
Abstract
D
An email from Dr. Chi received by the editorial office read, “My co-authors and I recently repeated the experiments and found some differences compared to the previous study results, which drew our great attention. For the rigor of scientific research, we have to [retract] our manuscript and need more time to verify the final results. We [are] sorry for this decision…”
After reviewing the circumstances and seeking all authors' agreement, the editorial leadership of the journal agreed to retract the article.
Human Gene Therapy
is committed to upholding the rigorous standards of scientific publishing and the veracity of the literature.
Introduction
A
Long noncoding RNAs (lncRNAs) have been documented to play a role in cancer progression. 4 The lncRNA HOXD-AS1 was shown to be an oncogenic regulator and is abnormally expressed in several human cancers. 5 In human hepatocellular carcinoma, HOXD-AS1 has been reported to be an important regulator of cell apoptosis and metastasis, suggesting that it can act as a positive regulator in human cancer. 6 Moreover, in gastric cancer, knockdown of HOXD-AS1 expression suppresses cell growth by inactivating the JAK2/STAT3 pathway. 7 This finding suggested that HOXD-AS1 can exert its oncogenic function by modulating expression of its downstream targets. Furthermore, HOXD-AS1 can affect expression of target miRNA to act as a tumor promoter. For example, HOXD-AS1 increased cell proliferation rates and invasion of ovarian cancer cells by targeting miR-133a-3p. 8
Based on these findings, we hypothesized that HOXD-AS1 could act as a tumor promoter in CC by targeting miRNA expression. We first investigated HOXD-AS1 expression patterns in CC tissues and cisplatin-resistant CC cells. We also conducted loss-of-function assays using two cisplatin-resistant CC cell lines (CaSki-DDP and HeLa-DDP) treated with various cisplatin concentrations. Bioinformatics analyses and luciferase reporter assays were conducted to assess HOXD-AS1 binding to miR-130a-3p. Finally, rescue assays were carried out to determine additional downstream targets of HOXD-AS1.
Materials and Methods
Specimens
A total of 63 pairs of CC tissues and nonmalignant tissues were obtained from patients with a diagnosis of terminal CC at the Department of Obstetrics and Gynecology at the First Affiliated Hospital of Soochow University (Soochow, China). Approval for this study was obtained from the ethics committee of the First Affiliated Hospital of Soochow University. All experiments were performed in accordance with relevant guidelines and regulations. 9 –11 All patients provided written informed consent prior to study enrollment.
Cell lines
CaSki and HeLa CC cells were purchased from the Tumor Cell Bank of the Chinese Academy of Medical Science (Shanghai, China). Cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 U/mL of ampicillin and 100 μg/mL of streptomycin at 37°C and 5% CO2. Two cisplatin-resistant CC cell lines (CaSki-DDP and HeLa-DDP) were separately derived from the parental cells (CaSki and Hela).
Real-time quantitative reverse-transcription polymerase chain reaction
Trizol reagent (Invitrogen, Carlsbad, CA) was used to isolate total RNA from tissues and cells according to the manufacturer's instructions. A PrimeScript RT kit (Takara, Dalian, China) was used for reverse transcription, and real-time quantitative reverse-transcription polymerase chain reaction (qRT-PCR) was performed using a SYBR Prime Script RT-PCR kit (Takara) according to the manufacturer's instructions. Relative quantification (2−ΔΔCt method) was used to calculate fold change in expression, and expression levels were normalized relative to GAPDH. All assays were performed in triplicate. The primer sequences used were HOXD-AS1 forward 5′-GGCTCTTCCCTAATGTGTGG-3′, reverse 5′-CAGGTCCAGCATGAAACAGA-3′; GAPDH forward 5′-CGCTCTCTGCTCCTCCTGTTC-3′, reverse 5′- ATCCGTTGACTCCGACCTTCAC-3′. All assays were performed at least three times.
Establishment of cisplatin-resistant CC cells
Cisplatin-resistant CC cells were established as previously described. 12 For highly cisplatin-resistant CC cells, CaSki and HeLa cells were cultured in continually increasing concentrations of cisplatin. In brief, cells were treated with cisplatin at different concentrations (1, 10, 50, and 100 μM) in growth media for about 4 h. Next, the culture media containing cisplatin was removed and replaced with fresh growth media for another 4 h. All cell cultures were incubated at 37°C and 5% CO2.
Plasmid construction and transfection
The miR-130a-3p and negative control (NC) mimics as well as the miR-130a-3p inhibitor and NC inhibitor were purchased from Applied Biological Materials (ABM, Vancouver, British Columbia, Canada). cDNA encoding HOXD-AS1 was PCR amplified and subcloned into the pLent-GIII-CMV-Puro vector (ABM), termed pLent-HOXD-AS1. A short small interfering RNA (shRNA) containing a sequence specifically targeted to HOXD-AS1 was obtained from Santa Cruz (Santa Cruz Biotechnology, Santa Cruz, CA). Specific siRNAs for HOXD-AS1 and zinc finger E-box homeobox 1 (ZEB1) were synthesized by ABM. The pLent-HOXD-AS1 plasmid was stably transfected in CC cells using a Lipofectamine 2000 kit (Invitrogen) according to the manufacturer's instructions and screened with puromycin (2 μg/mL) for 4 weeks. A lentivirus construct containing the desired vector was also transfected into CC cell lines, and screened with puromycin (2 μg/mL) for 28 days.
Dual luciferase reporter assay
Lipofectamine-mediated gene transfer was used to co-transfect PmirGLO, pmirGLO-HOXD-AS1-WT or pmirGLO-HOXD-AS1-MUT, and pmirGLO-ZEB1-WT or pmirGLO-ZEB1-MUT into HEK-293T cells with miR-130a-3p mimics or miRNA NC. The relative luciferase activity was normalized to Renilla luciferase activity 2 days after transfection.
Chemosensitivity assay
Chemosensitivity was assessed by MTT (Sigma, St. Louis, MO). Cells were cultured for 2 days in 96-well plates treated with cisplatin before MTT solution (5 mg/mL, 20 μL) was added to each well. After incubation for 4 h, the media was removed and 100 μL dimethyl sulfoxide was added to each well. The relative number of surviving cells was determined from the optical density (O.D.) at 560 nm of the cell lysates. The IC50 value represented the cisplatin concentration at which cell viability was 50%.
Colony formation assay
For colony formation assays, cells in six-well plates (5 × 10 3 cells/well) were incubated cells in RPMI 1640 containing 10% FBS at 37°C. After 2 weeks, the cells were stained with 0.1% crystal violet, and the number of visible colonies was counted.
Flow cytometry analysis to detect apoptosis
Cells were harvested 2 days after transfection with the indicated plasmid or NC. Flow cytometry analyses and the Annexin V:FITC Apoptosis Detection Kit (BD Biosciences, Franklin Lake, NJ) was used to measure the apoptosis rate according to the manufacturer's instructions. All samples were measured in triplicate.
Cell migration and invasion assays
Cell migration and invasion were separately measured using a transwell chamber (8-μm pore size, Corning, Corning, NY) and Matrigel invasion (Becton Dickinson, Franklin Lake, NJ), respectively. Forty-eight hours after transfection, cells that were previously cultured in serum-free media were placed into the upper chamber and coated with Matrigel (10 μg/mL). Media containing 10% FBS was placed in the lower chamber. After incubation for 2 days, cells remaining in the upper chamber were removed and cells that migrated or invaded were fixed in methanol. Crystal violet (0.1%) was used to stain the cells, and the stained cells were counted under a microscope. All experiments were repeated at least twice.
Immunofluorescence
Cells seeded on glass coverslips in six-well plates were fixed in 4% formaldehyde solution and 0.5% Triton X-100/PBS. The cells were blocked for 1 h at room temperature in 5% BSA/PBS before primary antibody was added and incubated with the cells at 4°C overnight. After washing, fluorescent-dye conjugated secondary antibody (Invitrogen) was added to the cells for 1 h before DAPI staining. The cells were imaged using an inverted fluorescence microscope. All assays were performed in triplicate.
Western blot analysis and antibodies
For Western blotting, total protein lysates were separated by 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes (Roche, Basel, Switzerland). Protein loading was estimated using mouse anti-GAPDH monoclonal antibody. Membranes were blocked for 2 h at room temperature with 10% nonfat milk in TBST, and then washed and incubated with antibodies against human E-cadherin, β-catenin, N-cadherin, vimentin, activated caspase-3, total caspase-3, activated caspase-9, total caspase-9, activated poly ADP-ribose polymerase (PARP), total PARP, and GAPDH at 4°C overnight. The membranes were then incubated with secondary antibody conjugated to horseradish peroxidase for 2 h at room temperature. An enhanced chemiluminescence system was used to detect the proteins. All antibodies were purchased from Abcam (Cambridge, United Kingdom). All experimental steps were repeated at least twice.
Statistical analysis
Data are displayed as the means ± SD of more than two individual experiments. All data were obtained from more than three independent experiments. All experiments were conducted in triplicate. Statistical analyses were carried out using SPSS17.0 software (SPSS Inc., Chicago, IL). A Mann-Whitney U-test was performed to make comparisons between groups. Correlations of HOXD-AS1, miR-130a-3p, and ZEB1 levels are expressed as Spearman's correlation coefficients. Significance was defined as p < 0.05.
Results
HOXD-AS1 expression is highly regulated in human CC tissues, cell lines, and cisplatin-resistant cell lines
Chemoresistance is a major difficulty in successful treatment of CC. To assess whether HOXD-AS1 plays a role in CC chemoresistance, we first examined HOXD-AS1 expression levels in 63 pairs of CC tissues and corresponding nonmalignant tissues using qRT-PCR. HOXD-AS1 expression levels were markedly higher in cancerous tissues compared to those in non-neoplastic tissue (Fig. 1A, B). Next, we separately examined HOXD-AS1 levels in two cisplatin-resistant CC cell lines (CaSki-DDP and HeLa-DDP) and the corresponding parental cell lines (CaSki and HeLa). As in the tissue samples, the cisplatin-resistant CC cell lines had higher expression of HOXD-AS1 compared to the parental cell lines (Fig. 1C). These results suggest that HOXD-AS1 could affect the likelihood of a chemoresistance phenotype in CC.
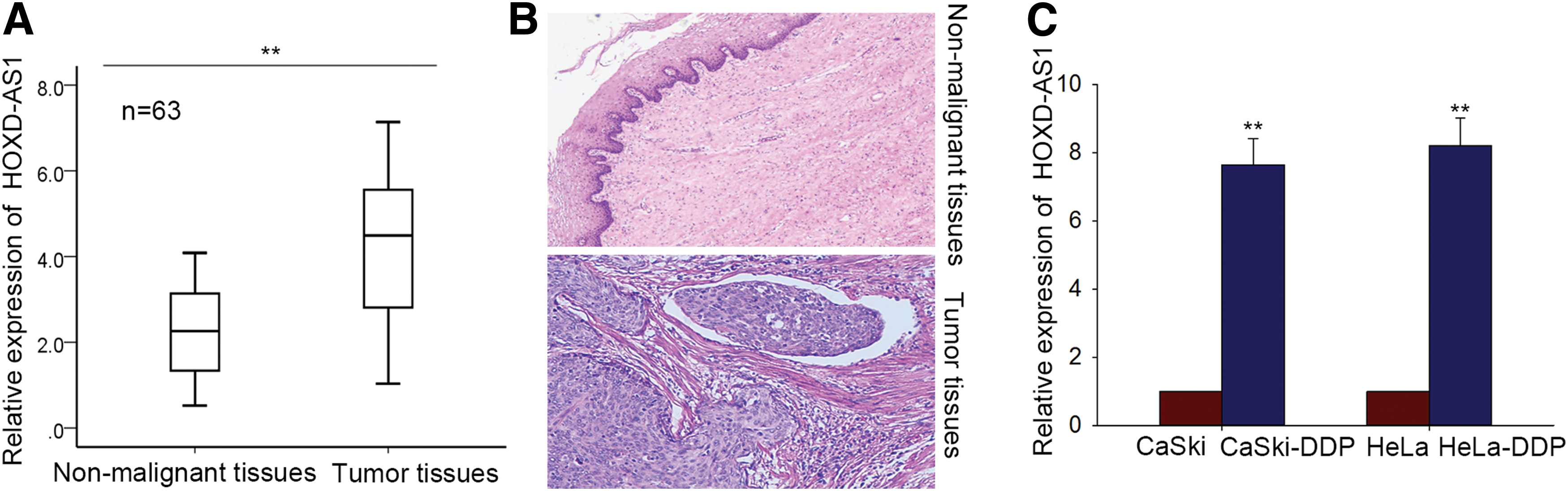
HOXD-AS1 is highly regulated in human cervical cancer (CC) tissues, cell lines, and cisplatin-resistant cell lines.
Effects of HOXD-AS1 expression on chemosensitivity of cisplatin-resistant CC cells
To explore the specific biological functions of HOXD-AS1 in CC cisplatin chemoresistance, cisplatin-resistant CaSki-DDP and HeLa-DDP cells were transfected with HOXD-AS1-specific siRNAs (si-HOXD-AS1#1 and si-HOXD-AS1#2). Nonspecific control siRNAs were used as a negative control (NC). At 48 h post-transfection, we selected the line that had the highest transfection efficiency for subsequent MTT cell viability assays (Fig. 2A). The MTT assay results indicated that downregulation of HOXD-AS1 expression decreased the cisplatin IC50 value of both the CaSki-DDP and HeLa-DDP cells (Fig. 2B). Colony formation assays using si-HOXD-AS1#2 based on its higher transfection efficiency showed that CaSki-DDP and HeLa-DDP cell proliferation was inhibited in the presence of HOXD-AS1 downregulation. Moreover, cells treated with higher concentrations of cisplatin had further reductions in proliferation (Fig. 2C). Flow cytometry analyses to assess the influence of HOXD-AS1 expression on apoptosis showed that HOXD-AS1 knockdown was associated with enhanced, dose-dependent apoptosis of CaSki-DDP and HeLa-DDP cells in the presence of cisplatin relative to cells transfected with NC siRNA (Fig. 2D). Downregulation of HOXD-AS1 expression increased levels of pro-apoptotic proteins, including activated caspase-3, caspase-9, and PARP in CaSki-DDP and HeLa-DDP cells, while the levels of total protein were similar between downregulated and control cells (Fig. 2E). Collectively, these data suggest that downregulation of HOXD-AS1 promoted CC chemosensitivity.

Effects of HOXD-AS1 expression on chemosensitivity of cisplatin-resistant CC cells.
Association of HOXD-AS1 expression with the epithelial-mesenchymal transition phenotype in cisplatin-resistant CC cells
Epithelial-mesenchymal transition (EMT) is a significant biological process that can significantly influence the malignant transformation of cancer cells. Although some studies have examined the EMT phenotype in cisplatin-resistant CC cells, 13 less is known about the function of HOXD-AS1 in the EMT. Here we used Western blotting and immunofluorescence to examine whether formation of the EMT phenotype was affected by HOXD-AS1 expression in cisplatin-resistant CC cells. The protein level of epithelial markers (e.g., E-cadherin, β-catenin) was reduced, whereas that of mesenchymal markers (e.g., N-cadherin, vimentin) was elevated in CaSki-DDP and HeLa-DDP cells compared with parental cells (Fig. 3A). Transwell assays further supported the metastatic capability of CC cells (Fig. 3B). In cisplatin-resistant cells, siRNA-mediated HOXD-AS1 expression knockdown enhanced and reduced levels of epithelial and mesenchymal markers, respectively (Fig. 3C). We also examined the protein level of the transcription factor ZEB1 and found a decrease in its expression in the presence of HOXD-AS1 knockdown. Immunofluorescence assays also showed similar changes in the protein level of ZEB1 (Fig. 3D). Similarly, CaSki-DDP and HeLa-DDP cells transfected with si-HOXD-AS1 showed relatively weak migration and invasion activity compared to the NC groups (Fig. 3E). Consequently, HOXD-AS1 could act as an important regulator of the EMT phenotype in cisplatin-resistant CC cells.
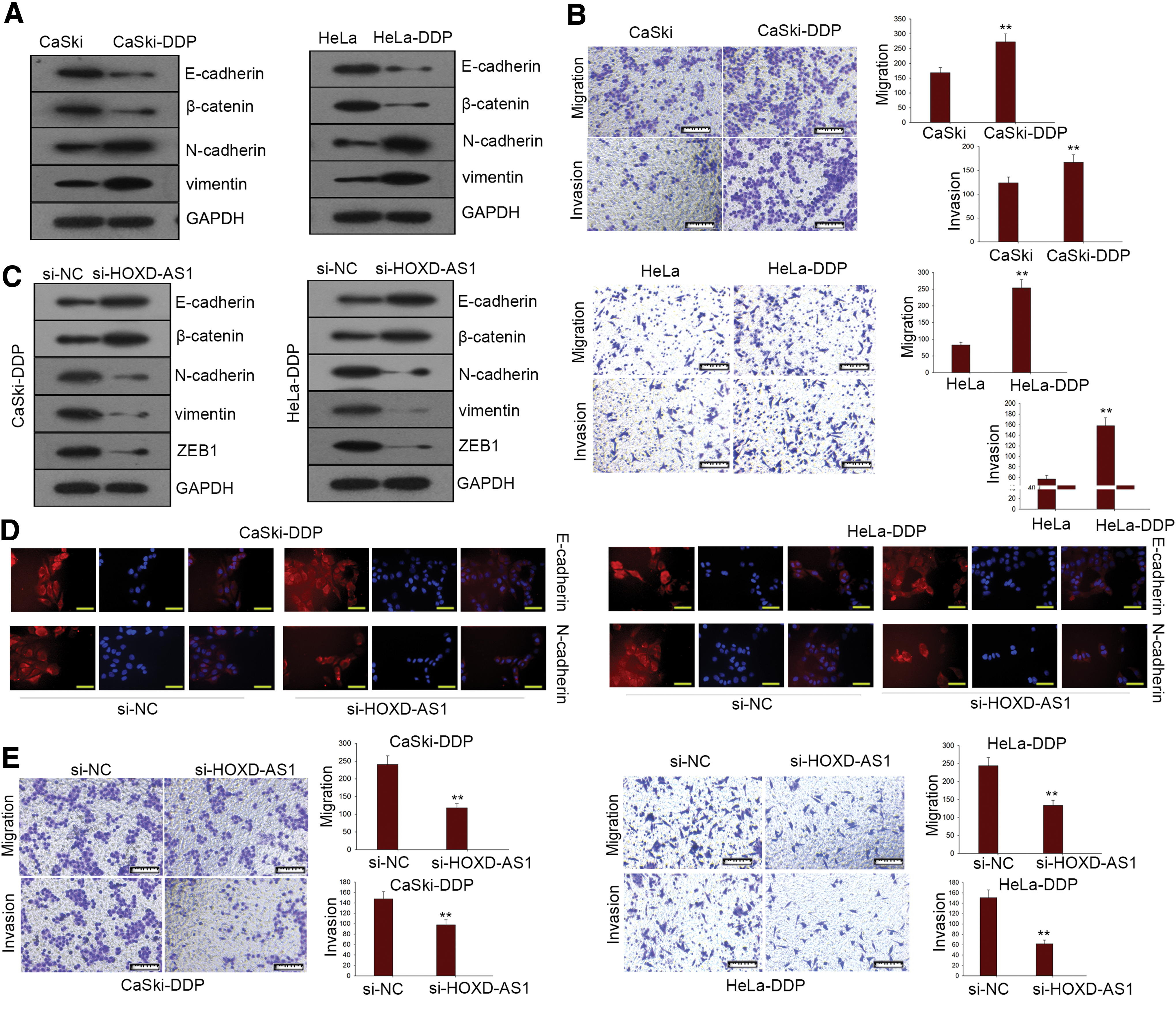
HOXD-AS1 expression is closely associated with formation of the epithelial-mesenchymal transition (EMT) phenotype in cisplatin-resistant CC cells.
HOXD-AS1 directly targeted miR-130a-3p in CC cells
lncRNAs can act as competing endogenous RNAs (ceRNAs) to bind specific miRNAs. Here, we searched for miRNAs that have complementary base pairing with HOXD-AS1 using starbase 2.0 (
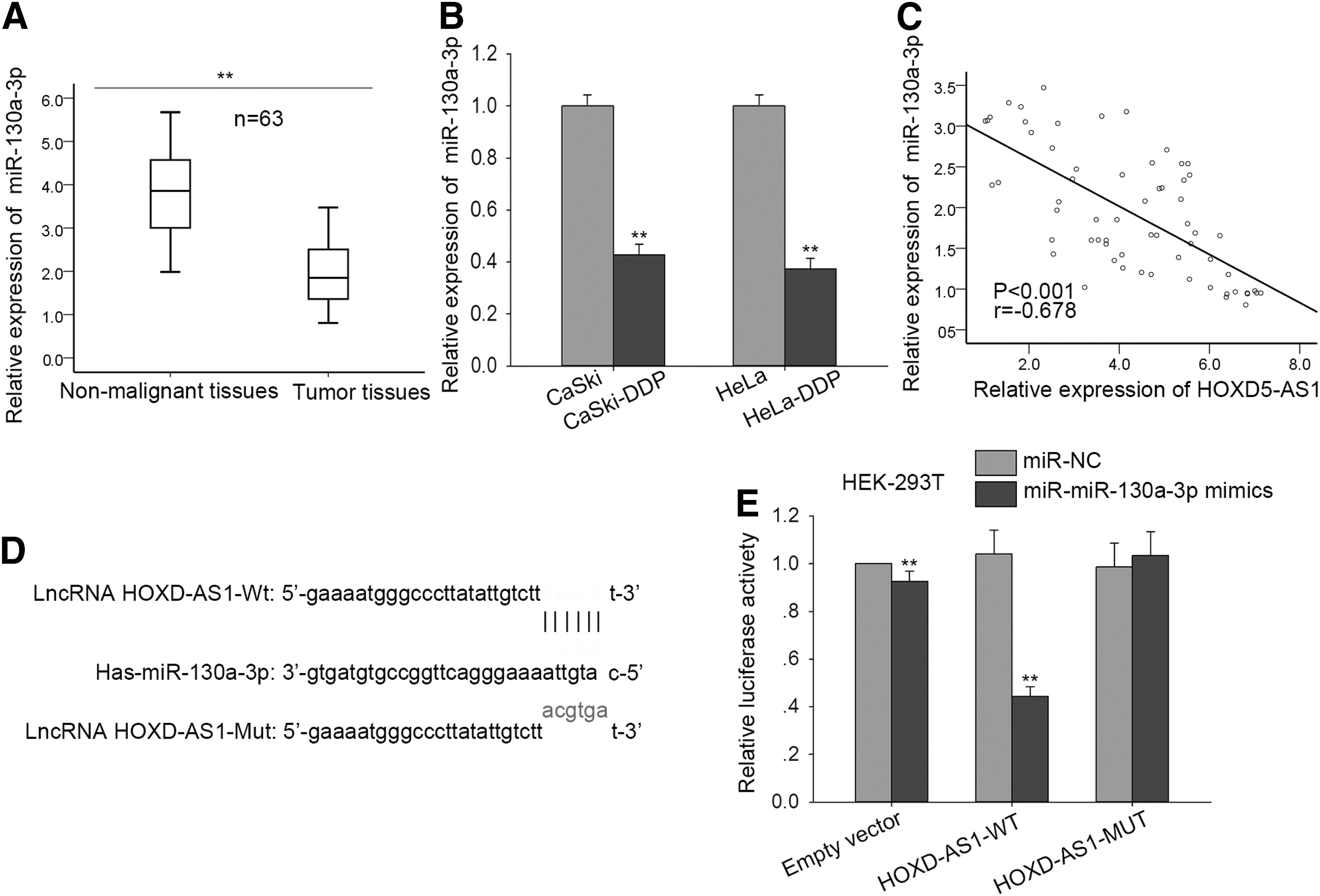
HOXD-AS1 interaction with miR-130a-3p.
Function of miR-130a-3p in chemoresistance of cisplatin-resistant CC cells
To explore the biological function of miR-130a-3p in CC cells, miR-NC or miR-130a-3p mimics were transfected into CaSki-DDP and HeLa-DDP cells. An MTT assay showed that upregulation of miR-130a-3p mimics decreased the cisplatin IC50 value of two cisplatin-resistant CC cells (Fig. 5A). A colony formation assay showed that in the presence of increasing concentrations of cisplatin, colony formation was decreased by miR-130a-3p mimics in the two cisplatin-resistant CC cell lines (CaSki-DDP and HeLa-DDP) (Fig. 5B). Furthermore, flow cytometry analysis of the CaSki-DDP and HeLa-DDP lines indicated that miR-130a-3p overexpression in the presence of cisplatin enhanced apoptosis (Fig. 5C). Western blot analysis showed that expression of epithelial and mesenchymal markers were up- and downregulated, respectively, following transfection of CaSki-DDP and HeLa-DDP cells with miR-130a-3p mimics (Fig. 5D). Immunofluorescence studies further supported a role for miR-130a-3p in the formation of EMT in CC cells (Fig. 5E). Together these results indicate that miR-130a-3p expression levels could regulate cisplatin chemosensitivity, proliferation, apoptosis, and EMT of CC cells.

miR-130a-3p in chemoresistance of cisplatin-resistant CC cells.
HOXD-AS1 positively modulates expression of the miR-130a-3p target gene (ZEB1) in CC tissues and cell lines
Bioinformatics analyses using searches of TargetScan, PicTar, and miRanda databases showed that ZEB1 is a miR-130a-3p target gene. The binding sites validated the binding between miR-130a-3p and ZEB1 (Fig. 6A), and a dual luciferase reporter assay in HEK-293T cells showed that luciferase activity associated with wild-type ZEB1 was reduced in the presence of miR-130a-3p mimics and could be rescued by pLent-HOXD-AS1 (Fig. 6B). To further demonstrate the relationship among HOXD-AS1, miR-130a-3p, and ZEB1, qRT-PCR and Western blot analyses were conducted in CaSki-DDP and HeLa-DDP cells (Fig. 6C, D). High mRNA and protein levels of ZEB1 were decreased by miR-130a-3p mimics, whereas this trend was reversed in the presence of pLent-HOXA-AS1. Strong ZEB1 expression was also detected in CC tissues by qRT-PCR analysis (Fig. 6E), and a Spearman's correlation analysis showed a negative relationship between miR-130a-3p and ZEB1 expression (Fig. 6F).
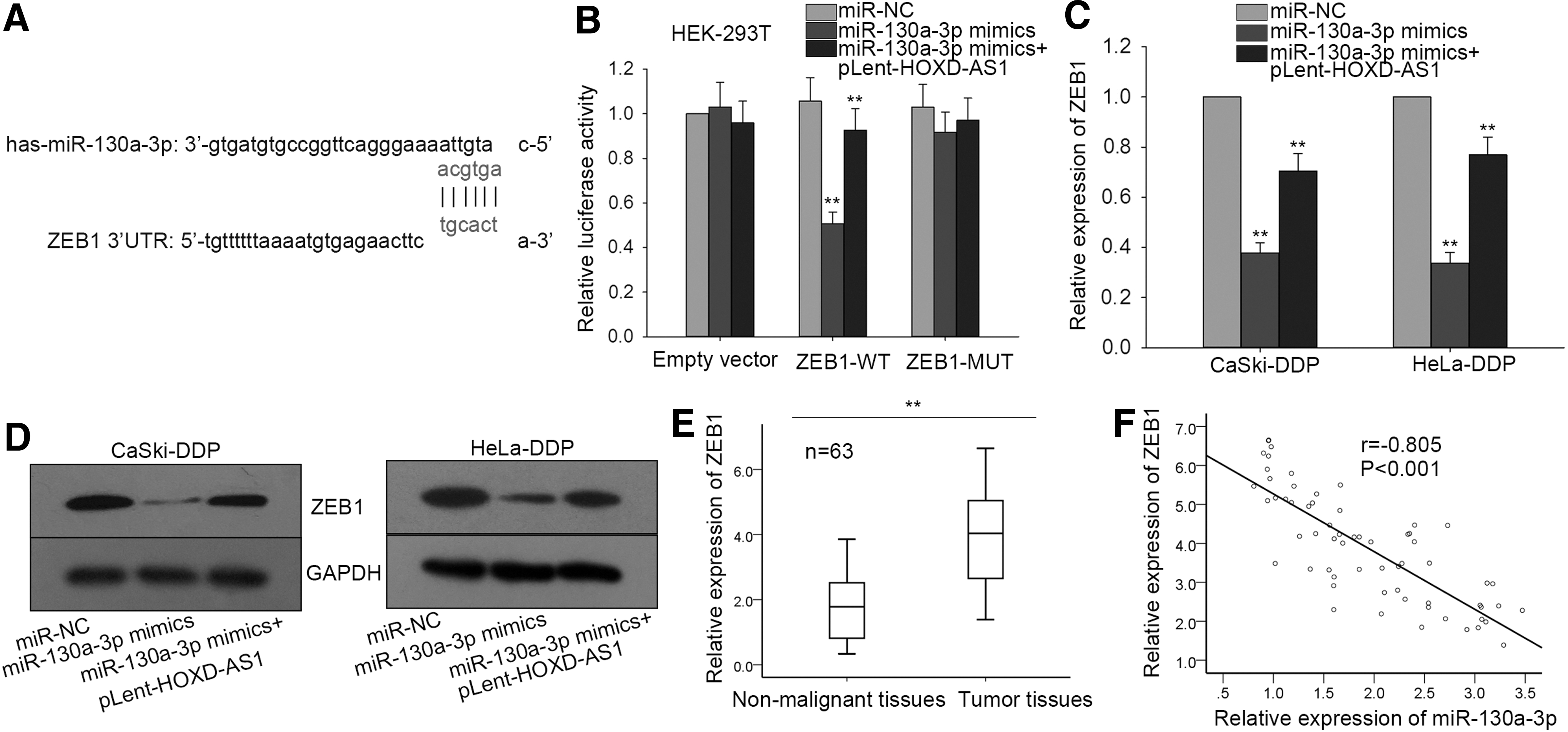
HOXD-AS1 positively modulates miR-130a-3p (zinc finger E-box homeobox 1 [ZEB1]) expression in CC tissues and cell lines.
Regulation of HOXD-AS1, miR-130a-3p, and ZEB1 expression affects proliferation, metastasis, and the EMT process
A rescue assay with CaSki-DDP cells was used to demonstrate the function of the HOXD-AS1-miR-130a-3p-ZEB1 axis and varying cisplatin concentrations (0, 10, 20 μg/mL). An MTT assay showed that decreases in the cisplatin IC50 value in the presence of sh-HOXD-AS1 could be reversed by a miR-130a-3p inhibitor, and the effects of this inhibitor could be reversed again by siZEB1 (Fig. 7A). A colony formation assay revealed that sh-HOXD-AS2-induced inhibition of cell proliferation could be reversed by the miR-130a-3p inhibitor. However, cell proliferation was decreased in the presence of si-ZEB1 (Fig. 7B). Flow cytometry to assess apoptosis levels showed that the amount of apoptosis induced by downregulation of HOXD-AS1 expression decreased in the presence of a miR-130a-3p inhibitor, but levels were restored when si-ZEB1 was added (Fig. 7C). Finally, Western blotting to evaluate EMT formation in CaSki-DDP and HeLa-DDP cells showed that EMT induced by HOXD-AS1 was reversed by down-regulation of HOXD-AS1 expression and restored in the presence of a miR-130a-3p inhibitor. Yet when ZEB1 levels were downregulated, the EMT process was reinitiated (Fig. 7D).

Regulation of expression of HOXD-AS1, miR-130a-3p, and ZEB1 affects CC cell proliferation, metastasis, and EMT process.
Discussion
Cisplatin resistance is one form of systemic chemoresistance in locally advanced CC. 14 Although various drugs are effective for CC treatment, drug resistance remains a significant obstacle. Overall, chemotherapy is most effective for early-stage CC. 15 Indeed, the 1-year survival rate of patients with advanced CC is only about 15% to 20%. 16 Therefore, investigation of the molecular mechanisms and biological processes associated with CC chemoresistance is needed.
With the advent of genome-wide sequencing analyses, there is increasing evidence that most noncoding RNAs (ncRNAs) in the mammalian genome are transcribed. 17 –19 Numerous reports have verified that one type of ncRNA, lncRNA, can significantly affect cell proliferation, differentiation, and immune response, 20,21 making these RNAs vital components of the cancer transcriptome. According to previous reports, lncRNAs can act as oncogenes or tumor suppressors in various human cancers. 22 –26 Moreover, lncRNAs can also regulate chemoresistance in human cancers, 27 –31 including CC. 32,33
The lncRNA HOXD-AS1 was shown to be a tumor facilitator in some types of malignancies. 8,34 –36 Here, we explored the specific biological functions and molecular mechanisms of HOXD-AS1 in CC. We first showed that HOXD-AS1 was upregulated in CC tissues and cisplatin-resistant CC cells. Loss-of-function assays revealed that downregulation of HOXD-AS1 reversed cisplatin resistance through regulation of proliferation and apoptosis. Moreover, HOXD-AS1 expression knockdown suppressed migration and invasion of cisplatin-resistant CC cells. Together, these results show that HOXD-AS1 exerts oncogenic functions and regulates chemoresistance in CC.
lncRNAs acting as ceRNAs can upregulate mRNAs by binding to miRNAs. 37 –39 Here, we hypothesized that HOXD-AS1 might be a ceRNA in CC. Bioinformatics analysis showed that miR-130a-3p was a potential target miRNA of HOXD-AS. We showed that miR-130a-3p was downregulated in CC tissues and cisplatin-resistant CC cells and that HOXD-AS1 and miR-130a-3p levels were negatively correlated, using Spearman's correlation analysis. Bioinformatics analyses and dual luciferase reporter assays were separately conducted to demonstrate the binding relationship between HOXDA-AS1 and miR-130a-3p and to show that miR-130a-3p expression affects chemoresistance of cisplatin-resistant CC cells. Gain-of-function experiments showed that overexpression of miR-130a-3p reversed chemoresistance of cisplatin-resistant CC cells. Subsequent bioinformatics analyses and dual luciferase reporter assays showed that ZEB1 was a target mRNA of miR-130a-3p. Similarly, Spearman's correlation analysis revealed a negative correlation between miR-130a-3p and ZEB1 levels. ZEB1 expression was also positively regulated by HOXD-AS1. Collectively, these results show that HOXD-AS1 acted as a ceRNA to upregulate ZEB1 expression via miR-130a-3p binding. Finally, rescue assays demonstrated the effects of HOXD-AS1-miR-130a-3p-ZEB1 axis on cisplatin-resistance of CC cells. Based on these findings, we concluded that HOXD-AS1 acted as a ceRNA to enhance the cisplatin resistance of CC cells through targeting of miR-130a-3p/ZEB1. The HOXD-AS1-miR-130a-3p-ZEB1 axis represents one pathway that can generate CC resistance, but other mechanisms can likely produce similar effects, and thus additional studies are needed to determine which other pathways are involved. Nonetheless, our findings support one pathway that can be targeted to avoid or reduce the likelihood of chemoresistance in CC.
Footnotes
Acknowledgments
The authors thank all laboratory members.
This study was supported by Suzhou basic application research fund program (52010103060004, SYS201604) and 2017 Suzhou industrial technology innovation projection (52010130010021, SYS201750).
Author Disclosure
No conflicts of interest to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.