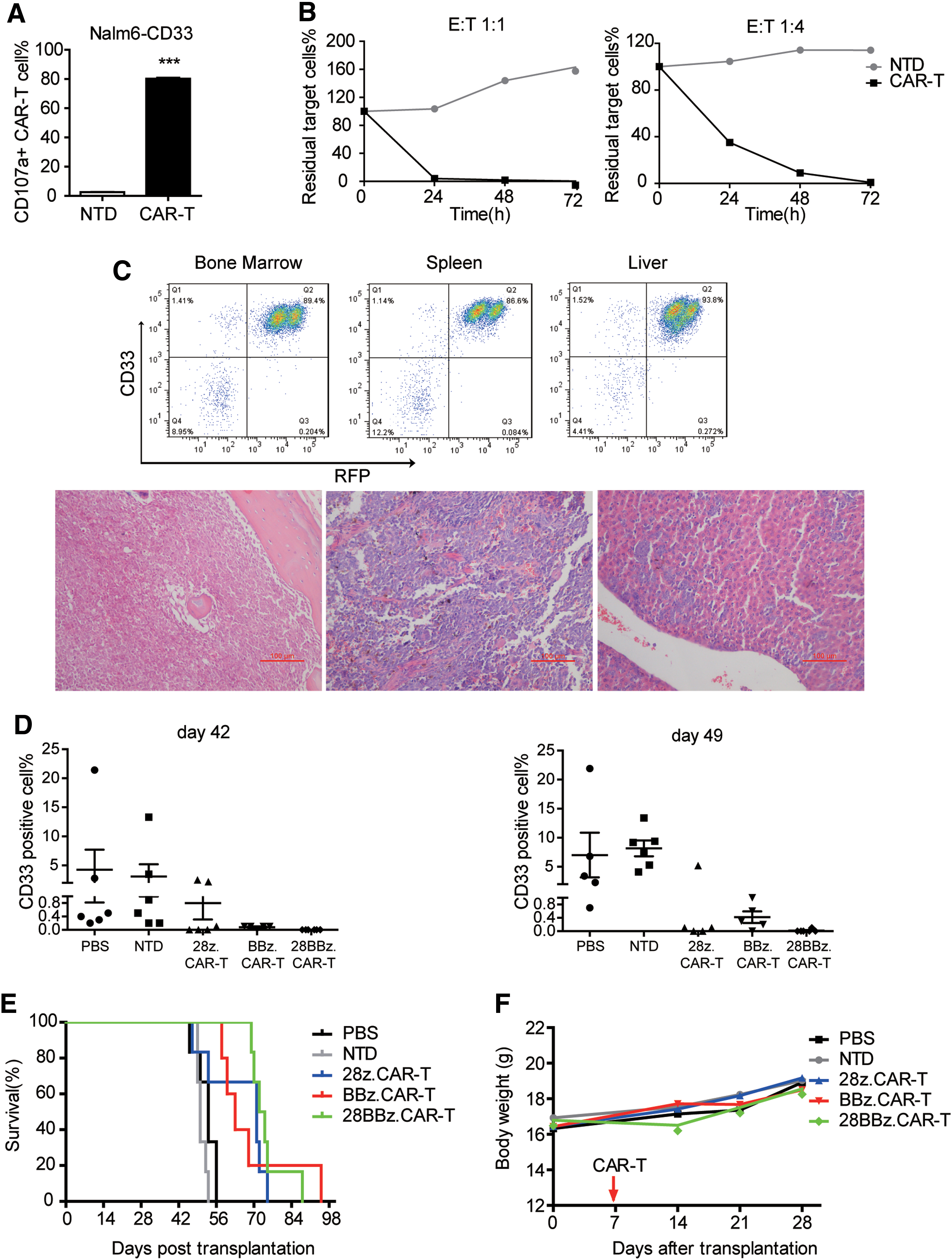
CD33 CAR-T cells revealed a significant antitumor effect in vivo.
Then, Nalm6-CD33 cells were inoculated into irradiated NOD/SCID mice to establish a CD33+ xenograft mouse model. All transplanted mice developed invasive leukemia, with CD33+ human cell infiltration confirmed by flow cytometry and pathological examination (Fig. 4C). After being treated with T cells, CD33+ leukemia cells in PB were dynamically detected. On days 42 and 49, CAR-T-treated mice had a lower burden of leukemia cells than PBS- and NTD-treated mice (Fig. 4D). This suggests that CD33 CAR-T cells exhibit an antitumor effect in vivo. Compared to PBS and NTD, treatment with CAR-T cells significantly prolonged the mean survival time of mice (Fig. 4E). The mean survival times were 64.83 ± 4.79, 68.80 ± 6.76, and 74.67 ± 2.82 days in mice treated with 28z.CAR-T, BBz.CAR-T, and 28BBz.CAR-T cells, respectively, while the survival times for mice treated with PBS or NTD cells were 52.33 ± 1.56 and 50.50 ± 0.67 days, respectively. All three types of CAR-T-treated mice had a longer survival time compared to the control group (p < 0.05). This demonstrates that CD33 CAR-T cells had an anti-leukemia effect in vivo. The body weight of the mice did not change significantly (Fig. 4F). Other symptoms such as diarrhea or lipsotrichia were not observed in PBS-, NTD-, or CAR-T-treated groups, suggesting that treatment with CAR-T cells was safe to some extent.
Incorporation of different co-stimulators affected CAR-T cell phenotype
The above studies verified that CD33 CAR-T cells were effective for CD33+ leukemia cells. Differences between the three types of CAR-T cells were investigated further during ex vivo expansion, since studies have shown that the co-stimulator component of the CAR structure may affect the function and phenotype of CAR-T cells, and then influence long-term anti-leukemia efficiency. 16
The phenotype assays of CD33 28z.CAR-T, CD33 BBz.CAR-T, and CD33 28BBz.CAR-T cells showed that BBz.CAR-T cells were biased to CD4+ T cells, while 28z.CAR-T cells were predominantly CD8+ (Fig. 5A). In order to explore whether different co-stimulators may affect the phenotypes of CAR-T cells during in vitro expansion, three types of CAR-T cells were analyzed by memory markers CCR7 and CD45RA (Fig. 5B), which were reported to be related to T-cell persistence in vivo. BBz.CAR-T cells significantly increased the CCR7+CD45RA– central memory compartment (TCM) compared to that of 28z.CAR-T and 28BBz.CAR-T. The mean values were 32.18% versus 12.91% (p < 0.01) and 17.11% (p < 0.01) of CD4+ TCM, and 27.35% versus 6.30% (p < 0.01) and 10.46% (p < 0.01) of CD8+ TCM, respectively (Fig. 5C). The TCM subtype of BBz.CAR-T cells even exceeded that of NTD cells (32.18% vs. 28.58% [p = 0.1819] of CD4+ TCM, and 27.35% vs. 6.63% [p < 0.01] of CD8+ TCM; Fig. 5C).
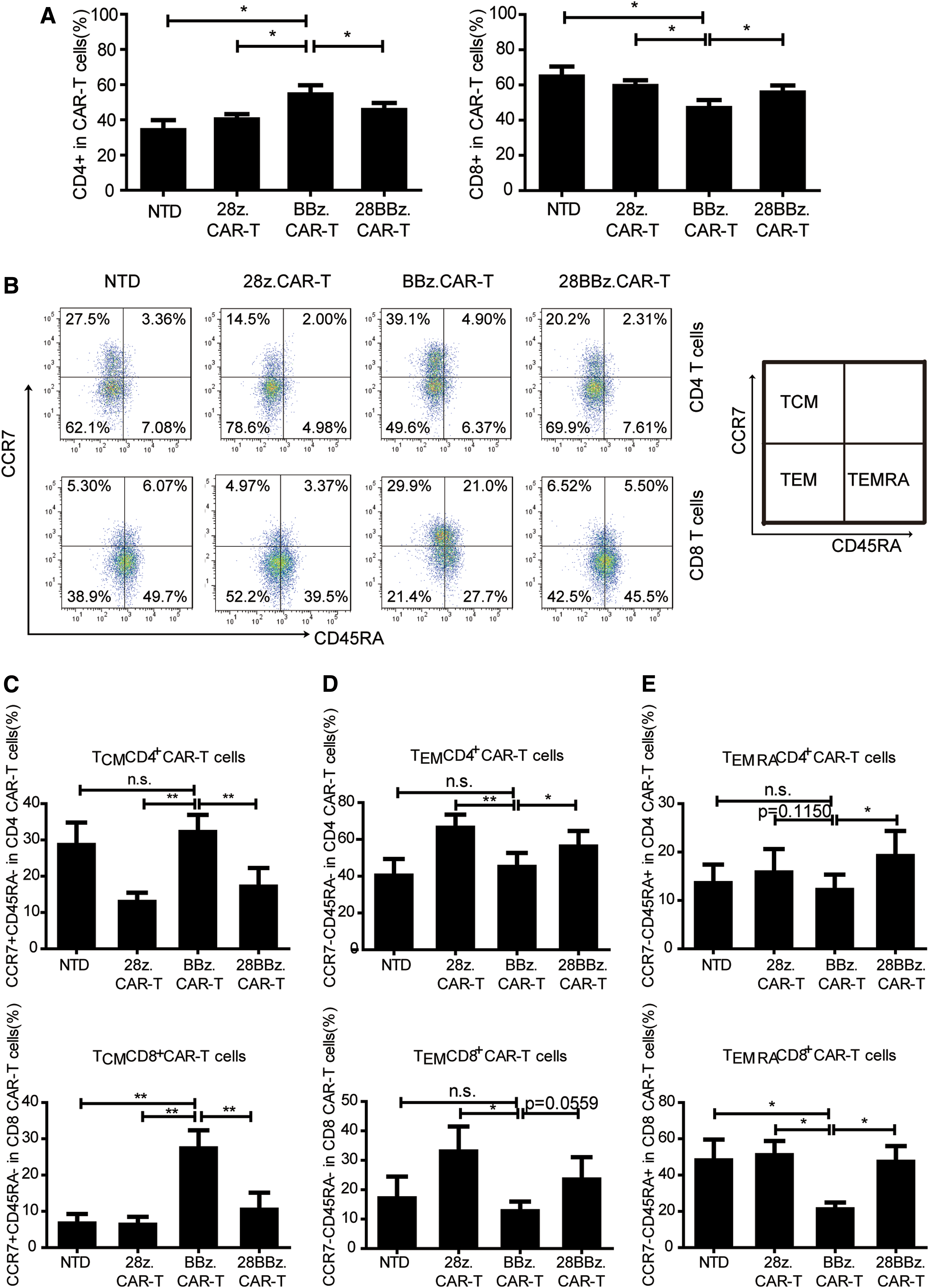
CAR-T cells with different co-stimulators showed varying phenotypes.
In contrast to the TCM, the proportion of effector memory T cells (TEM, CCR7-CD45RA–) in BBz.CAR-T cells decreased compared to that of 28z.CAR-T and 28BBz.CAR-T. The mean values were 45.03% versus 66.35% (p < 0.01) and 56.08% (p < 0.05) of CD4+ TEM, and 12.71% versus 32.98% (p < 0.05) and 23.48% (p = 0.0559) of CD8+ TEM, respectively (Fig. 5D). In addition, compared to 28z.CAR and 28BBz.CAR-T cells, BBz.CAR-T cells seemed to have a decreased proportion of CCR7-CD45RA+ effector memory T cells (TEMRA), the terminally differentiated T cells, especially for CD8+ TEMRA. The mean values for BBz.CAR-T, 28z.CAR-T, and 28BBz.CAR-T were 21.13% versus 50.98% (p < 0.05) and 47.35% (p < 0.05) of CD8+ TEMRA, respectively (Fig. 5E). Similarly, the CD8+TEMRA subtype in BBz.CAR-T cells was less than that of NTD cells (21.13% vs. 48.08%; p < 0.05).
CD33 28z.CAR-T cells were prone to exhaustion during ex vivo expansion
In addition to T-cell type, efficacy and persistence of CAR-T cells, especially efficacy in vivo, may also be affected by T-cell status when transfused into the body. Therefore, the markers representative of T-cell exhaustion were detected during ex vivo expansion. On days 4–14 after infection, CAR-T cells with co-stimulator CD28 displayed a phenotype consistent with exhaustion, including higher expression of cell surface exhaustion–associated biomarker PD-1, LAG-3, and Tim-3 (Fig. 6A), with a higher median fluorescence intensity specific to isotype (Fig. 6B) on day 7. The transcription factors T-bet and Blimp-1, which are considered to reflect T-cell exhaustion, were also elevated in these CD28z.CAR-T cells (Fig. 6C). These phenotypic and transcriptional analyses demonstrated that during the same ex vivo culture condition, CD33 28z. CAR-T cells tended to be exhausted, while 4-1BB molecule may be conductive for CAR-T cells to resist exhaustion.
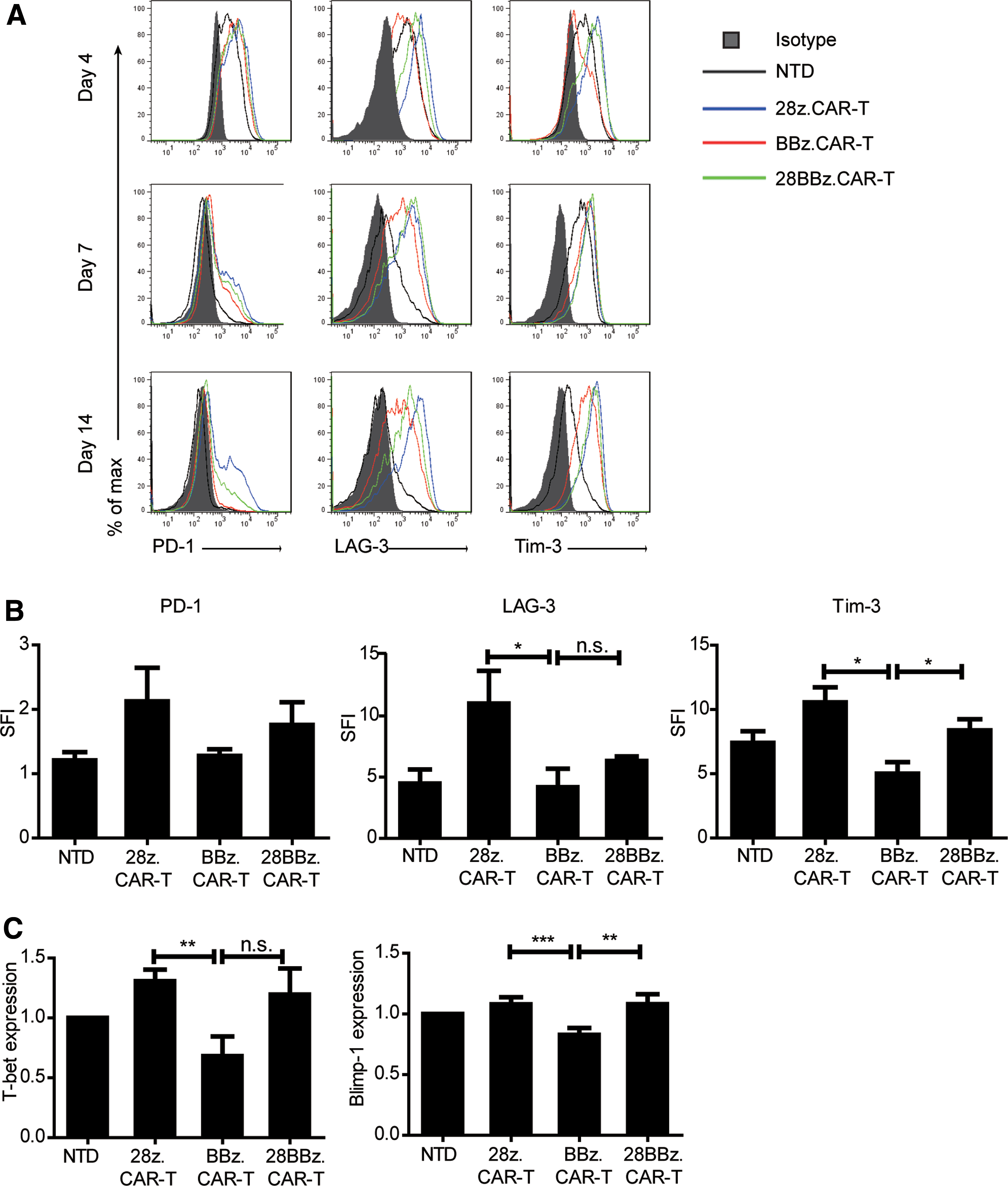
CD33 28z.CAR-T tended to be exhausted during ex vivo expansion.
Discussion
The above results demonstrate the preclinical activity of CD33 CAR-T cells. Three different CD33 CARs, including second-generation CARs with CD28 or 4-1BB as co-stimulator and a third-generation CAR with both co-stimulators, with a scFv derived from self-developed mouse hybridoma cells, were constructed. CD33 CAR-T cells exhibited profound proliferation and potent effector functions against AML cell lines, including specific killing at low E:T ratios of 1:1 or even as low as 1:8, degranulation, and robust cytokine production, as well as prolonged survival time for the CD33+ leukemia mouse. The CD33 CAR-T cells had less of an influence on CD34+ cells, which was confirmed by the colony-forming assay.
There have been several studies on the preclinical effect of CD33 as immunotherapy target, but the efficiency of CAR-T cells varied greatly. In a study of anti-CD33 CAR CIK cells (cytokine-induced killer cells), the E:T ratio was 5:1. 20 In a study of anti-CD33 chimeric receptor expressing EBV-CTL, the E:T ratio was higher at 50:1. 21 O'Hear et al. reported the anti-leukemia effect of CD33 CAR-T cells at lower E:T ratios of 1:20. 22 The cytotoxic efficiency of CAR-T cells may be influenced by a number of variables, such as the avidity of antigen recognition domain derived from the antibody, the structure of CARs, especially the involved co-stimulators, the transduction method, and the T-cell stimulation mode. It is worth noting that recently, CIK-CD33-CAR-T cell was used to treat an AML patient in the Chinese PLA General Hospital. Although the patient gained short-term benefit from CD33 CAR-T, slight fluctuations in bilirubin was the only observed adverse effect, indicating the safety of this therapy to treat AML patients. 23 As with the CD33 CAR-T cells constructed in this study, in vitro studies have shown potent effector functions that were tested by killing effect against AML cell lines, and the cytokines IL-2, IFN-γ, and TNF-α had a sharp increase within 24 h of co-culture. These probably induce some adverse effects, in particular cytokine release syndrome. This suggests that when transfused, the CD33 CAR-T cells should be delivered at a low dose and in a dose escalation manner.
With regard to AML, many other targets have been tried for immunotherapy, such as CD123, CD44v6, and Le-Y. The expression levels of these antigens on AML blast cells and normal tissues are different. Immunotherapies targeting CD123, CD44v6, or Le-Y also achieved good effects on refractory AML. 20,24 –28 The myeloid differentiation antigen CD33, on account of its expression feature, could also be an ideal target for AML immunotherapy.
Studies have shown that CAR-T cells' function and phenotype may be affected by the co-stimulator component. CD19 CAR-T cells containing the co-stimulator 4-1BB had superior anti-leukemic efficacy and improved persistence. 16 In this study, the differences between the three types of CD33 CAR-T cells in terms of phenotype and exhaustion, which were reported to be related to T-cell persistence in vivo, were compared. It was found that in the same culture condition, BBz.CAR-T cells were biased to CD4+ T cells, while 28z.CAR-T cells were predominantly CD8+. Meanwhile, BBz.CAR-T cells had an increased central memory compartment, while 28z.CAR-T cells were inclined to differentiation toward effector memory T cells. The 28z.CAR-T cells exhibited a slightly stronger killing effect in vitro (Fig. 2B), probably because they contained more TEM and TEMRA, which are known for high effector capacity and strongly express important genes in CD8 T-cell effector function. 29 However, TEMRA are terminally differentiated cells characterized by a lack of proliferative capacity, 30 which may result in 28z.CAR-T cells lasting for a relatively shorter duration in vivo.
During expansion, higher expression of Tim-3, LAG-3, PD-1, and transcription factors T-bet and Blimp-1 suggested that 28z.CAR-T cells were more inclined to become exhausted. T-cell exhaustion is considered to be a state of hyporesponsiveness caused by continuous stimulation of external antigens. 31 However, in the culture system for ex vivo expansion used in this study, CAR-T cells appeared to be exhausted without antigen stimulus. The emergence of exhaustion markers was directly related to the difference between co-stimulators. Long et al. also demonstrated that 4-1BB co-stimulation ameliorated T-cell exhaustion induced by tonic signaling of GD2 CAR. 32 It is pivotal to overcome T-cell exhaustion for stronger therapeutic potency of engineered T cells. Eyquem et al. used CRISPR/Cas9 technology to install a CD19-specific CAR to the T-cell receptor α constant (TRAC) locus. Then, the tonic CAR signaling was averted, and differentiation and exhaustion of effector T cells were delayed. 33 In this study, incorporation of the 4-1BB molecule exhibited an anti-exhaustive effect, as shown in Fig. 6, expression levels of Tim-3, T-bet, and Blimp-1 in CD33 BBz. CAR-T cells were lower than in NTD cells. Kawalekar et al. illustrated that different signaling domains have different influences on the state of T-cell metabolism, which can alter the mitochondrial biogenesis and persistence. They found that 4-1BB can enhance the persistence of CAR-T cells and increase the differentiation of CD8+ CAR-T cells toward central memory phenotype, 34 which is in accordance with the present research. These results highlight that incorporation of 4-1BB as a co-stimulator in CARs may improve the long-term effect of CAR-T cells, especially when transfused to the human body.
With regard to anti-leukemic effects, no significant difference was observed between the three types of CAR-T cells in in vitro killing effects or the in vivo anti-leukemia effect. On the one hand, this may be because of the rapid and potent capacity of CAR-T cells that were constructed. On the other, perhaps because it was difficult for human T cells to persist and proliferate efficiently in the mouse environment. To confirm this possibility, CAR-T cell persistence in mice was monitored by detecting their existence in PB. The result showed that CAR-T cells could only exist for 5–7 days after CAR-T cell transfusion, and could hardly be detected in mouse PB 7 days later (data not shown). So, the persistence advantages of CD33 BBz. CAR-T cells observed in the in vitro expansion failed to materialize in in vivo treatment in mice, which resulted in similar in vivo anti-leukemia effects in the three types of CAR-T cells. Repeated CAR-T cell infusion might be administered to acquire better efficacy.
Since CD33 is also expressed on myeloid progenitors, a colony-forming assay was conducted to assess potential on-target off-tumor effects using CD34+ cells sorted from three samples of human UCB. After being co-cultured with 28z.CAR-T cells, the number of CFU generally decreased. For samples 1 and 2, the number of CFU was significantly reduced. However, for sample 2, the number of CFU was comparable between the vector T co-cultured group and the CAR-T co-cultured group, implying that the reduced CFU may be due to the incompatibility of T cells with sample 2 but not the off-tumor effect of the CD33 CAR. For sample 3, co-culture with 28z.CAR-T cells had no obvious influence on the number of CFU (Fig. 2G). The above results suggest that the CD33 CAR-T cells had less of an influence on normal CD34+ cells. In addition, according to the research by O'Hear et al., although the number of CFU was significantly reduced following incubation with anti-CD33 CAR T cells, there were still colonies present, implying a myelosuppressive not myeloablative effect of the CAR-T cells. 22 Therefore, the possible toxicities of CD33-specific CAR-T cells such as hematopoiesis inhibition and agranulocytosis are worth noting and investigating, since there may be individual differences in response to CD33 CAR-T cells.
Preclinical trial results from CD19 CAR-T show that BBz.CAR-T has more persistence in patients and strives for longer remission time in patients with relapsed refractory acute lymphoblastic leukemia (unpublished data). These results suggest that 4-1BB appears to be a better co-stimulator for CARs to produce superior CAR-T cells.
Footnotes
Acknowledgments
This work was accepted as a poster abstract by the ASH Program Committee at the 59th Annual Meeting and Exposition, December 9–12, 2017, Atlanta, GA. This work was supported by the National Natural Science Foundation of China (81430004 and 81570147); the National Key Research and Development Program for Precision Medicine (2017YFC0909800); CAMS Initiative Fund for Medical Sciences (2016-I2M-1-007); PUMC Graduate Innovation Fund (2015-1002-01-19); and PUMC Youth Fund and the Fundamental Research Funds for the Central Universities (2017320031).
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.