
Abstract
Successful translation of chimeric antigen receptor (CAR) T cells designed to target and eradicate CD19+ lymphomas has emboldened scientists and physicians worldwide to explore the possibility of applying CAR T-cell technology to other tumor entities, including solid tumors. Next-generation strategies such as fourth-generation CARs (CAR T cells redirected for universal cytokine killing, also known as TRUCKs) designed to deliver immunomodulatory cytokines to the tumor microenvironment, dual CAR designs to improve tumor control, inclusion of suicide genes as safety switches, and precision genome editing are currently being investigated. One major ongoing goal is to determine how best to generate CAR T cells that modulate the tumor microenvironment, overcome tumor survival mechanisms, and thus allow broader applicability as universal allogeneic T-cell therapeutics. Development of state-of-the-art and beyond viral vector systems to deliver designer CARs coupled with targeted genome editing is expected to generate more effective off-the-shelf CAR T cells with activity against a greater number of cancer types and importantly solid tumors.
Introduction
O
Advances in tumor cell characterization have led to increasing numbers of potential CAR targets. Tumor-specific genomic alterations such as single nucleotide variants, insertions, or deletions can be readily identified by genomic approaches such as whole-exome or transcriptome sequencing, and this information can be used to predict potential tumor-specific antigens and neoantigens. 4 Proteomic approaches such as mass spectrometry may identify tumor-specific fusion proteins that are generated due to chromosomal translocations or proteins that exhibit altered posttranslational modifications specifically in tumor cells and thus may result in expression of novel epitopes that can be targeted by CAR T cells. 5 To exploit this knowledge properly to increase the therapeutic efficacy of novel CAR T cells, it will be important to evaluate on- and off-target activities carefully to balance therapeutic benefit against potential toxic side effects. CAR expression patterns with respect to dose and duration are another critical component of immunotherapies, which may be fine-tuned by empirically testing promoter and posttranscriptional regulatory sequences.
For CAR T cells to be most effective, they should come into contact with cancer cells that express the antigen targeted by the CAR. Due to ease of administration, systemic injection (intravenous) is the preferred delivery modality of CAR T cells. However, as T cells, including CAR T cells, may have difficulty trafficking into solid tumors (e.g., due to physical barriers such as the tumor stroma), improved therapeutic efficacy might be obtained by local administration of CAR T cells either directly into the tumor or intraperitoneally. 6,7 T or CAR T cells delivered intravenously may remain several hours in the lungs before redistribution to the liver, spleen, and lymph nodes, and pulmonary toxicity may develop if activated T cells are not cleared quickly enough from the lungs. In a murine tumor model, CAR T cells delivered intravenously only poorly penetrated tumor tissue, while intraperitoneally or subcutaneously delivered cells remained in close proximity to tumors. 6
The activity of T cells that manage to penetrate the tumor tissue may be compromised by certain characteristics of the tumor microenvironment (TME), such as hypoxic conditions that present a metabolic challenge to T-cell activity. In contrast to most healthy cells, cancer cells were shown by Warburg to use aerobic glycolysis as the main source for energy production. 8 As a consequence of aerobic glycolysis, tumor cells produce and secrete lactic acid. Inhibition of human cytotoxic T-cell proliferation and cytokine production by tumor cell-derived lactic acid could be reversed by blocking tumor cell lactic acid production. 9 Another study found that lower local pH due to lactic acid secretion diminished cytotoxic T-cell activity, whereas hypoxic conditions did not negatively affect cytotoxic T cells. 10 Immune suppressor cells (e.g., tumor-associated macrophages [TAMs], regulatory T cells [Tregs], and myeloid-derived suppressor cells) in the TME also downregulate cytotoxic activity of immune cells, including CAR T cells (Fig. 1). 11,12 TAMs may indirectly suppress antitumor immune functions through secretion of chemokines, such as CCL17, CCL18, and CCL22, which recruit Tregs to the TME, as well as secretion of IL-10 and prostaglandin E2 (PGE2), which induce Tregs, all of which lead to downregulation of cytotoxic T-cell activity. 13 Additionally, TAMs and tumor cells can exploit self-protective functions of immune cells via expression of ligands that stimulate the immune checkpoint function, an important component of immune cell capacity to distinguish between normal “self” cells and foreign or transformed cells. 14,15 For example, expression of the immune checkpoint receptors cytotoxic T lymphocyte-associated 4 (CTLA-4) or programmed cell death 1 protein (PD-1) may be important for T-cell function and contribute to prevention of autoimmune diseases. Tumors can evade the immune response via expression of CTLA-4 ligands CD80/CD86 or PD-1 ligands programmed cell death ligand 1/2 (PD-L1, PD-L2; Fig. 1), and PD-L1 expression on TAMs can contribute to tumor cell immune escape. 15,16 Importantly, PD-L1 and PD-L2 expression correlated with poor prognosis in different tumor types, including breast cancer, urothelial cancer, renal cancer, gastric cancer, melanoma, glioma, ovarian cancer, and lung cancer. 17 –20
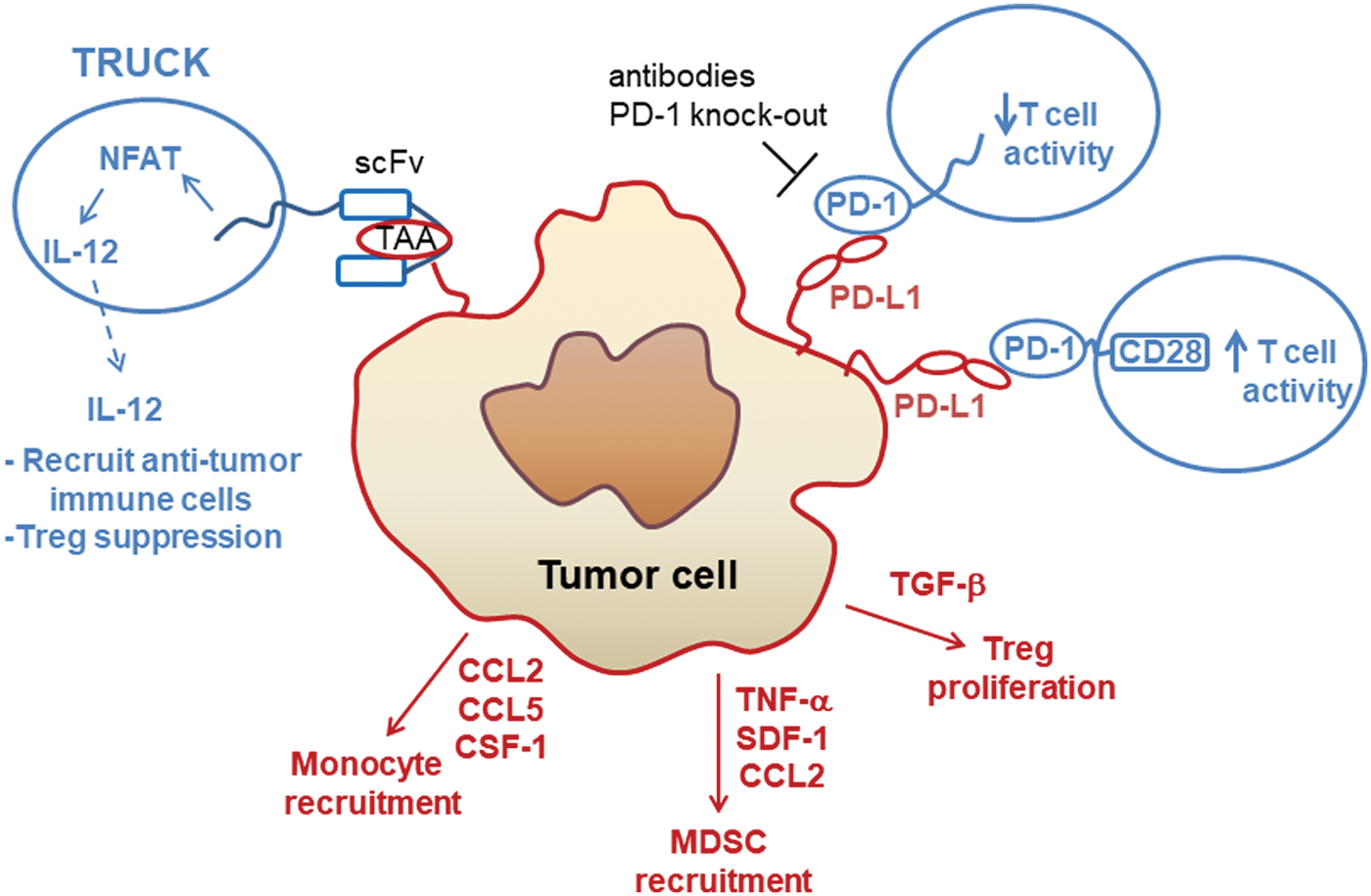
Strategies to overcome the immunosuppressive signaling from tumor cells and the tumor microenvironment. Cancer cells can secrete cytokines and growth factors to suppress immune cell activity by various mechanisms, including support of regulatory T (Treg) cell proliferation or recruitment of myeloid-derived suppressor cells (MDSC). Fourth-generation chimeric antigen receptor (CAR) T cells or T cells redirected for universal cytokine mediated killing (TRUCKs) are engineered to express and release interleukin (IL)-12 inducibly upon recognition of the tumor-associated antigen (TAA) by the single-chain variable fragment (scFv) of the CAR. IL-12 release causes recruitment of antitumor immune cells such as macrophages for additional killing of tumor cells. Many types of tumor cells express the PD-1 ligand (PD-L1), which binds to PD-1 expressed on CAR T cells and leads to decreased T-cell cytotoxicity. This immune checkpoint can be inhibited by therapeutic antibodies or genome editing to disrupt PD-1 expression in T cells. The PD-1/PD-L1 immunosuppressive signaling axis can also be overcome by genetic modification of T cells to express a PD-1-CD28 fusion protein, which stimulates T-cell activity upon binding to PD-L1 on the tumor cell. CAR T cells and their receptors are shown in blue; tumor receptors are in red. Color images available online at
Tumor heterogeneity can present an additional mechanism of tumor cell escape from CAR T-cell therapies, as not all cells of a tumor might express sufficient levels of the targeted antigen to be recognized and eliminated by the CAR T cells. This phenomenon is called “antigen loss,” and relapses due to antigen loss may be prevented by simultaneously targeting two tumor antigens. 21 Thus, improved comprehension of the many mechanisms that cancer cells can employ to evade CAR T cells will aid development of more efficacious strategies for clinical application of CAR T cells and improved patient care.
Car T Cells in the Clinic
Clinical effectiveness of CAR T cells engineered to target CD19 has been demonstrated in patients with acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL), B-cell non-Hodgkin's lymphoma, diffuse large B-cell lymphoma (DLBCL), and follicular lymphoma.
22
–30
In 2017, the Food and Drug Administration (FDA) approved the anti-CD19 CAR T-cell therapies Kymriah™ for refractory or relapsed B-cell ALL and Yescarta™ for refractory large B-cell lymphomas. Encouragingly, the CAR principle was also successfully used to target other antigens in liquid tumor patients, such as anti-CD20 CAR T cells in refractory advanced DLBCL and refractory B-cell non-Hodgkin lymphoma patients (
A Phase I/II dose-escalation (1 × 10
4
to 1 × 10
8
CAR T cells/m2) clinical study treating patients with recurrent or refractory human epidermal growth factor receptor 2 (HER2)-positive sarcomas found HER2-CAR T-cell application to be well tolerated, with no dose-limiting toxicities observed in patients who received >1 × 10
6
/m2 HER2-CAR T cells (
A Phase I dose-escalation trial investigating lymphodepletion via non-myeloablative chemotherapy prior to co-administration of low dose interleukin (IL)-2 with a first-generation CAR containing the CD3ζ signaling domain and designed to target the prostate-specific membrane antigen (PMSA) in six prostate cancer patients with recurrent or metastatic disease demonstrated anti-PSMA-CAR T cell engraftment efficiencies of 2.5–56%, with >20% of all patients who received 10 10 activated T cells containing at least 10% anti-PSMA-CAR T cells. 45 Of interest, the two patients who achieved partial responses as measured by reduced prostate-specific antigen levels received the lowest cell dose (10 9 CAR T cells) and had the lowest percentages of activated CAR T cells (5% and 12%), suggesting that the administered IL-2 dose (75,000 IU/kg/day for 4 weeks) was insufficient for those patients exhibiting greater CAR T-cell engraftment. 45 The authors observed no dose-limiting toxicities, no cytokine storm, and no apparent off-tumor CAR T cell–induced cytotoxicity in normal tissues that express PSMA (e.g., no central nervous system [CNS] or renal toxicity).
Access of immune cells, including CAR T cells, to the CNS is usually low since migration of these cells into the CNS is controlled by the endothelial blood–brain barrier and the epithelial blood–cerebrospinal fluid barrier.
46
A Phase I study in one patient whose highly aggressive glioblastoma had failed to respond to other therapies was treated by intracranial infusion of IL13Rα2-CAR T cells into the resected tumor cavity (CAR T-cell doses: initial dose 2 × 10
6
, followed by five doses of 10 × 10
6
), which provided local tumor control but did not prevent occurrence of new metastatic lesions in the spine (
Clinical experience with CAR T cells to treat solid tumors suggests that improvements in cancer cell recognition as well as CAR T-cell persistence and activity will lead to increased efficacy. More complete recognition of tumor cells can be achieved by engineering T cells to express CARs that target more than one tumor antigen. However, while the risk of on-target off-cancer effects that may cause unintended destruction of healthy tissue seems to be manageable with anti-CD19 CARs that also ablate the normal B-cell population, on-target off-cancer effects may be more challenging in other cases, such as with CAR T cells that target multiple TAA or depletion of cells with stem-cell markers. In some cases, CAR T-cell persistence and antitumor function can be improved by co-administration of immune checkpoint inhibitors. 44 An additional challenge to CAR T-cell therapy against solid tumors is the release of chemokines and cytokines that recruit immunosuppressive cells to the TME. Modification of CAR T cells to overcome or avoid these immunosuppressive signals would improve the antitumor activity of CAR T cells. To achieve success rates such as those obtained in patients with liquid tumors, strategies to surmount challenges presented by solid tumors seem to be necessary. State-of-the-art vector systems can be engineered to deliver designer CAR modalities that allow more complete recognition of tumor cells (e.g., via expression of more than one CAR from a single vector), improved control of the TME (e.g., CAR T cells redirected for universal cytokine killing [TRUCK] concept), increased cell persistence and/or functionality (e.g., checkpoint inhibition), decreased alloreactivity (e.g., genomic editing), and the possibility for in vivo selection and/or elimination of CAR T cells (e.g., co-delivery of truncated receptors or suicide cassettes). In the following sections of this review, an overview is provided of the vector technologies that can be employed for CAR delivery and the exciting possibilities of achieving improved cancer control when CAR T-cell design is coupled with advanced genomic editing technologies.
Vector Developments for Cars
Retroviral vectors are particularly suitable for delivery of therapeutic transgene cassettes, as they can stably anchor their genetic cargo into the host cell genomic DNA of hematopoietic cells, including lymphoid cells, and thus allow a long-term therapeutic benefit. In particular, murine leukemia virus (MLV)-derived gammaretroviral and human immunodeficiency virus (HIV)-derived lentiviral vectors were successfully used in a number of clinical gene therapy trials for the treatment of severe combined immunodeficiencies (SCID; including X-linked SCID, 47 ADA-SCID, 48,49 and Wiskott–Aldrich Syndrome 50 ), HIV infection, 51,52 metabolic diseases (metachromatic leukodystrophy 53 and adrenoleukodystrophy 54 ), as well as cancer therapy. 22 –28,30,55,56
Conceptually, two vector architectures are frequently used in clinical CAR trials: (1) long terminal repeat (LTR)-driven retroviral vectors, and (2) so-called self-inactivating (SIN) vectors, which harbor a large deletion in the U3 promoter/enhancer regions of the LTRs. LTR-driven vectors employ the U3 promoter region to drive CAR expression, often MLV, myeloproliferative sarcoma virus (MPSV) 57 –59 or spleen focus-forming virus (SFFV) 60 derived, which are very potent and strong promoters in hematopoietic cells, including T cells. To boost expression further, intronic sequences are included in the 5′ untranslated region (UTR). While these LTR-driven vectors mediate strong and robust CAR expression, SIN vectors (including extensive deletions in the U3 region) are dependent on an internal promoter of choice. This allows fine-tuning of CAR expression, which may be necessary to balance the overall avidity of the CAR to a given application/disease setting, as well as on- and off-target activities. Thus, this needs to be carefully tested empirically in appropriate preclinical models and decided on a case-by-case basis. Commonly used internal promoters with moderate activity in lymphoid cells include promoters from constitutively expressed cellular genes, for example elongation factor 1α (EF-1α) and phosphoglycerokinase (PGK). 21,61,62 Direct comparison of several promoters (cytomegalovirus, EF-1α, ubiquitin C, and PGK) in CD4+ and CD8+ T cells demonstrated mostly stable transgene expression in both cell subtypes for >2 weeks based upon enhanced green fluorescent protein expression. 63 There are different strategies available to allow co-expression of multiple genes from a single vector, including incorporation of multiple promoters (e.g., one for each gene to be expressed), bidirectional promoters, or use of internal ribosomal entry site (IRES) elements or 2A sequences to allow expression of multiple transgenes from a single promoter. 64
Posttranscriptional regulatory elements (PRE), for example the Woodchuck hepatitis virus (WHV) PRE, can be embedded into the 3′ UTR to stabilize and improve CAR expression. 57,65 –68 There are several WHV PRE elements available (600 vs. 900 bp), which—to varying degree in a context dependent fashion—enhance CAR mRNA nuclear export and stability, and therefore expression and viral titer. 57,68 However, in concert with the 5′ promoter driving the CAR expression cassette, the choice of a PRE element needs to be carefully balanced to achieve the appropriate level of CAR expression. In addition to PRE elements, mRNA stability and export may also be improved by codon-optimization and/or upstream polyadenylation enhancer elements. 69
In addition to gammaretroviral and lentiviral vectors, novel SIN vectors based on alpharetroviral Rous sarcoma virus and foamy virus 70,71 have also been created, which tend to have a more neutral integration behavior with respect to localization close to promoter/enhancer elements and gene transcription units, 72 and are thus considered to be safer tools for genetic modification. 73,74 Other interesting modalities for delivery of CARs were successfully developed and applied in clinical trials, including mRNA delivery 75,76 and Sleeping Beauty transposons. 77 While lentiviral, gammaretroviral, and alpharetroviral vectors can be used to deliver transgene cassettes up to approximately 8–10 kb in size, 78 –81 the non-viral Sleeping Beauty transposon/transposase system can efficiently transfer transgene cassettes >10 kb in length, although with lower efficiency than transfer of 7 kb transgenes. 82,83 In contrast to gammaretroviral and lentiviral vectors, Sleeping Beauty gene transfer exhibits a rather neutral integration pattern in primary T cells. 77 The amount of genetic material that can be efficiently transferred to T cells is an important consideration, depending upon the complexity of the CAR construct, and may help determine which delivery system is most appropriate for the intended application.
Currently, two CAR T-cell products have market authorization: the lentiviral Kymriah™ and the gammaretoviral Yescarta™. However, several lentiviral 23 –25,84 and gammaretroviral 85 vectors as well as Sleeping Beauty 77 constructs have been used to generate CAR T cells that were tested in clinical trials. Transduction efficiencies of CAR vectors into T cells can depend on T-cell sources but may also depend upon the strategy used to modify the cells. Use of lentiviral vectors to transduce T cells with CARs was reported to proceed with a median of 20% (range 4.7–39.2%) in end-product cells 84 and a median of 39.8% (range 4.4–69.3%) of CD3+ T cells in patients with responses were found to express the CAR construct. 24 A median of 11.6% (range 5–14.2%) was reported for gammaretroviral CAR transfer into T cells by one group, 86 while others detected a median of 24% (range 5–60%) CAR-positive T cells. 87 Sleeping Beauty CAR transfer into T cells was >80% (range 59–97%), with an anti-CD19-specific CAR as quantified following selective expansion of electroporated T cells on γ-irradiated artificial antigen-presenting cells. 77
As clinical studies showed that some relapsed tumors after CAR T-cell therapy do not express the antigen targeted by the CAR, 7,23,24 T cells expressing more than one CAR to target additional tumor antigens may be more effective in tumor control. In this regard, bispecific anti-CD20-CD19 CAR T cells outperformed anti-CD20 CAR T cells and anti-CD19 CAR T cells in a mouse model of mixed phenotype CD19+CD20+/– pediatric acute lymphocytic leukemia. 88 Tandem CAR T cells designed to target both HER2 and IL13Rα2 from a single CAR molecule exhibited improved T-cell activity without increased T-cell exhaustion and had better tumor control in a mouse glioblastoma model, likely due to reduced antigen escape of tumor cells. 89 The feasibility of creating a CAR T-cell therapy, in which two entire CAR molecules are expressed in each CAR T cell, was recently demonstrated by transduction with two lentiviral constructs designed to express second-generation CARs containing a CD8 hinge, 4-1BB costimulatory domain, and a CD3ζ signaling domain via the elongation factor-1α (EF-1α) promoter. 21 Dual CART123/CART19 cells showed greater control of B-ALL than either CAR T-cell alone or a mixture of both CAR T-cell types. These findings have direct impact for clinical strategies, as loss of antigen expression (or CAR T-cell selection for antigen-negative tumor cells) is observed in tumors from relapsed patients. In this study, CD19+ and CD19− B-ALL cells were efficiently depleted with the dual CART123/CART19.
A similar approach to express two CARs in CAR T cells for better tumor control was tested in multiple myeloma. Here, the authors used a single lentiviral construct to express two entire second-generation CARs: one against BCMA and the second against CS1, each containing a CD8 hinge and transmembrane domain, the 4-1BB costimulatory domain, and a CD3ζ signaling domain. 90 The strong SFFV promoter was used for dual CAR expression, and the self-cleaving P2A peptide was incorporated between the two CAR sequences to allow expression of each CAR on the T cells. Compound CAR T cells exhibited superior in vitro and in vivo anti-myeloma activity compared to CAR T cells that targeted a single antigen. This strategy may advance myeloma cell therapeutic approaches and help abrogate relapses observed in earlier CAR T cell–treated myeloma patients. 35 The compound CAR T-cell paradigm may be applicable to additional tumor entities and represents a promising advance in anticancer therapy, but the potential side effects must be carefully assessed with each new CAR construct.
The non-viral Sleeping Beauty transposon/transposase system was used to generate anti-CD19 CAR T cells for Phase I trials that used these CAR T cells during minimal residual disease after hematopoietic stem cell transplantation in non-Hodgkin lymphoma and ALL patients (
Safety switches can be engineered into CAR vectors to allow the opportunity for controlled elimination of CAR T cells in the case of nonspecific cytotoxic activity. Depletion of cells via iCasp9 is caused by application of AP1903, a clinically allowed small molecule dimerizer that activates iCasp9 and thus induces apoptosis of the expressing cells. 93 An alpharetroviral SIN vector with a MPSV promoter to express a third-generation CAR (CD28, 4-1BB, CD3ζ) directed against CD123 and an iCasp9 suicide cassette via an IRES element was successfully used to generate CAR T cells from cord blood–derived CD34+ cells. 94 This work demonstrated that use of the elongation-factor-1-short-form (EFS) to express iCasp9 resulted in only minor levels (<30%) of induced cell apoptosis upon addition of dimerizer agent, while iCasp9 expressed from the MPSV promoter eliminated >90% of transduced T cells, suggesting inadequate iCasp9 expression from the EFS promoter. 94 It would be of interest to replace the IRES component with a self-cleaving 2A sequence to explore whether this would allow greater iCasp9 expression in this setting. As an alternative safety switch, the herpes simplex virus thymidine kinase can be engineered into the CAR vector, and CAR T-cell elimination can be induced by application of ganciclovir. 95 –97 Furthermore, CAR T cells can be engineered with 2A self-cleaving sequences to co-express CARs with truncated EGFR or truncated CD19 to provide the possibility of eliminating the CAR T cells by administration of depleting antibodies. 7,98
While alpharetroviral vectors have yet to be tested in clinical trials, they were successfully used to transduce T and natural killer (NK) cells efficiently in preclinical settings. 99 –101 In addition, it was demonstrated that alpharetroviral SIN vectors can be produced in stable human 293-based packaging cell lines to achieve robust, high-titer alpharetroviral production for >17 weeks. 100 Furthermore, to avoid the problem of superinfection of human packaging cells during long-term culture and virus production, a CRISPR-Cas9-based nuclease approach was developed to stop productive retroviral entry into human 293 packaging cells. 100 These two developments (i.e., the generation of stable packaging lines and the lack of superinfection) will enable a valuable perspective to solve potential production bottlenecks in case cost-effective large-scale manufacturing is needed to make gene therapy accessible to larger patient cohorts.
Compared to viral vectors, production of Good Manufacturing Practice (GMP)-compliant plasmid vectors for use in the Sleeping Beauty gene transfer system is cheaper and can be relatively easily scaled up, and Sleeping Beauty vectors may have a longer shelf life. 92 In short, integrating retroviral (including lentiviral) and Sleeping Beauty vectors are suitable systems to transduce primary human T cells, even with complex CAR constructs designed to express suicide genes (e.g., inducible Caspase-9, which is around 1,250 nucleotides long), selection markers (e.g., truncated CD19, which is approximately 1,002 nucleotides long), in addition to CAR sequences, which are generally >2,000 nucleotides in length. As discussed above, some integrating vectors seem to have more random integration profiles (e.g., alpharetroviral and Sleeping Beauty vectors) and thus might be considered to be inherently safer than other integrating systems. 73,77 However, advances in retroviral vector design such as the SIN architecture and incorporation of physiologic promoters to drive transgene expression have alleviated the adverse events due to proto-oncogene activation in transduced cells observed in some cases when earlier generation vectors were used to transduce hematopoietic stem cells. 47 Furthermore, the cellular context is an important factor here, with stem-cell populations being more susceptible to insertional mutagenesis compared to terminally differentiated somatic cells such as T cells. Of note, even in T cells modified with LTR-driven vectors, there have been no reports of insertional mutagenesis. As an increased safety precaution, the integration capacity of retroviral vectors can be deleted to allow use of the gene transfer function without permanent modification of the target cell genome. 102,103 Anti-CD19 CAR T cells generated with a non-integrating lentiviral vector containing a scaffold/matrix attachment region (S/MAR) element exhibited similar CAR expression levels and cytotoxic activities as T cells transduced with an integrating lentiviral anti-CD19 CAR vector. 104
State-of-the-art vector systems can be used to control CAR expression, as well as to deliver complementary strategies to improve CAR T-cell function. As described in the following sections, the anticancer activity of CAR T cells can be enhanced by various approaches to disable the immunosuppressive activity that the tumor cell niche exerts on T cells.
Next-Generation Strategies to Address Challenges to Immunotherapy
Vector systems can be engineered to create CAR T cells that also exhibit enhanced functions to counteract various challenges presented by the immunosuppressive TME described above. Remodeling the tumor niche by elimination of cells that promote tumor escape mechanisms or by promotion of antitumor immune cell recruitment has been shown. For example, CAR T cells retargeted against CD123 killed Hodgkin lymphoma cells and TAMs, demonstrating the effectiveness of CAR T therapy to overcome the immunosuppressive nature of the TME via elimination of TAMs. 105
CAR T cells can also be used to deliver cytokines to modulate the TME (Fig. 1). Abken et al. developed a promising strategy they termed CAR T-cell redirected for universal cytokine killing (TRUCKs), which can be used to modulate the immunological balance between pro-tumor and pro-immune response. 106,107 In this work, the authors designed CAR T cells to express IL-12 via an engineered switch through NFAT signaling that is activated upon recognition of the tumor antigen. These CAR T cells deliver IL-12 to the tumor and avoid toxicities that would be caused by systemic IL-12 application. This approach resulted in improved antitumor response via recruitment and activation of innate immune cells, such as antitumor macrophages to the microenvironment, elegantly demonstrating the potential therapeutic significance of CAR T cells engineered to modify the TME specifically. The importance of this anticancer tactic is further underscored when the heterogeneous nature of tumors is considered. Different tumor cells may exhibit variable antigen expression levels, and those expressing low amounts or none of the antigen targeted by CAR T cells could otherwise escape being eradicated if IL-12 (or other cytokines) were not delivered to recruit the anticancer effects of additional innate immune cells. For example, it was recently demonstrated that infusion of CAR T cells that release IL-18 into the TME upon antigen recognition elicited superior tumor control than CAR T cells not engineered to secrete IL-18. 108 IL-18 delivery to the tumor was observed to elicit several antitumor activities, including decreased numbers of immune suppressive cells (Tregs, CD103+ dendritic cells, M2 TAMs), increased numbers of antitumor immune cells (CD206− M1 macrophages, NKG2D+ NK cells), and interruption of immune checkpoint signaling by downregulation of FoxO1 and, subsequently, PD-1 expression on T cells. 108
As introduced earlier, interference with immune checkpoint function is another way to increase the antitumor activity of CAR T cells. Due to the putative relevance of the PD-1–PD-L1/PD-L2 signaling axis for cancer treatment, several anti-PD-1/PD-L1 blocking antibodies were designed, including the anti-PD-1 antibodies pembrolizumab (Keytruda) and nivolumab (Opdivo) and the anti-PD-L1 antibodies atezolizumab (Tecentriq), avelumab (Bavencio), and durvalumab (Imfinzi). A randomized, open-label international Phase III study of non-squamous non–small cell lung cancer (NSCLC) patients whose disease progressed during or following platinum-based chemotherapy found that the anti-PD-1 antibody nivolumab provided superior overall survival compared to docetaxel (
A retrospective study demonstrated the feasibility of allogeneic hematopoietic stem-cell transplantation in relapsed or refractory lymphoma patients who previously received PD-1 inhibitors. However, patients treated with PD-1 inhibitor had a decreased ratio of Tregs to conventional CD4 and CD8 T cells and may have elevated risk of early immune toxicity due to PD-1 inhibitor-induced immunologic changes. 111 Another adverse event observed in NSCLC patients treated with PD-1 inhibitors is pneumonitis. 112 As systemic PD-1 inhibition is also associated with toxicities due to increased activation of autoreactive T cells, 113 other strategies to block immune checkpoints to improve anticancer activity of CAR T cells may be needed for clinical application. For example, engineering CARs with different costimulatory or signaling domains may be an alternative to PD-1 ablation. 114 Interestingly, CAR T cells designed to express a PD-1-CD28 switch receptor, in which the extracellular domain of PD-1 is fused to the CD28 transmembrane and signaling domains, exhibited enhanced activity against solid tumors (Fig. 1). 115 This work demonstrates the feasibility of employing CAR technology to change an immune inhibitory signal into an anticancer immune activity, and represents a novel strategy that may find broader use for generation of modified T cells that exhibit improved tumor control.
Genome Editing to Create Designer Cars
Modern molecular biological technologies using nucleases such as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), or clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) provide the possibility of precisely editing the genome and specifically knocking out unwanted genes. These nucleases cause DNA double-strand breaks, which can be repaired by non-homologous end-joining (NHEJ) or homology-directed repair. NHEJ can result in the generation of insertion and deletion mutants, which can be exploited to disrupt target gene transcription or translation. ZFN and TALEN approaches are more laborious, as they utilize protein–DNA interactions for gene targeting, while the CRISPR-Cas9 system allows greater flexibility, as an RNA guide sequence directs the genetic editing to a specific DNA sequence. 116 However, all three techniques were successfully used to edit CAR T-cell genomes.
CRISPR-Cas9 was successfully employed to make CAR T cells less susceptible to the immunosuppressive effects of immune checkpoint by knockout of lymphoid activation gene-3 (LAG-3), a negative regulator of T-cell activity. 117 LAG-3 expression on intratumoral T cells from follicular lymphoma patients was observed mainly in the PD-1+ population and identified functionally exhausted T cells. 118 Perhaps clinically important, LAG-3 expression correlated with poor outcome in these patients. 118
CRISPR-Cas9 was also applied to interfere with the PD-1/PD-L1 signaling axis and thus improve CAR T-cell effectiveness and possibly treatment safety by eliminating the need to employ anti-PD-1 antibodies. Nucleofection of CRISPR-Cas9 ribonucleoprotein to disrupt PD-1 in CAR T cells resulted in anti-CD19 CAR T cells with better tumor control in vitro and in an in vivo murine subcutaneous tumor xenograft model, raising the possibility that CAR T-cell activity can be improved by exploiting the mechanisms through which PD-1/PD-L1 signaling inhibit CAR T-cell function or signaling. 114 Several studies have explored the possibility of using CRISPR-Cas9 for multiplex genome editing to generate CAR T cells with improved function. T-cell receptor α chain (TRAC) and beta-2 microglobulin (B2M, essential for HLA-I heterodimer cell surface expression) were targeted to create double-knockout CAR T cells, while TRAC, B2M, and PD-1 were disrupted to make triple knockout CAR T cells. 119 The authors of this study tested whether the order of T-cell modification influenced the efficiency of the entire procedure. They found that lentiviral transduction of T cells with the CAR construct first followed by electroporation of Cas9 protein and in vitro transcribed gRNA for genome editing led to superior gene editing efficiency and CAR T-cell viability. Although similar gene editing efficiencies were obtained with single-, double-, or triple-knockout experiments, enrichment of gene edited CAR T cells via negative selection resulted in purity of >95% for double knockout cells but ranged from 57% to 90% for triple-knockout cells, indicating that editing strategies involving more than two knockouts was suboptimal for clinical application using these settings. Furthermore, gene edited CAR T cells expanded about 100-fold, and non-edited CAR T cells expanded about 300-fold. Regarding functional activity of gene edited CAR T cells, the TRAC/B2M double knockout CAR T cells exhibited better tumor control than non-edited CAR T cells in a lymphoma xenograft mouse model. 119 Similarly, another study using primary human T cells demonstrated that simultaneous disruption of the endogenous TCR, B2M, and PD-1 resulted in CAR T cells with greater in vivo anticancer activity and reduced alloreactivity with no evidence of graft-versus-host-disease (GvHD). 120 Here, stimulated T cells were transduced with lentiviral CAR vectors and then electroporated to deliver Cas9 mRNA and gRNAs. This “hit and run” transient CRISPR-Cas9 approach to disrupt the endogenous TCR seems to provide an acceptable balance between genome editing efficiency and safety, although subsequent T-cell expansion of edited cells was reduced likely due to toxicity of electroporation. 120 Importantly, no off-target mutations were detected when high-fidelity eSpCas9 was used. High-fidelity Cas9 variants were developed to reduce SpCas9 off-target activities to increase the specificity, and thus the safety, of this genome editing technology. 121,122 Precise gene editing with the one-shot system was reported using the high-fidelity Cas9 variant eSpCas9 to generate allogeneic universal CAR T cells via knockout of TCR and HLA-I with a double-knockout efficiency of 47%. 123 The tumor necrosis factor α family of death receptors includes the Fas receptor, which was implicated in activation induced CAR T-cell death. 124 TCR/HLA-1/Fas triple CAR19 T-cell knockouts exhibited increased longevity due to resistance against Fas-induced death. 123 Here, the most efficient protocol to generate gene edited CAR T cells was first to transduce T cells with a lentiviral vector to deliver the CAR construct and guide RNA and then delivery of Cas9 mRNA via electroporation 2 days later. This method provided higher genome editing efficiency than the TALEN approach, but efficiencies decreased with increased numbers of gene targets (e.g., targeted disruption of three genes was more efficient than disruption of four genes). 123 This might be overcome by multiple delivery of Cas9, but this may also cause increased off-target effects. The authors also noted that the amount of genetic cargo that can be packaged by lentiviral vectors designed to deliver guide RNAs and a CAR was a limiting factor when more than four genes are targeted for editing.
While genetic modification of allogeneic T cells to express tumor-specific CARs can improve cancer control, CAR T cells still have the risk to induce GvHD due to the endogenous αβ T-cell receptor (TCR). To ameliorate this potential hazard, ZFN were designed to eliminate αβTCR expression by targeted disruption of the α or β constant region sequences (TRAC and TRBC, respectively) in CAR T cells. 125 The Sleeping Beauty vector system was used to express an anti-CD19 CAR stably followed by several rounds of selective propagation via stimulation with γ-irradiated CD19+ artificial antigen-presenting cells to achieve >90% CAR-expressing T cells. CAR T cells were then electroporated with mRNA coding for TRAC or TRBC ZFNs. Since ZFNs can permanently disrupt gene expression, transient ZFN application is expected to improve safety by decreasing the likelihood of off-target gene editing events. 125 Another approach used CRISPR-Cas9 to insert a CD19-specific CAR into the TRAC locus, which resulted in uniform CAR expression and enhanced activity of modified human T cells in an ALL mouse model. 126 Cas9 mRNA and gRNA were delivered into stimulated T cells by electroporation to achieve approximately 70% TRAC knockout, and the CAR was delivered 2 h later via transduction with an adeno-associated virus vector with an efficiency >40% and a multiplicity of infection of 10 6 . 126
In a similar approach, Qasim et al. designed TALENs to disrupt expression of the αβTCR by targeting the constant region of the TRAC to create universal CAR19 T (UCART19) cells with a reduced risk of GvHD.
127
The authors used another TALEN pair to target the CD52 gene to endow UCART19 cells simultaneously with resistance to Alemtuzumab (Campath®), a monoclonal antibody used to target and eradicate CD52+ lymphocytes (e.g., B cells, T cells, and NK cells). Alemtuzumab has U.S. FDA approval for treatment of B-cell CLL patients who were treated with alkylating agents and who failed fludarabine therapy.
128
In their landmark study, Qasim et al. demonstrated the feasibility of using UCART19 cells as a bridge-to-transplantation strategy in two infant patients (aged 11 months and 16 months) with high-risk B-ALL, both of whom failed to respond to the bispecific T-cell engager blinatumomab that targets CD19 and CD3.
127
The UCART19 approach is currently being tested in Phase I clinical trials in children (
While recombinant DNA technologies, including ZNF, TALENs, and CRISPR-Cas9, may be successfully used to modify human cells to improve treatment of diseases, there are also risks and potential ethical conflicts associated with these procedures. 129 –131 The possibility of using nucleases to create DNA double-strand breaks at selected sites in the genome can be harnessed to treat disease, but it is important to remember that double-strand DNA breaks are also recombinogenic and mutagenic. 132 The potential to acquire cooperative oncogenic events increases with each genetic manipulation to which a cell is exposed. Thus, multiple genetic manipulations are expected to carry higher risks than strategies involving only a single genetic insult. Considering that ZFNs, TALENs, and Cas9 can exert their effects directly on the genome to cause permanent genome editing and in the worst case even translocations, transient delivery methods, such as electroporation of mRNA encoding for the nuclease or integration-deficient retroviral vectors, may be safer modalities to limit potential adverse effects of long-term nuclease expression via integrating vectors. 120,125,127 Regardless, the potential for genotoxicity of engineered CAR T cells should be considered and should be incorporated into a rational risk–benefit assessment.
In summary, the relative difficulty of engineering ZFNs with domains that exhibit sufficiently high specificity and affinity seems to be a limitation for broader use of this technology in T cells, although ZFN can be successfully delivered into T cells by electroporation or integration deficient lentiviral vectors. 125,133 In the case that more than one gene is to be targeted, the individual ZFNs should be delivered sequentially to limit off-target activity due to possible formation of undesired ZFN dimer pairs. 133 Compared to ZNF, TALENs exhibit higher genome editing activity 134 with less nuclease-associated toxicity and lower off-target effects. 135 Disadvantages of TALENs include the highly repetitive sequence and larger size compared to ZFN. Indeed, lentiviral delivery of TALENs failed due to recombination events in the TALE repeat array as a consequence of the error-prone reverse transcriptase and RNase H activity used by retroviruses to generate the proviral chromosomal insertion (but was successfully accomplished with adenoviral vectors). 136 CRISPR-Cas9 technology is currently the most easily programmable genomic editing tool and best suited for multiplexing to allow simultaneous disruption of several target genes. 120,123 However, as discussed above, efficiency of gene editing decreases when more than three genes are targeted. 123
Recently engineered Cas9 variants exhibit improved specificity for genomic target sequences, 121,122 and CRISPR-Cas9 gene editing seems to be very specific in T cells. 120 However, the risk of unintended genomic modification cannot be completely ruled out, and the possibility of undesired off-target activity may increase as the number of genes targeted for editing increases. Therefore, it is critical that state-of-the-art technologies such as deep sequencing are applied and that results of these tests are carefully analyzed to detect any potential off-target genomic editing prior to administrating modified cells to patients. Furthermore, the safety of CRISPR-Cas9-edited CAR T cells can be estimated in suitable animal models such as nonhuman primates. The safety of clinical use of cell products that contain multiple edited genes remains to be determined.
Discussion and Perspective
The recent dramatic clinical success in treatment of B-cell malignancies led to the approval of the first CAR T-cell therapies in 2017. The combination of state-of-the-art vector systems and advanced genomic engineering technologies have greatly aided development of CAR T cells with greater therapeutic potency. Continued development of improved methods for targeted genome modification, such as high-fidelity Cas9 variants that exhibit less off-target activity, will increase the safety of genome editing with the goal of generating CAR T cells with increased clinical efficacy. Such genome editing strategies can be used to generate “universal” CAR T cells to replace the currently used patient-specific CAR T cells. This would help expedite delivery of CAR T-cell therapies to patients, as CAR T cells could be prepared and stored as an off-the-shelf medicine without the patient having to wait for their T cells to be isolated, modified, and expanded. Off-the-shelf CAR T cells would also eliminate the uncertainty of CAR T-cell delivery, as the risk of failure to produce suitable numbers of patient-specific CAR T cells would be mitigated. Additionally, this would be expected to reduce production costs greatly, especially when coupled with viral production via stable packaging cell lines.
However, there are still some hurdles that have to be overcome to broaden the applicability of CAR T-cell therapy to additional cancers, including solid tumors. Advances in genome editing applications to improve CAR T-cell activity by disruption of immunosuppressive signaling axes are particularly promising. Additional strategies such as fourth-generation CARs (TRUCKs) designed to deliver immunomodulatory cytokines to the TME were also shown to overcome tumor survival mechanisms by modulation of the TME via recruitment of additional antitumor immune cells.
Efforts to curtail the possibility of antigen escape by tumors that exhibit heterogeneous expression of antigens targeted by CAR T cells resulted in improved tumor control with tandem, dual, and bispecific CARs, which target two different TAA. As the number of targeted antigens increases, the potential risk of off-target cytotoxicity also increases. Therefore, incorporation of safety mitigation strategies, such as inclusion of inducible suicide genes or truncated receptors that allow antibody-mediated removal of CAR T cells, are important considerations for CAR vector design.
One factor limiting more rapid progress in the field of CAR T-cell therapy is the current lack of suitable viral vector production facilities. Several academic and industrial laboratories are available for production of GMP-grade viral vectors (including Batavia Biosciences, EUFETS/BioNTech, King's College, Indiana University, Virxsys, Généthon, Molmed, Beckman Research Institute, and Oxford BioMedica), but the global demand for vector production has greatly increased, largely due to the recent clinical success of CAR T-cell therapeutics. As the waiting time for production of some new gene therapeutic vectors can be several years, 137 more viral vector production sites, as well as improved technologies to allow more efficient production of the therapeutic vectors and/or modified T cells, are critically needed.
As cell surface proteome analyses continue to advance, identification of novel neoantigens is awaited that can be targeted by CARs to allow greater tumor specificity and decreased loss of healthy tissue. More detailed understanding of the TME and how it affects immune cell function will also lead to enhanced CAR T-cell therapies. The recent authorization of CAR T-cell therapies to treat different types of liquid tumors has further raised expectations for the possibilities of expanding immunotherapeutic approaches to treat additional types of cancer, including solid tumors. Here, it is important to assess soberly and apply the lessons learned from the multitude of earlier studies to devise strategies to convert additional challenges presented by solid tumors successfully into therapeutic opportunities that will eventually allow delivery of efficacious CAR T-cell therapies to a greater number of patients.
Footnotes
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB738, Cluster of Excellence REBIRTH [EXC 62/1]), the Bundesministerium für Bildung und Forschung (BMBF, Joint Research Project IFB-Tx, PID-NET), From CARs to TRUCKs (Krebshilfe-Priority Program in Translational Oncology), and the European Union (FP7 projects PERSIST, CELLPID, and Horizon 2020 projects SCIDNET, RECOMB).
Author Disclosure
The authors declare that they have no conflict of interest, except that A.S. is co-inventor on a patent application describing alpharetroviral SIN vectors.