
Abstract
Colorectal cancer (CRC) is among the cancers with the highest incidence globally, and it currently ranks as the fourth leading cause of cancer-related deaths worldwide. Novel strategies for the treatment of advanced CRC are urgently needed, and adoptive transfer of allogeneic natural killer (NK) cells represents an attractive option. In this study, we successfully expanded NK cells from umbilical cord blood (UCB) with membrane-bound interleukin (IL)-21, termed eUCB-NK cells. eUCB-NK cells efficiently lysed CRC cell lines in vitro and secreted significantly higher levels of interferon-γ, tumor necrosis factor-α, granulocyte-macrophage colony stimulating factor, and chemokine ligand 3 compared with IL-2–stimulated NK cells. Adoptive transfer of these NK cells significantly inhibited the growth of HT29 xenografts, whereas LoVo tumors were not effectively controlled with eUCB-NK cells. Higher numbers of NK cells inside HT29 tumors, not seen in LoVo tumors, might contribute to the differences in response to eUCB-NK cells. Bevacizumab increased extravasation of adoptively transferred NK cells into LoVo tumors and improved the therapeutic activity of eUCB-NK cells. These results justify clinical translation of UCB-derived NK cell–based therapeutics, used alone or in combination with bevacizumab, as a novel treatment option for patients with CRC.
Introduction
C
Recent progress in immune checkpoint inhibitors and chimeric antigen receptor T cells has led to a paradigm shift in the treatment of tumors, and great interest has been expressed by scientists and physicians searching for novel therapeutics that harness a patients' own immune system. 4,5 Natural killer (NK) cells, broadly defined in humans as the CD56+ CD3− lymphocyte subset, compose the first barrier of innate immunity that has an important role in the elimination of pathogens and tumors. 6 NK cells express a series of activating and inhibitory receptors that can discriminate cells of self and nonself origins in the absence of antigen priming. 7 Upon activation, NK cells directly destroy sensitive malignant cells and secrete a broad array of cytokines that can stimulate the subsequent adaptive immune response. 8 Additionally, NK cells are the main effector cells mediating antibody-dependent cell-mediated cytotoxicity. 8 Harnessing NK cells for therapeutic purposes has attracted substantial attention.
Dysregulation of NK cells, including decreased cell number and reduced cell activity, has been well described in CRC and is considered to be one of the most important contributing factors to disease progression. 9 –11 Approaches aimed at restoring or replacing dysfunctional NK cells may serve as a novel focus of cancer therapy for CRC. Adoptive cell transfer of autologous NK cells was initially developed for clinical application. Despite a very remarkable in vitro antitumor effect and increased numbers of circulating NK cells in vivo, autologous NK cell therapy has failed to demonstrate a significant clinical effect. 12 Following the discovery of the killer-cell immunoglobulin-like receptor/human leukocyte antigen (KIR/HLA) mismatch, infusions of allogeneic NK cells were increasingly used for adoptive cellular therapies because these cells were less likely to be inhibited by the self-HLA expressed on the tumor cells. 13 Adoptive transfer of allogeneic NK cells has been shown to be safe and to improve clinical outcome in hematological malignancies. 14,15 There is also increasing evidence that allogeneic NK cells have activities against solid tumors. 16,17 However, their potential utilities have rarely been explored in CRC treatment.
Until recently, the majority of clinical studies used NK cells from peripheral blood in adoptive immunotherapy. Umbilical cord blood (UCB) provides an alternative source for NK cells, and the use of UCB-derived NK cells is associated with several advantages, including a higher percentage of NK cell progenitors, faster engraftment and immune reconstitution, less stringent requirements for HLA matching, and a lower risk of graft versus host disease. 18 Obtaining NK cells from UCB through the use of several cytokine-based culture methods has been described. 19 –21 However, further research is needed to provide more mature and highly functional NK cells applicable for cellular therapy. In the present study, we explored the potential of a genetically engineered K562 cell line with membrane-bound expression of interleukin (IL)-21 for ex vivo generation of mature and functional NK cells from UCB. We next assessed the in vivo and in vitro efficacy of the expanded UCB-derived NK (eUCB-NK) cells against CRC cells and further determined whether bevacizumab can synergize with this NK cell–based practice to better suppress CRC.
Materials and Methods
Cell lines and cell culture
Human CRC cell lines HT29 and SW480 were obtained from the American Type Culture Collection (Manassas, VA). SW620, LoVo, and K562 cell lines were derived from Shanghai cell bank of Chinese Academy of Sciences. K562 cells were lentivirally transduced with virus encoding human membrane-bound IL-21 (K562-mbIL21). Cells were grown in RPMI-1640 medium (Hyclone, Logan, UT) or Dulbecco's modified Eagle's medium (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin and streptomycin (Sigma, St. Louis, MO). All cells were cultured at 37°C in a humidified atmosphere containing 5% CO2.
Isolation and expansion of UCB-derived NK cells
Human UCB samples were obtained from the First Affiliated Hospital of Nanchang University under a protocol approved by the Ethical Committee Department, and written informed consent was obtained from each donor. UCB mononuclear cells (UCB-MNCs) were separated from the buffy coats by Ficoll-Hypaque density gradient centrifugation. The UCB-MNCs were resuspended at 1.5 × 10 6 cells/mL with 1 × 10 6 cells/mL lethally irradiated K562-mbIL21 feeder cells in the presence of 300 IU/mL recombinant human IL-2 (rhIL-2) (PeproTech, Rocky Hill, NJ). Half of the culture medium was changed every 2–3 days and fresh rhIL-2 was added every other day. At approximately 6 days of expansion, cells were harvested and restimulated with the irradiated K562-mbIL21 and fresh rhIL-2 for an additional week. Culture medium was added based on the cell density. For functional assays, NK cells were purified using an NK cell isolation Kit (Miltenyi, Bergisch Gladbach, Germany), obtaining NK cell populations with a purity of more than 95%.
Flow cytometry analyses
The purity of NK cells was measured using flow cytometry by anti-human CD3–fluorescein isothiocyanate (CD3-FITC) and CD56-phycoerythrin (CD56-PE) antibodies (BD Biosciences, Franklin Lakes, NJ). The expression of NK cells markers was determined before and after 13 days of cell expansion using mouse anti-human antibodies to CD16, CD56, NKG2D, NKp30, NKp44, NKp46, TRAIL, 2B4, NKG2A, KIR2DL1, KIR2DL2/3, KIR3DL1, CD96, TIGIT, PD-1, TIM-3, LAG-3, CXCR3, and CXCR6. Antibodies to TRAIL, KIR2DL1, KIR2DL2/3, KIR3DL1, PD-1, TIM-3, and LAG-3 were purchased from BD Biosciences, and others were obtained from eBiosciences (San Diego, CA). For detection of degranulation, eUCB-NK cells or IL-2–stimulated NK cells from the same UCB donors were co-cultured with HT29 cells at an effector-to-target (E:T) ratio of 5:1 for 4 h, and FITC-conjugated antibody to CD107a (BD Biosciences) was added at the beginning of stimulation along with monensin (BD Biosciences). The cells were washed and analyzed using a Navios flow cytometer (Beckman Coulter, Brea, CA).
In vitro cytotoxicity assays
The cytolytic activities of eUCB-NK cells were measured with the lactate dehydrogenase–releasing assay using the Cytotoxicity Assay Kit (Promega, Madison, WI). Briefly, CRC cell suspensions (target cells, 2 × 10 5 /mL) were incubated with eUCB-NK cells or IL-2–stimulated NK cells from the same UCB donors (effector cells, 1 × 10 6 /mL) at an E:T ratio of 5:1 in a total volume of 100 μL. The cells were incubated in the wells of 96-well V-bottom plates for 6 h at 37°C in a humidified atmosphere containing 5% CO2. Afterward, the supernatants were obtained by centrifugation for 5 min at 200 × g, and 50 μL of supernatant was then transferred to a microtiter 96-well plate. Lactate dehydrogenase solution (50 μL) was added to the supernatant, and the mixture was then measured at 490 nm using the microtiter plate reader. The spontaneous release of effector and target cells and the maximum release of target cells were obtained from individual cultures, respectively. The mean cytotoxicity of effector cells against target cells was calculated as follows: cell lysis (%) = 100 × (experimental release − target spontaneous release − effector spontaneous release)/(maximal release − spontaneous release).
Enzyme-linked immunosorbent assay
eUCB-NK cells or IL-2–stimulated NK cells from the same UCB donors were co-cultured with different CRC cells at an E:T ratio of 5:1 for 24 h at 37°C in a humidified atmosphere containing 5% CO2. Cell-free supernatants were collected for interferon-γ (IFN-γ), tumor necrosis factor (TNF-α), granulocyte-macrophage colony stimulating factor (GM-CSF), and chemokine ligand 3 (CCL3) secretion by ELISA (R&D Systems, St. Paul, MN).
Mouse models
All animal experiments were authorized by the Medical Experimental Animal Care Commission of the First Affiliated Hospital of Nanchang University and carried out in accordance to the approved protocols. HT29 (5 × 10 6 /mouse) and LoVo (5 × 10 6 /mouse) cells were subcutaneously implanted in the right flank of 6-week-old NOD-scid IL2Rgamma null (NSG) mice. When the tumor volumes were approximately 100 mm3, mice were randomly allocated to receive (1) phosphate-buffered saline (PBS); (2) eUCB-NK cells (5 × 10 6 /mouse); or (3) IL-2–stimulated NK cells (5 × 10 6 /mouse). To evaluate the combined effect of eUCB-NK cells and bevacuzimab, LoVo tumor xenografts were established as described above. When tumor volumes reached an average of approximately 100 mm3, we treated the mice with (1) PBS; (2) bevacizumab at 5 mg/kg; (3) eUCB-NK cells (5 × 10 6 /mouse); or (4) eUCB-NK cells (5 × 10 6 /mouse) 48 h after bevacizumab 5 mg/kg injection. All treatments were administrated twice intravenously with a 1-week interval. The tumor volumes were measured regularly with a metric caliper and calculated using the following formula: length × width 2 × 0.5. Mice were euthanized when signs of discomfort were detected.
NK cell infiltration
When animal studies were terminated, all mice were euthanized, and tumor tissues were surgically excised. Some were fixed with formalin, some were reserved in liquid nitrogen, and the others were collected, cut into small pieces, and digested for 45 min in RPMI-1640 medium containing 1.5 mg/mL collagenase D and 500 μg/mL DNase I. All cells were then passed through a 40-μm nylon cell strainer (BD Biosciences) to make single-cell suspensions. The cells were stained with anti-human CD3-FITC and CD16/CD56-PE antibodies (BD Biosciences) for 45 min and washed before undergoing flow cytometry.
Quantitative real-time PCR
Tumor tissues stored in liquid nitrogen were used for RNA extraction. Total RNA was isolated using the TRIzol reagent (Invitrogen, Carlsbad, CA) and reverse transcribed into cDNA using PrimeScript RT Reagent Kit (TaKaRa, Dalian, China). Real-time PCR was performed with 20 ng of RNA on a SYBR Green real-time PCR detection system according to the manufacturer's instructions. Primers purchased from Genewiz (Soochow, China) are shown in Supplementary Table S1 (Supplementary Data are available online at
Immunohistochemistry analysis
Tumor tissues that had been fixed with formalin were embedded in paraffin and serially sectioned at a 4-μm thickness. Specified sections were immunostained for analysis of NK infiltration with mouse anti-human CD56 antibody (1:500 dilution, Abcam, Cambridge, United Kingdom).
Statistics
Data are presented as the means ± standard deviation (SD), and the statistical calculations were performed by Graphpad Prism 5.0. The analyses were performed using paired Student's t-tests or two-tailed Student's t-tests or two-way ANOVA with Bonferroni post tests. Differences were considered statistically significant at p < 0.05, p < 0.01, and p < 0.001.
Results
Selective expansion of NK cells from UCB
Several cytokine-based culture methods have been defined to generate sufficient NK cells from UCB. 19 –21 Efficacy and ex vivo expansion of NK cells with free cytokines were variable, however, and the final product had substantial T-cell contamination. Highly purified NK cells can be expanded ex vivo with K562 cells expressing membrane-bound IL-15 and 4-1BB. 22 Although NK cells expanded using this approach are highly functional in vitro and in vivo, their proliferation is nonetheless limited by NK cell senescence resulting from telomere shortening, which has restricted their therapeutic potential. Genetically modified feeder cells expressing membrane-bound IL-21 have been reported to promote NK cell proliferation with longer telomeres and less senescence. 23 However, whether this method can be used for the ex vivo generation of highly active NK cells from UCB remains unclear. In this study, we first generated K562-mbIL21 feeder cells. Cell surface expression of membrane IL-21 was confirmed by flow cytometry (Fig. 1A). We found it feasible to efficiently generate a high dose of NK cells from UCB-MNCs with stimulation from lethally irradiated K562-mbIL21 cells and rhIL-2. Figure 2B displays the representative proportion of increasing NK cells (CD56+ CD3−) in UCB-MNCs stimulated with K562-mbIL21 cells in the presence of IL-2 at various times. NK cells could be selectively expanded to greater than 500-fold within 14 days of culture with purity approaching to 90%, whereas their counterpart T cells remained basically unchanged over time (Figs. 1C, 1D). Collectively, we were able to successfully expand NK cells from UCB-MNCs using the K562-mbIL21 system.
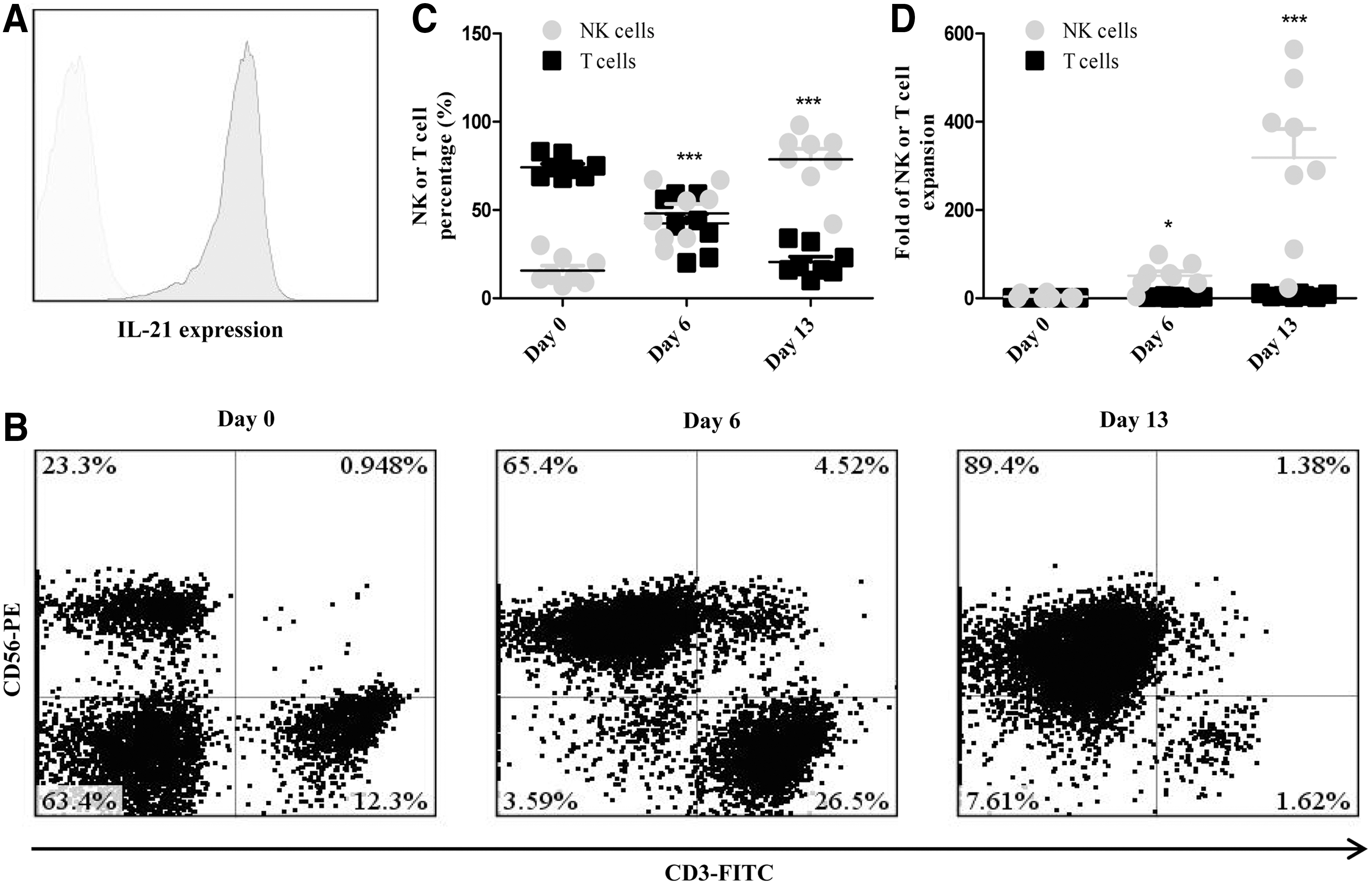
Selective expansion of natural killer (NK) cells from umbilical cord blood (UCB) in the presence of lethally irradiated K562-mbIL21 cells.
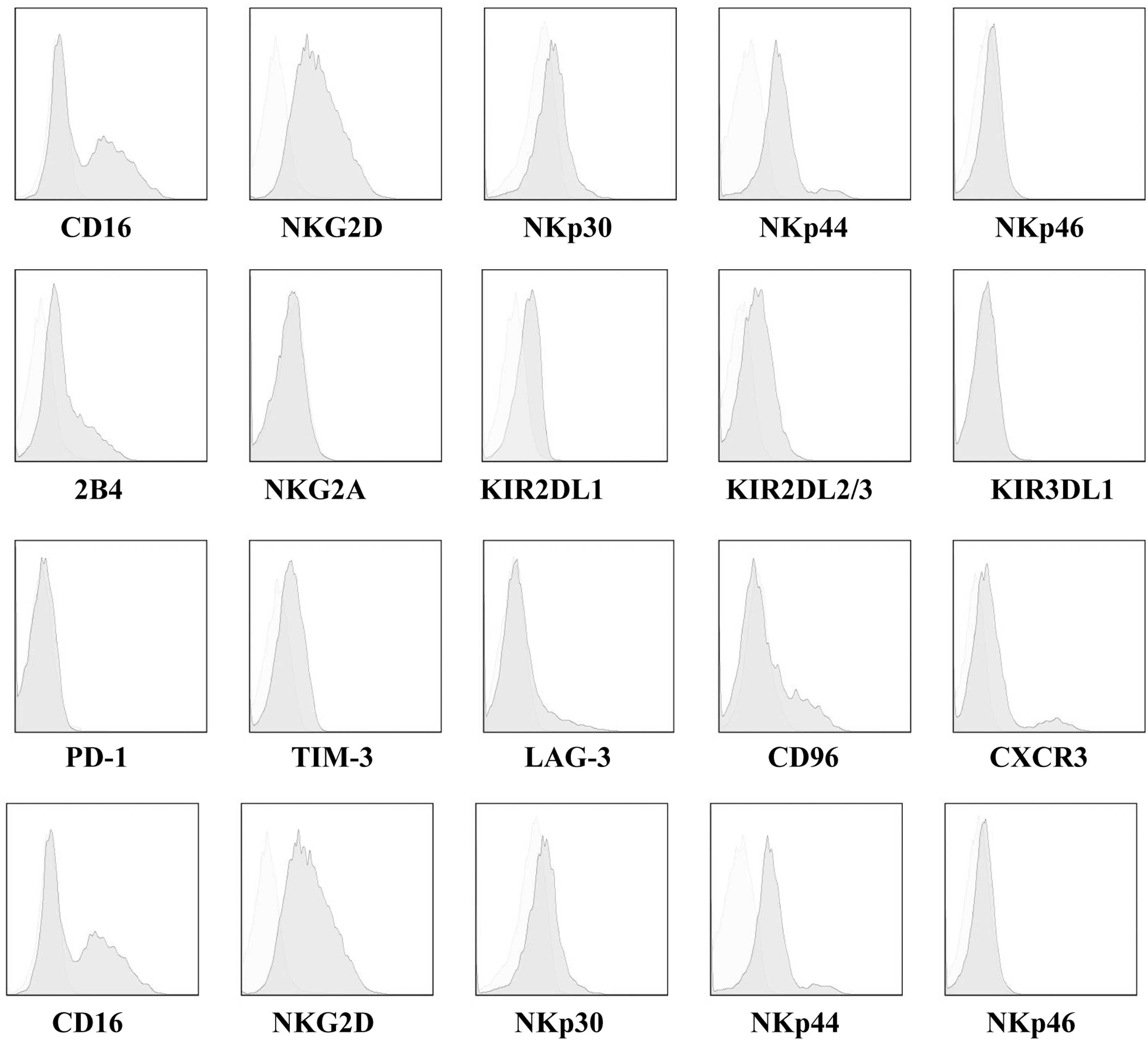
Phenotypic characteristics of expanded UCB-derived NK (eUCB-NK) cells. Flow cytometry illustrating the cell surface density of the activating receptors including CD16, CD56, NKG2D, NKp30, NKp44, NKp46, TRAIL, and 2B4; the inhibitory receptors including NKG2A, KIR2DL1, KIR2DL2/3, and KIR3DL1; the inhibitory molecules including CD96, TIGIT, PD-1, TIM-3, and LAG-3; and chemokine receptors including CXCR3 and CXCR6. Gating was based on the IL-2–stimulated NK cells staining with isotype-matched antibody. Light and dark gray represent the IL-2–stimulated NK or eUCB-NK cell staining with the specific antibodies, respectively. Data shown are the representative of experiments with similar results.
Phenotypic characterization eUCB-NK cells
Next, we investigated whether the expansion procedure could change the NK cell phenotype. We used flow cytometry to compare the expression of activating receptors including CD16, CD56, NKG2D, NKp30, NKp44, NKp46, TRAIL, and 2B4; inhibitory receptors including NKG2A, KIR2DL1, KIR2DL2/3, and KIR3DL1; inhibitory molecules including CD96, TIGIT, PD-1, TIM-3, and LAG-3; and chemokine receptors including CXCR3 and CXCR6 on the surface of eUCB-NK cells and IL-2–stimulated NK cells. Compared with IL-2–stimulated NK cells, the expression of almost all activating receptors were upregulated on eUCB-NK cells. Among the receptors, NKG2D and TRAIL, which are strongly associated with NK cell activity, had the greatest upregulation, indicating a highly functional active phenotype of these generated NK cells (Fig. 2). No significant variation in inhibitory receptors was observed before and after expansion (Fig. 2). Surface expression of the inhibitory molecules CTLA-4, PD-1, TIGIT, CD96, LAG-3, and TIM-3, associated with exhaustion of lymphocytes, was also measured. Similar surface levels were found in IL-2–stimulated NK cells, indicating that expanding procedure would not predispose NK cells to exhaustion (Fig. 2). Additionally, slightly higher expression of CXCR3 and CXCR6 was observed on eUCB-NK cells, indicating that these generated NK cells might have a better homing capacity (Fig. 2).
Enhanced cytotoxicity of NK cells against CRC cell lines
We next evaluated the cytotoxic activity of eUCB-NK cells against CRC cell lines. Four-hour cytotoxicity assays were performed with eUCB-NK cells from different donors at an E:T ratio of 5 (n = 6). NK cells derived from the same donors without expansion were used as controls. As shown in Fig. 3A, eUCB-NK cells were able to efficiently kill CRC cells over the course of 6 h, while their paired IL-2–stimulated NK cells failed to elicit potent cell lysis. We determined whether eUCB-NK cells also increased their cytotoxicity against normal health untransformed cells. Allogeneic peripheral blood mononuclear cells (PBMCs) served as target cells. As shown in Fig. 3B, cytotoxicity remained low at an E:T ratio of 2:1, regardless of whether NK cells were expanded or not (n = 6). We further analyzed the cell surface expression of the lysosomal-associated membrane protein-1 (LAMP-1 or CD107a), which has been shown to be a sensitive marker for NK cell functional activity. CD107a degranulation was significantly enhanced in eUCB-NK cells compared to counterpart IL-2–stimulated NK cells in response to HT29 cells (Figs. 3C, 3D). These results indicated that the expansion procedure could augment the cytotoxicity of eUCB-NK cells against CRC cells without triggering attack towards healthy non-transformed cells.
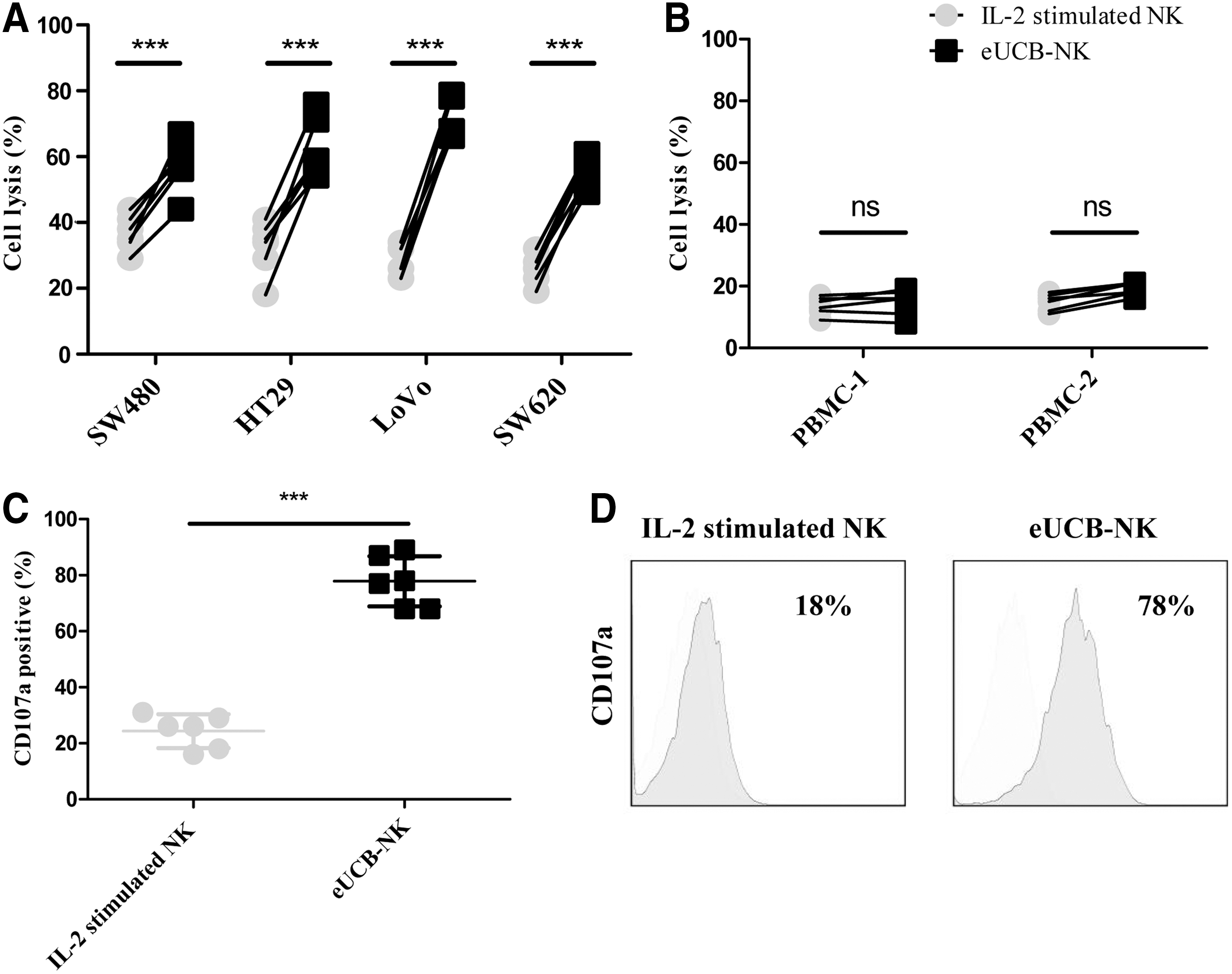
Enhanced cytotoxicity of eUCB-NK cells against colorectal cancer (CRC) cell lines.
Elevated cytokines of NK cells against CRC cell lines
To make our findings more clinically relevant, we investigated whether the NK cell–mediated killing was accompanied by increased cytokine secretion. We incubated eUCB-NK cells with CRC cell lines at an E:T target of 5:1 for 24 h and the supernatants were assessed with ELISA. The results showed significantly higher levels of IFN-γ, TNF-α, GM-CSF, and CCL3 production by eUCB-NK cells compared with IL-2–stimulated NK cells (Fig. 4).
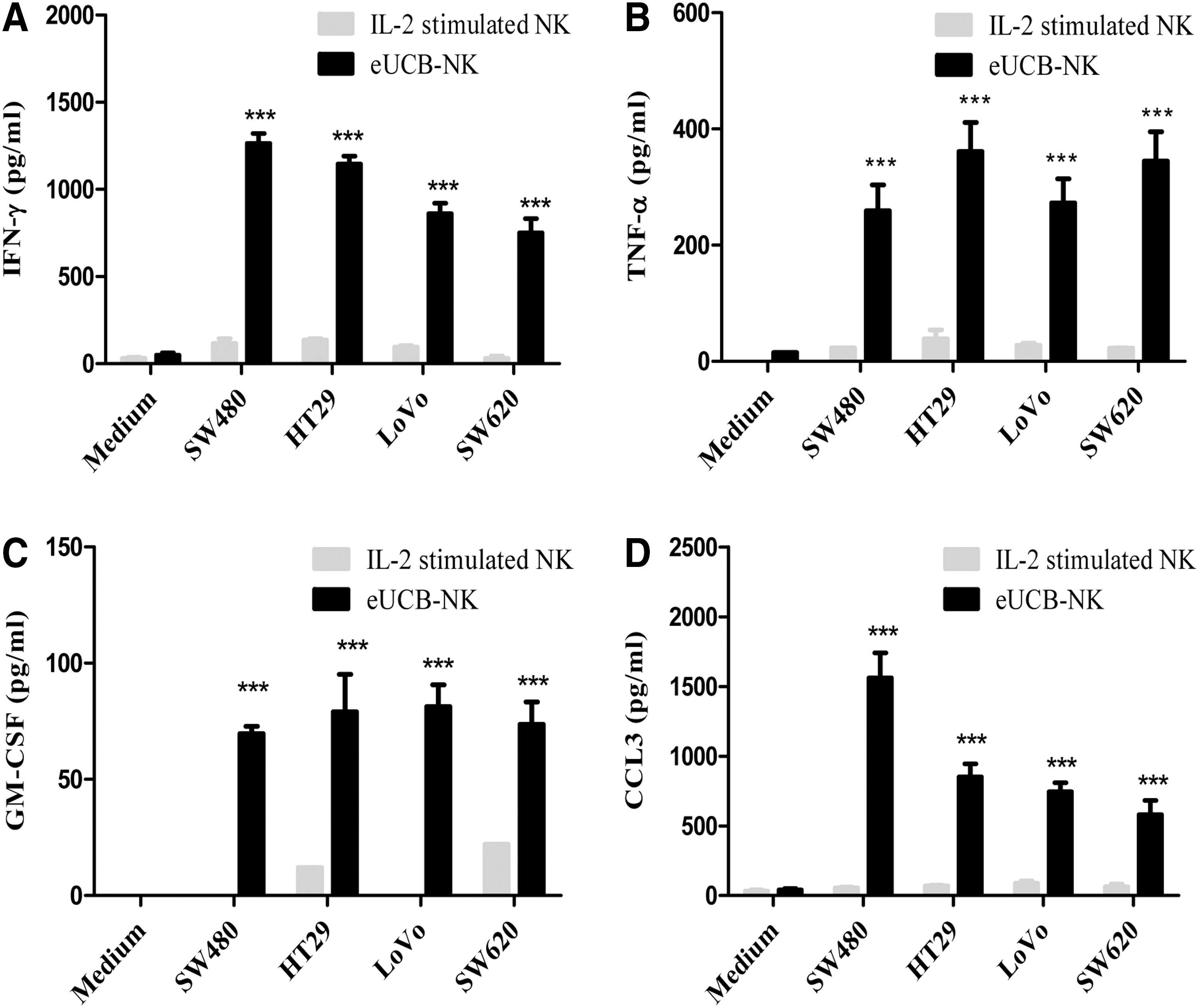
Cytokine production of eUCB-NK cells against CRC cell lines.
Therapeutic efficacy of NK cells against established CRC xenografts
Having demonstrated that eUCB-NK cells potently kill CRC cell lines in vitro, we next assessed whether they similarly suppressed the growth of established CRC xenografts in vivo. NSG mice bearing subcutaneous HT29 and LoVo xenografts were established (n = 6). About 2 weeks after tumor cell incubation and when the tumor volumes reached an average of 100 mm3, mice were allocated to receive PBS, eUCB-NK, or IL-2–stimulated NK cells. Mice bearing HT29 tumors treated with eUCB-NK cells showed dramatic responses, whereas they did not show significant response to the treatment with IL-2–stimulated NK cells or PBS (Fig. 5A). In contrast, mice bearing LoVo tumors did not show significant tumor shrinkage with eUCB-NK cells and the tumor burdens were not controlled by any group of treatment (Fig. 5B). Since HT29 and LoVo cells have similar sensitivity to NK cell–mediated cytotoxicity in vitro (Fig. 3A), other factors that are present may explain this lack of responsiveness to NK cell therapy in LoVo tumors. The infiltration of a sufficient number of NK cells should be a prerequisite for successful NK cell therapy. We examined the tumors for the presence of NK cell infiltration. Seven days after T-cell injections, tumors from mice were analyzed by flow cytometry for CD56+ NK cells. Results revealed that HT29 tumor xenografts, but not LoVo tumor xenografts had appreciable numbers of detectable NK cells in the sections of tumors (Fig. 5C-D). These data suggest that a lack of NK cell infiltration inside LoVo tumors might have contributed to their unresponsiveness to eUCB-NK cells.
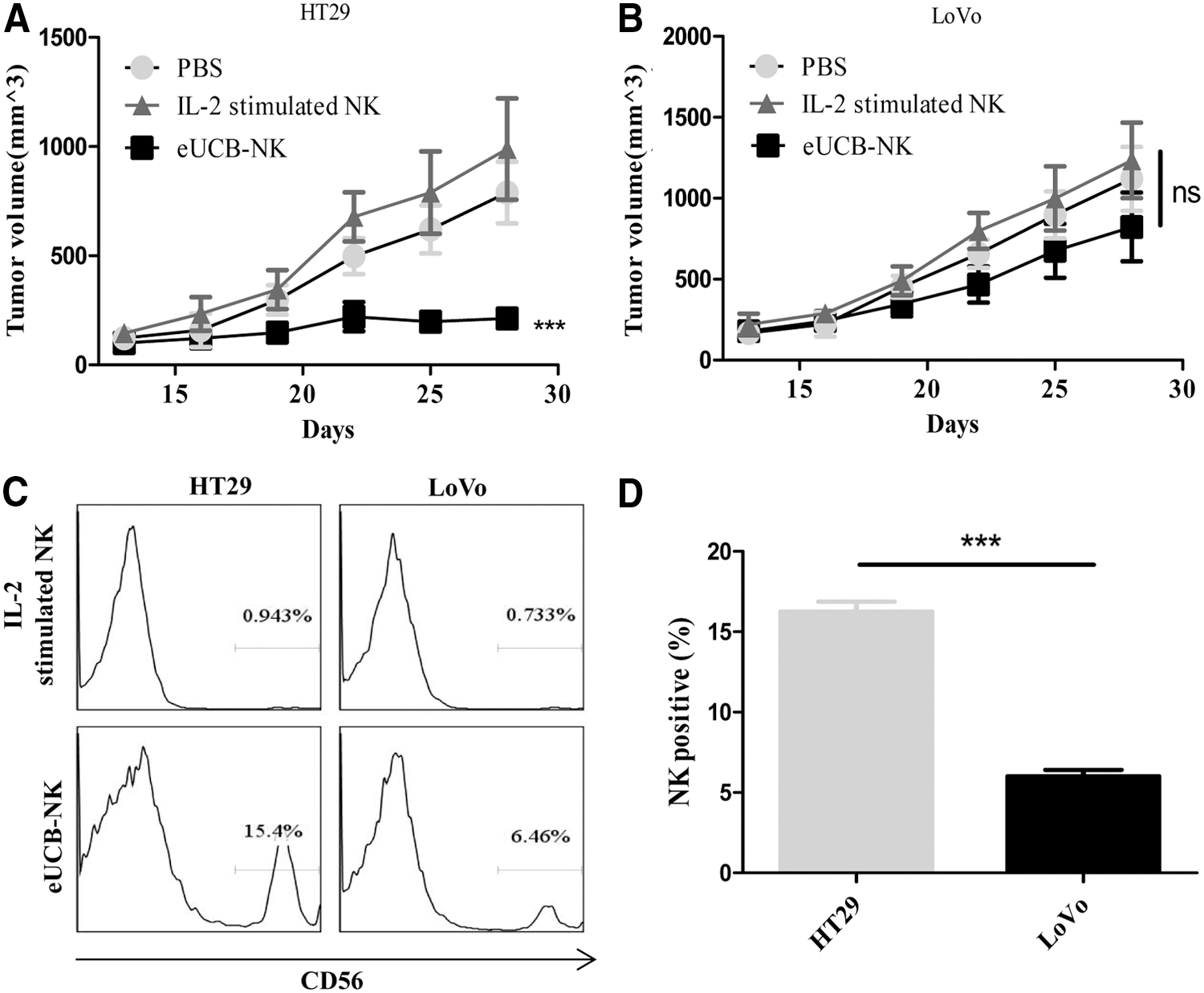
Therapeutic efficacy of NK cells against established CRC xenografts. Growth curve of
Low NK cell infiltration might result from abnormal tumor vasculature
Emerging data indicate that vascular abnormalities, driven by proangiogenic factors secreted by tumor cells, foster a microenvironment that favors tumor progression and facilitates immune evasion. 24,25 Total RNA from HT29 and LoVo tumor specimens was extracted, and quantitative PCR was performed to examine whether the antiangiogenic signals were associated with the level of NK cell infiltration. Data revealed that several of the antiangiogenic markers mRNA, including VEGF, VEGFR-1, VEGFR-2, CD31, and vWF were significantly lower in HT29 tumors compared to that in LoVo tumors, whereas the NK cell marker CD56 mRNA was significantly higher in HT29 tumors than that in the LoVo tumors (Fig. 6A-F). These data indicate that insufficient NK cell infiltration in LoVo tumor xenografts might have resulted from a high level of tumor neoangiogenesis.
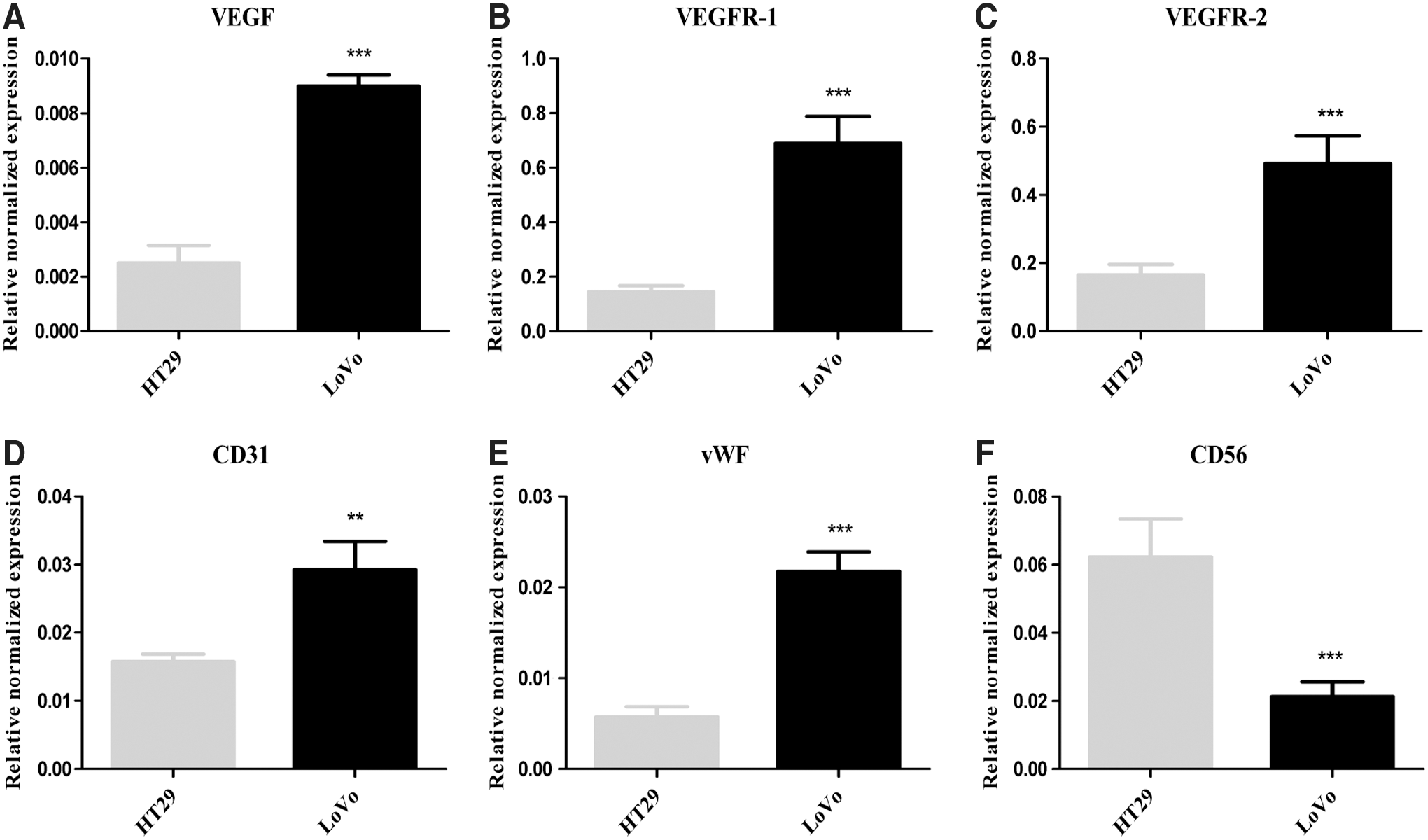
Low NK cell infiltration might result from abnormal tumor vasculature.
Combination with bevacizumab enhanced NK cell-based therapy efficacy
Antiangiogenic agents can normalize the abnormal tumor vasculature and increase the infiltration of immune effector cells into tumors. 24,26 Here we investigated whether the combined administration of antiangiogenic agents and eUCB-NK cells to immunodeficient mice xenografted with human LoVo tumors improved NK cell migration and invasion to the tumor site. Bevacizumab, as the most widely prescribed antiangiogenic drug, has an important role in the treatment of CRC either used alone or combined with standard cytotoxic therapy. 27 We applied bevacizumab in this study and found that LoVo tumor growth was dramatically inhibited by eUCB-NK cells in combination with bevacizumab, while the eUCB-NK cells or bevacizumab alone has no such superior effect on tumor growth (Fig. 7A). Furthermore, the tumor weights in mice receiving a combined treatment were significantly less than those in mice receiving a single treatment (Fig. 7B). Significantly increased NK cell infiltration was observed in the tumor bed harvested from mice receiving bevacizumab and eUCB-NK cells (Fig. 7C-D). Collectively, bevacizumab has the potential to increase NK cell infiltration and improve the therapeutic activity of eUCB-NK cells in LoVo tumors that were primarily unresponsive to eUCB-NK cells.
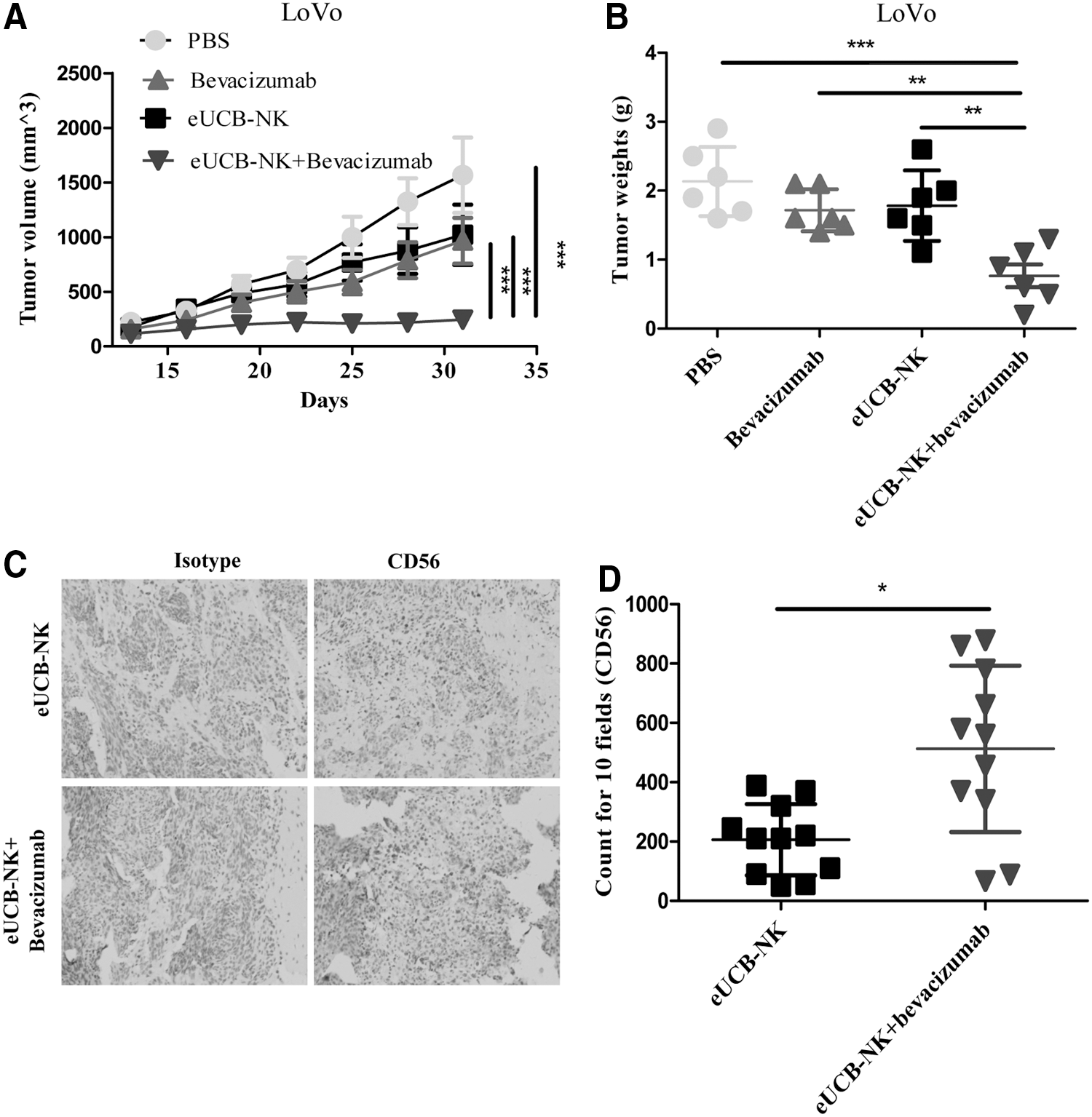
Combination with bevacizumab-enhanced NK cell–based therapy efficacy.
Discussion
Convincing evidence suggests that chronic inflammation plays a pivotal role in the clinical course of CRC. 28 The induced inflammatory microenvironment recruits more immune cells including T helper, cytotoxic T lymphocytes, mast cells, plasma cells, and macrophages. 29 NK cells are among the most scarce immune cell populations found in the tumor bed, leading to debate about whether NK cells have an effect on CRC tumor progression. 9 A typical chemically induced mouse model involved in colorectal carcinogenesis detected persistent dysregulation of NK cells starting from an early stage of CRC, suggesting that dysfunction of NK cells might contribute to cancer progression in the early phases. 30 In patients with CRC, peripheral or tumor-infiltrating NK cells are often functionally impaired and NK cells show decreased numbers, which are correlated with poorer outcome. 31,32 In addition, the phenotypic features of NK cells from patients with CRC are altered, with a drastic reduction in the expression of NK cell-activating receptors and a significant increase of PD-1 expression on peripheral and tumor-infiltrating NK cells. 33 –35 The increased PD-1 expression on NK cells is associated with poorer prognosis in some digestive cancer types, and PD-1 blockade markedly enhanced NK cell functions and greatly suppressed the growth of tumor xenografts. 33 Although somewhat unilaterally, these studies provided compelling evidence for demonstrating critical roles of the marginally explored and widely inhibited NK cells against CRC, and immunotherapies based on NK cells might be promising for the future development against CRC.
Obtaining sufficient numbers of NK cells is a priority for robust preclinical and clinical evaluations. Denman and colleagues previously developed a robust NK cell expansion platform using the K562-mbIL21 system. 23 This methodology expanded NK cells 1,000-folds and produced highly functional NK cells with longer telomeres and less senescence. In the present study, we successfully applied this method to propagate large quantities of NK cells from UCB donors, despite the efficacy remaining variable due to individual donor differences. Two approaches are widely used to for determining NK cell function—degranulation assay and cytotoxicity assay. Compared with the IL-2–stimulated NK cells, the eUCB-NK cells were highly toxic against diverse CRC targets as measured by the degranulation and cytotoxicity assay. Additionally, the eUCB-NK cells secreted higher levels of IFN-γ, TNF-α, GM-CSF, and CCL3. The phenotypes of eUCB-NK and IL-2–stimulated NK cells were also compared. Data revealed that eUCB-NK cells had increased expression of activating receptors, particularly TRAIL and NKG2D, which are strongly associated with NK cell activity. The increase in expression of activating receptors and molecules likely contributes to their enhanced cytotoxicity, and blocking experiments are needed to define the exact contribution of individual pathways to augmented NK cell cytolytic function. It is unlikely that the change in NK cell inhibitory receptors and molecules played any role in augmenting NK cell cytotoxicity because no significant variation of these receptors and molecules was observed following NK cell expansion.
Since eUCB-NK cells can efficiently target CRC cell lines in vitro, we investigated whether intravenous infusions of those NK cells can eliminate human CRC xenografts in immunodeficient NSG mice. Administration of eUCB-NK cells markedly inhibited the growth of the HT29 tumor xenograft but did not affect the growth of LoVo tumors. Since HT29 and LoVo cells have similar sensitivity to NK cell–mediated activity in vitro, we hypothesized that HT29 and LoVo might have different tumor microenvironments that could contribute to the differences in response to eUCB-NK cells. We compared the levels of CD56+ lymphocyte infiltration, finding that HT29 tumors have much higher amounts of NK cells (CD56+) than LoVo tumors. These data raise the possibility that the lack of sufficient NK cells make LoVo tumors less responsive to eUCB-NK cells, and agents that could promote lymphocyte infiltration into microenvironments might be an alternative to overcome resistance to eUCB-NK cells in LoVo tumors.
Abnormal tumor blood vessels foster an immunosuppressive tumor milieu that plays a crucial role in impeding lymphocyte cell infiltration. 26,36 In this study, we found evidence that the higher abnormal levels of tumor vascular factors (VEGF, VEGFR-1, VEGFR-2, CD31, and vWF) were correlated with low NK cell infiltration (CD56) at the mRNA level, indicating that insufficient NK cell infiltration in LoVo tumor xenografts might result from a high level of tumor neoangiogenesis. We thus sought to combine antiangiogenic agents with eUCB-NK cells to enhance the cells' therapeutic activity in LoVo tumors. Bevacizumab, the most widely prescribed antiangiogenic drug, has an important role in the treatment of CRC either used alone or in combination with standard cytotoxic therapy. 27 Data revealed that neither eUCB-NK cells nor bevacizumab alone exerted significant anti-tumor activity, whereas the combination of eUCB-NK cells and bevacizumab dramatically suppressed tumor growth. Moreover, increased NK cell infiltration was observed in the tumor bed harvested from mice receiving bevacizumab and eUCB-NK cells, suggesting that bevacizumab should improve NK cell–based therapies by increasing the extravasation of adoptively transferred NK cells into tumors. An unexpected finding was that bevacizumab administrated alone failed to elicit significant responses. This finding might be explained by LoVo cells expressing high levels of EGFR, which are associated with and may contribute to resistance to bevacizumab treatment. 37 So far, few studies have explored the effect of bevacizumab in the LoVo tumor–bearing mouse model, and whether bevacizumab treatment can inhibit LoVo tumors and by how much has yet to be fully determined.
In conclusion, we demonstrated that eUCB-NK cells have potent in vitro and in vivo antitumor activities, which might be applicable to patients with advanced CRC. Unresponsiveness to NK cell therapy may be due to insufficient NK cell infiltration, and bevacizumab may increase NK cell infiltration and improve the efficacy of NK cell–based therapy. These results provide a rationale for exploring eUCB-NK cell–based therapeutics, either alone or together with bevacizumab, in the treatment of human CRC.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China under grant numbers NSCF 81402401 and 81702922. Jiaping Hu and Taiyuan conceived the idea, designed the research and revised the manuscript; Chen Xu designed subsequent experiments, performed most of the in vitro and in vivo work, and wrote the manuscript; Fan Zhuo and Huankui Sun helped perform the in vivo experiments; Dongning Liu and Zhixin Chen assisted with interpretation of data and helped to perform the in vitro and in vivo work.
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.