
Abstract
CRISPR/Cas9 is an adaptive immune system where bacteria and archaea have evolved to resist the invading viruses and plasmid DNA by creating site-specific double-strand breaks in DNA. This study tested this gene editing system in inhibiting human immunodeficiency virus type 1 (HIV-1) infection by targeting the viral long terminal repeat and the gene coding sequences. Strong inhibition of HIV-1 infection by Cas9/gRNA was observed, which resulted not only from insertions and deletions (indels) that were introduced into viral DNA due to Cas9 cleavage, but also from the marked decrease in the levels of the late viral DNA products and the integrated viral DNA. This latter defect might have reflected the degradation of viral DNA that has not been immediately repaired after Cas9 cleavage. It was further observed that Cas9, when solely located in the cytoplasm, inhibits HIV-1 as strongly as the nuclear Cas9, except that the cytoplasmic Cas9 does not act on the integrated HIV-1 DNA and thus cannot be used to excise the latent provirus. Together, the results suggest that Cas9/gRNA is able to target and edit HIV-1 DNA both in the cytoplasm and in the nucleus. The inhibitory effect of Cas9 on HIV-1 is attributed to both the indels in viral DNA and the reduction in the levels of viral DNA.
CRISPR/Cas9 is an adaptive immune system where bacteria and archaea have evolved to resist the invading viruses and plasmid DNA by creating site-specific double-strand breaks in DNA. This study tested this gene editing system in inhibiting human immunodeficiency virus type 1 (HIV-1) infection by targeting the viral long terminal repeat and the gene coding sequences. Strong inhibition of HIV-1 infection by Cas9/gRNA was observed, which resulted not only from insertions and deletions (indels) that were introduced into viral DNA due to Cas9 cleavage, but also from the marked decrease in the levels of the late viral DNA products and the integrated viral DNA. This latter defect might have reflected the degradation of viral DNA that has not been immediately repaired after Cas9 cleavage. It was further observed that Cas9, when solely located in the cytoplasm, inhibits HIV-1 as strongly as the nuclear Cas9, except that the cytoplasmic Cas9 does not act on the integrated HIV-1 DNA and thus cannot be used to excise the latent provirus. Together, the results suggest that Cas9/gRNA is able to target and edit HIV-1 DNA both in the cytoplasm and in the nucleus. The inhibitory effect of Cas9 on HIV-1 is attributed to both the indels in viral DNA and the reduction in the levels of viral DNA.
Introduction
Human immunodeficiency virus (HIV) still infects more than 35 million people worldwide according to the Global Health Observatory data (updated November 2017). Currently, highly active antiretroviral therapy (HAART) is able to suppress HIV infection below detectable levels in HIV patients. 1,2 However, HAART is limited in its high cost, patient compliance, side effects from long-term therapy, and emergence of drug resistance. Most of all, it does not cure HIV infection. 3,4 Therefore, there is a continuous need to develop more effective therapeutics and cure strategies for HIV infection.
The clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 system in bacteria and archaea protects the host from invading genetic elements, such as viruses and plasmid DNA. 5 –15 Through recognizing a short trinucleotide protospacer adjacent motif (PAM), Cas9 is assisted by a guide RNA (gRNA) to recognize and cleave the specific double-stranded DNA, resulting in deletions or insertions (indels) upon repair by the non-homologous end joining (NHEJ) machinery. 16 –19 Cas9 cleaves the target dsDNA by its RuvC and HNH nuclease domain. 16,20 CRISPR/Cas9 has been proven a versatile tool for genome engineering in many organisms. 21 –26 Meanwhile, CRISPR/Cas9 has also been explored as an antiviral tool to control viral infections, including adenovirus, 27 herpes simplex virus type 1 (HSV-1), 27 –30 Epstein–Barr virus (EBV), 30 –33 human cytomegalovirus (HCMV), 30 human papillomavirus, 34 –38 vaccinia virus, 39 hepatitis B virus (HBV), 31,40 –49 JC virus (JCV), 50,51 and HIV type 1 (HIV-1). 52 –65
In the case of HIV-1, several genome editing strategies have been employed to inhibit HIV-1. One strategy is to use CRISPR/Cas9 to edit HIV-1 co-receptor CCR5, CXCR4, or other cellular factors essential for HIV-1 replication, thus protecting the genetically modified primary CD4+ T cells from HIV-1 infection. 66 –74 Alternatively, CRISPR/Cas9 has been employed to inactivate HIV-1 DNA by designing gRNAs to target viral long terminal repeat (LTR) promoter DNA or essential viral genes. 53,54,56,58 –60,64,65,75 Ebina et al. first reported that HIV LTR-driven reporter gene expression was inhibited by Cas9 and an LTR-targeting gRNA. Further sequencing analysis showed that CRISPR/Cas9 treatment not only caused indels in the LTR, but also excised the viral DNA between the 5′ and 3′ LTRs. 52 Subsequent studies confirmed and further expanded the utility of CRISPR/Cas9 in inactivating and removing latent HIV-1 DNA in various cell lines and primary cells. 54,56,58,60,75 –77 Further, Kaminski and Yin used the recombinant adeno-associated virus (AAV) vector to express and deliver gRNAs and saCas9 into mice, and demonstrated efficient editing of HIV-1 DNA in various HIV transgenic mouse models. 65,78 Along this line of study, one important progress is the elucidation of the unique molecular mechanism that leads to HIV-1 escape from CRISPR/Cas9 attack. 61,62,79 –81 These studies showed that the Cas9-resistant viral mutations are mainly derived from the indels that are created by the NHEJ machinery when repairing the double-stranded DNA breaks caused by Cas9, in addition to the contribution by the error-prone viral reverse transcriptase. Fortunately, HIV-1 escape from CRISPR/Cas9 attack can be surmounted with two gRNAs that are properly designed to target essential viral genes. 60,77
In addition to exploiting the DNA nuclease activity of Cas9, the catalytically inactive dCas9 has been engineered by fusing with transcription activator domains and used to stimulate viral gene expression from latent HIV-1 reservoirs, providing a promising means for the “shock and kill” cure strategy. 82 –89 Taken together, CRISPR/Cas9 has provided a novel and powerful molecular tool that has been explored by many research groups to suppress and, hopefully, eradicate HIV-1 infection in cultured cells and in mouse models. Most of these studies have examined the integrated HIV-1 DNA for indels to evaluate the editing effect of Cas9. It is also unclear to what extent Cas9 affects that the newly synthesized HIV-1 DNA in the cytoplasm. This study further explored the molecular mechanisms by which CRISPR/Cas9 suppresses HIV-1 infection, and found that Cas9 not only caused indels in viral DNA, but also diminished the levels of the late viral DNA products and the integrated viral DNA. The data also demonstrate that Cas9, when present only in the cytoplasm, is able to edit the newly synthesized HIV-1 DNA and inhibit viral infection.
Materials and Methods
Plasmid construction
pX330-U6-Chimeric_BB-CBh-hSpCas9 was purchased from Addgene (plasmid # 42230). The hCas9 without nuclear location signal (NLS) plasmid was constructed by deleting the two NLS sequences. HIV-1NL4-3-▵E-YFP viral DNA was kindly provided by Dr. David Levy. 90
To design gRNA targeting HIV-1 DNA, the HIV-1 genome (NCBI reference sequence: NC_001802.1) was searched for unique CRISPR target sites (N20NGG, where N is any nucleotide), and selected gRNAs that are located at HIV-1 LTR and conserved viral gene sequences. The off-target possibilities were also excluded by blasting the human genome. gRNAs were designed to target GFP DNA and renilla luciferase DNA, as reported previously.
56
All the targets were screened to avoid off-target possibilities, especially in functional genes in human genome, using the online CRISPR design tools.
91
The Cas9-gRNA expression plasmids were constructed, as described previously.
91
gRNA sequences are provided in Supplementary Table S1 (Supplementary Data are available online at
Cell culture and transfection
Human embryonic kidney 293T (HEK-293T) cells and HEK-293 cells were maintained in the Dulbecco's modified Eagle's medium (DMEM; Invitrogen). HEK-293 LTR-GFP cells were obtained by transfecting HEK-293 cells with HIV-1-based retroviral vector containing the Tat and GFP open reading frames both under the control of the HIV promoter in the 5′ LTR. J-Lat 10.6 (J-Lat Full Length Clone 10.6) cells were maintained in RPMI 1640 medium (Invitrogen). All cells were supplemented with 10% fetal bovine serum (Life Technologies), 100 IU/mL of penicillin, and 100 mg/mL of streptomycin (Solarbio; cat. no. P1400) at 37°C with 5% CO2 incubation. J-Lat10.6 expresses GFP and produces incomplete viral particles after treatment with tumor necrosis factor alpha (TNF-α; Pepro Tech; cat. no. 300-01A). 56 293T cells were seeded onto plates 16 h prior to transfection. Cells were transfected with polyethylenimine (PEI; Sigma–Aldrich) according to the manufacturer's instructions. J-Lat 10.6 cells were transfected with hCas9/gRNA by Neon (Life Technologies) according to the manufacturer's instructions. Briefly, J-Lat 10.6 cells (2 × 105) were mixed with 2 μg of plasmid DNA in 10 μL of resuspension buffer R and electroporated with the following settings: pulse voltage, 1325 V; width, 10 ms; and number, 2. Cells were cultured in complete growth medium with no antibiotics for 24 h. Then, the cells were incubated with TNF-α (10 ng/mL) for 16 h and analyzed by flow cytometry. 56
Cell viability assay
293T cells were cultured on 96-well plates at a concentration of 1 × 103 cells per well and transfected with plasmid DNA using Lipofectamine (Invitrogen). The cells were incubated for 48 h before being examined with the CellTiter-Glo® Luminescent Cell Viability Assay Kit (G7570; Promega). Briefly, 100 μL of CellTiter-Glo® Reagent was added to 100 μL of medium containing cells and mixed for 2 min on an orbital shaker to induce cell lysis. The luminescence of each well was recorded using a Modulus Microplate Reader (Turner Biosystems).
HIV-1 pseudovirus preparation and infection
Viral stocks were generated by co-transfection of HEK-293T cells with 8 μg of HIV-1NL4-3-▵E-YFP and 2 μg vesicular stomatitis virus glycoprotein (VSV-G) plasmids on 10 cm plates using PEI. Two days after transfection, the supernatants were collected and filtered (0.45 μM). After hCas9/gRNA transfection, 293T cells were infected with VSV-G pseudotyped virus. Thirty-six hours later, cells were harvested and analyzed by flow cytometry and Western blotting for HIV-EYFP expression.
Flow cytometry
Cells were detached using Trypsin-EDTA (0.25%; Life Technologies) and re-suspended in complete growth media. Cells were washed with phosphate-buffered saline (PBS; Hyclone; cat. no. SH30256.01) and suspended in PBS containing 1% paraformaldehyde (Solarbio; cat. no. P1110). The cell membrane was permeabilized with 0.2% Triton X-100 (bio-rexd). hCas9 was labeled by 647 conjugate Flag antibody (Cell Signaling Biotechnology; cat. no. 3916s). YFP and hCas9 expression were determined using the FACSCanto II and CELLQuest software.
Western blotting
Western blotting analysis was performed, as previously described. 92 In brief, 36 h post infection of 293T cells with the HIVNL4-3-▵E-YFP virus, which had been transfected with hCas9/gRNA plasmid, cells were lysed in RIPA buffer (0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate, 150 mM of NaCl, 10 mM of Tris [pH 7.5], 1 mM of EDTA). Equal amounts of cell lysates were separated in SDS-12% PAGE. Proteins were transferred onto nitrocellulose membranes (Whatmann). The membranes were probed with CA-p24 antibody (Sino Biological; 11695-V08E), Flag antibody (Sigma–Aldrich; F3165), actin antibody (Proteintech; 60008-1-Ig), and GFP antibody (Proteintech; 66002-1-Ig), followed by incubation with IRDyeTM secondary antibodies (1:20,000). Protein bands were visualized on a LiCor Odyssey instrument.
CA-P24 Western blotting
Twenty-four hours post transfection and infection, 293T cells were detached from the six-well plate and seeded onto 10 cm plates. Eighteen hours later, 293T cells were transfected with 3 μg VSV-G plasmid to package the newly produced viruses. Forty-eight hours post transfection of 293T cells with VSV-G plasmid, the supernatants were collected and filtered (0.45 μM). The virus particles were pelleted by ultracentrifugation through a 20% sucrose cushion using volume-equal supernatant, and the number of virus particles was quantified by CA-p24 Western blotting. HEK-293 cells were infected with CA-p24 equal viruses. Forty-eight hours after infection, cells were harvested and analyzed by CA-p24 Western blotting and YFP flow cytometry.
SURVEYOR nuclease assay
HEK-293T cells were transfected with hCas9/gRNA vector and infected with virus. Cells were incubated at 37°C for 36 h post infection prior to extraction of total cellular DNA. Total cellular DNA was extracted using QIAamp DNA mini kit (Qiagen; cat. no. 51304). The viral genomic region flanking the CRISPR target site was amplified by polymerase chain reaction (PCR; Supplementary Table S2). The PCR products were purified using QIAquick Gel Extraction Kit (Qiagen) following the manufacturer's protocol. NHEJ events were measured by performing the SURVEYOR assay 17 according to the manufacturer's instructions (Transgenomics). First, a total of 100–400 ng of the purified PCR products were subjected to a reannealing process to enable heteroduplex formation: 95°C for 10 min; 95°C to 85°C ramping at −2°C/s; 85°C to 25°C at – 0.3°C/s; and held at 25°C for 1 min. After reannealing, products were treated with SURVEYOR nuclease and SURVEYOR enhancer S (Transgenomics) following the manufacturer's recommended protocol, and were analyzed on 10% TBE polyacrylamide gels. Gels were stained with GelRed for 30 min and imaged with a Gel Doc gel imaging system (Bio-Rad Laboratories). The frequency of indels was calculated following methods previously described. 91
TA cloning and sequencing
The DNA extracted from cells was amplified by PCR, and the products were cloned into the pMD18-T vector (TaKaRa). Primers that amplify 400–600 bp around the target in HIV-1 genome were designed (Supplementary Table S2). Thirty-fifty clones of each PCR product were sequenced. 56
Measuring viral DNA synthesis
Levels of early and late viral reverse transcription products, as well as the levels of integrated viral DNA, were measured by real-time PCR, as previously reported. 93 –95 Briefly, equal amounts of DNase-treated virions (100 ng of CA-p24) were used to infect 5 × 106 293T cells that had been transfected with hCas9/gRNA plasmids. Following 2 h of incubation at 4°C, the cells with bound virus were washed twice with PBS and maintained with fresh DMEM. At 12 and 24 h, cells were collected, and cellular DNA was extracted with the QIAamp DNA mini kit (Qiagen; cat. no. 51304). Using equal amounts of cellular genomic DNA, early (R-U5) and late (U5-gag) minus-strand reverse transcripts were quantitated by real-time PCR. The integrated proviruses were quantitated by TaqMan based Alu-LTR real-time nested PCR. The primer sets used to detect each product are shown in Supplementary Table S3. Real-time PCR was conducted with the Power Up SYBR Green Master Mix (Applied Biosystems; cat. no. A25742) or Taqman Universal PCR Master Mix (Applied Biosystems; cat. no. 4440040) in accordance with the manufacturer's instructions.
Results
Inhibition of HIV-1 replication by Cas9/gRNA
First, sgRNAs were designed that target HIV-1 LTR, gag, pol, tati, and rev regions (Fig. 1a and Supplementary Table S1). A gRNA targeting both YFP and GFP DNA (called gYFP or gGFP [GTGAACCGCATCGAGCTGAA]) was included as a positive control. YFP DNA is inserted in HIV-1NL4-3-▵E-YFP. The gRNA targeting renilla luciferase DNA (called gRL [GTAGCGCGGTGTATTATACC]) was utilized as a negative control. Then, HEK-293T cells were transfected with plasmid DNA expressing hCas9-NLS and sgRNAs. These Cas9/sgRNA-expressing cells showed cell viability similar to that of the control cells (Supplementary Fig. S1a). Six hours after transfection, 293T cells were infected with the HIV-1NL4-3-▵E-YFP virus that was pseudotyped with VSV-G. To quantify HIV-1 infection, YFP expression was measured by flow cytometry, and HIV-1 Gag protein expression was determined by Western blotting. The number of YFP-positive cells was reduced by 57% to 89% when HIV-1 sgRNAs were used, compared to transfection of control sgRNA (Fig. 1b and c). Furthermore, results of Western blots showed that expression of Gag proteins was also significantly diminished in cells that were transfected with HIV-1 sgRNAs (Fig. 1d). Overall, sgRNAs targeting the LTR and the tat gene inhibit HIV-1 infection more strongly than the other sgRNAs tested.

Suppression of human immunodeficiency virus type 1 (HIV-1) gene expression by Cas9-NLS/gRNA.
Targeted viral DNA fragments were then isolated from these cells, amplified by PCR using the specific primers (Supplementary Table S2), and further examined by sequencing. Sequence analysis of the viral region showed that HIV-1 DNA contained various mutations (Supplementary Fig. S1b). Meanwhile, SURVEYOR assay was performed to measure the frequency of indels as a result of the targeted Cas9 cleavage of HIV-1 DNA. The frequency ranged from 4% to 25% (Supplementary Fig. S1c). The data verified the generated indels and the mutations that had been introduced at the Cas9 cleavage site of HIV-1 DNA.
Production of HIV-1 is inhibited by Cas9/gRNA
Next, the effect of Cas9/gRNA on HIV-1 production was examined. To this end, the progeny viruses were harvested from 293T cells that had been transfected with Cas9-NLS/gRNA and infected with VSV-G pseudotyped HIV-1NL4-3-▵E-YFP. After 48 h, cells were also transfected with the VSV-G vector to provide the newly produced HIV-1 with the VSV-G envelope protein (Fig. 2a). The progeny viruses were pelleted by ultracentrifugation through a 20% sucrose cushion and quantified by CA-p24 Western blotting. Virus production was reduced by 44% to 76% in the presence of the Cas9/gRNA (Fig. 2b). The 293 cells were then infected with HIV-1NL4-3-▵E-YFP of equal CA-p24 amount. Results of flow cytometry analysis showed that the Cas9/gRNA edited progeny viruses generated 73% to 94% fewer YFP-positive cells compared to the control virus (Fig. 2c and d). Levels of Gag proteins were also greatly reduced (Fig. 2e). It is noted that the reduction in the number of YFP-positive cells and levels of Gag expression from the infection by Cas9/gRNA-edited HIV-1 were greater compared to the infection of Cas9-gRNA expressing cells by HIV-1NL4-3-▵E-YFP, which suggests defects in the genome of these progeny viruses as a result of Cas9/gRNA editing (Supplementary Fig. S2). Together, these results further confirm that CRISPR/Cas9 is able to suppress HIV-1 infection by mutating viral DNA.

Inhibition of the production of progeny viruses by Cas9-NLS/gRNA.
CRISPR/Cas9 inhibits HIV-1 reverse transcription and integration
Steps of infection by HIV-1 involve entry of the viral core into cells, reverse transcription to form the linear viral DNA, and integration of viral DNA into the cellular chromosome. It was hypothesized that in addition to inactivating HIV-1 by causing indels in viral DNA, CRISPR/Cas9 may also diminish the levels of viral DNA during HIV-1 infection because the Cas9-cleaved HIV-1 DNA, if not immediately repaired, may undergo degradation. To test this, 293T cells were transfected with Cas9-NLS/gRNA targeting the LTR R-U5 region. They were then infected with VSV.G-HIV-1NL4-3-▵E-YFP. At different time points post infection, equal aliquots of cells were collected, and total cellular DNA was extracted. Then, utilizing the specific primers, real-time PCR was performed to determine the levels of newly synthesized HIV-1 DNA, including the early DNA, late DNA, and the integrated DNA products. 93 –95 The results showed a three- to fivefold reduction of integrated viral DNA in cells expressing Cas9/gRNA, a twofold decrease in late viral DNA, and no significant change in the early viral DNA products (Fig. 3). It was expected that the Cas9/sgRNA LTR3, which targets the tRNA3 Lys binding site in HIV-1 DNA, would not affect the early reverse transcription because Cas9/gRNA cannot cleave dsRNA. 96 However, the early viral DNA products were affected by neither Cas9/sgRNA LTR1 or LTR2, which may have been because Cas9 contains nuclear location signal and is mainly located in the nucleus, 17 and thus does not have the opportunity to cleave the early reverse transcription products that are only found in the cytoplasm. Together, these results demonstrate that CRISPR/Cas9 not only inactivates HIV-1 DNA by causing indels, but also decreases the level of proviral DNA likely as a result of DNA degradation. In addition to NHEJ repair after CRISPR/Cas9 cleavage, which facilitates virus escape, 61, 62,80,81 the data indicate another mechanism through which CRISPR/Cas9 cleavage leads to DNA degradation.
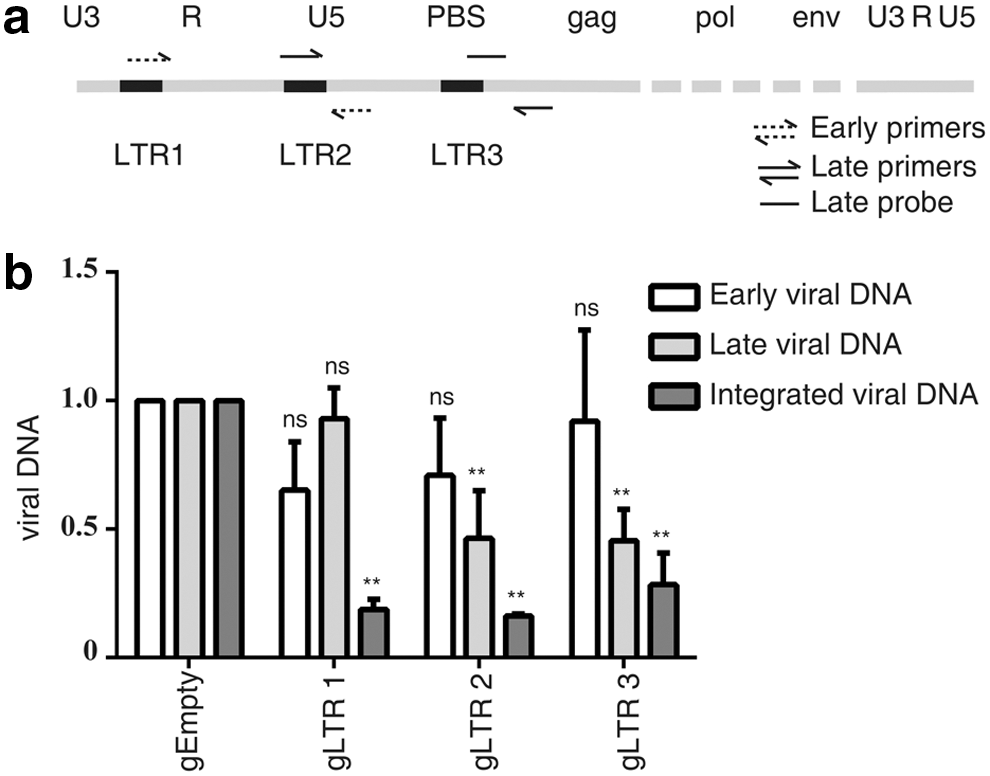
Inhibition of HIV-1 DNA synthesis by Cas9/gRNA.
Nuclease activity is required for Cas9 to inhibit HIV-1 infection
Cas9 contains two nuclease domains, HNH and RuvC, which are necessary for Cas9 to cleave the two DNA strands. 16,20 Next, the study tested the catalytically inactive Cas9-NLS (dCas9-NLS, D10A, and H840A) for its ability to inhibit HIV-1. The results showed that HIV-1 infection was not inhibited by dCas9-NLS when associated with HIV-1-specific gRNAs, except for gLTR1 that targets HIV-1 tat responsive stem-bulge-loop structure (TAR) and moderately diminished HIV-1 infection (Fig. 4a). This might be a result of dCas9/gLTR1 binding to the transcription initiation site and occluding RNA polymerase to start transcription. Results in Fig. 4b further show that levels of the integrated viral DNA were not affected in cells that expressed dCas9-NLS/gRNA. Together, these results demonstrate that the nuclease activity is essential for Cas9 to inhibit HIV-1 infection.
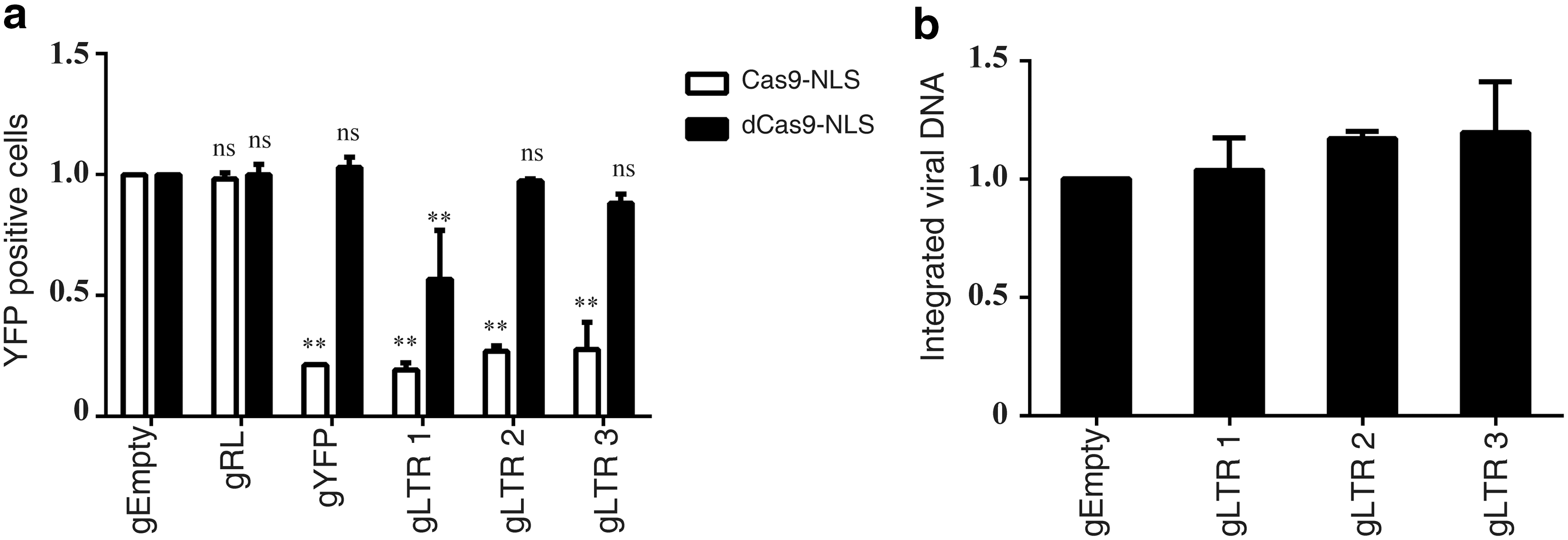
The nuclease activity of Cas9-NLS is essential for inhibiting HIV-1 infection.
CRISPR/Cas9 is able to edit and inactivate HIV-1 DNA in the cytoplasm
Since HIV-1 reverse transcription takes place in the cytoplasm, the study asked whether a Cas9 without NLS (Cas9-delNLS), which is not located to the nucleus, is also able to cleave the newly synthesized HIV-1 DNA in the cytoplasm and thus inhibit HIV-1 infection. The results of immunofluorescence imaging experiments showed that the Cas9-delNLS was strictly located in the cytoplasm, as opposed to the distribution of Cas9-NLS in both the cytoplasm and the nucleus (Fig. 5a). As expected, Cas9-delNLS/gRNA inhibited HIV-1 gene expression to a similar extent as Cas9-NLS/gRNA did (Fig. 5b–d). Furthermore, results of real-time PCR showed that both Cas9-delNLS/gRNA and Cas9-NLS/gRNA reduced the levels of the late viral DNA and the integrated viral DNA. Moreover, Cas9-delNLS/gRNA reduced the levels of the late viral DNA and the integrated viral DNA to similar levels, indicating that the decrease in proviral DNA is a result of the decrease in the late viral DNA, while the level of integrated viral DNA was lower than the level of late viral DNA edited by Cas9-NLS/gRNA. Additionally, Cas9-delNLS/gRNA moderately diminished the level of the early viral DNA products (Fig. 5e). These data demonstrate that Cas9-delNLS is able to inhibit HIV-1 infection by cleaving and degrading HIV-1 DNA in the cytoplasm.

Cas9/gRNA edits HIV-1 DNA in the cytoplasm.
In order to demonstrate further that Cas9-delNLS cleaves HIV-1 DNA only in the cytoplasm not in the nucleus, experiments were performed in 293 LTR-GFP cells that bear the GFP DNA that is controlled by the HIV-1 LTR promoter. The HIV-1 LTR and GFP DNA is integrated in the cellular DNA, and is thus only present in the nucleus. When these LTR-GFP reporter cells were transfected with Cas9-delNLS/gRNA or Cas9-NLS/gRNA and GFP expression was monitored by flow cytometry 5 days after transfection, a significant reduction of GFP expression was observed only with Cas9-NLS/gRNA, not with Cas9-delNLS/gRNA (Supplementary Fig. S3), suggesting that Cas9-delNLS, which is in the cytoplasm, does not act on DNA in the nucleus. This observation was further confirmed in experiments that were performed with the JLat10.6 HIV-latent cells. 97 JLat10.6 cell line harbors the HIV-1 DNA that is transcriptionally silent in the absence of external stimulation. Treatment with cytokines such as TNF-α activates viral gene expression, which can be measured by monitoring the expression of GFP that has been inserted into HIV-1 DNA as a reporter. Several gRNA clones were constructed that targeted the integrated HIV-1 LTR region, including the two gRNAs (gJkat LTR3 and gJkat LTR4) that were previously reported. 56 The results showed that the number of GFP-positive JLat10.6 cells decreased significantly with Cas9-NLS/gRNA, in contrast to Cas9-delNLS/gRNA that exerted no effect (Fig. 6a and b). These data suggest that Cas9 without NLS is able to target newly infected HIV-1 but not the integrated provirus. All together, these results demonstrate that CRISPR/Cas9 is able to target and edit HIV-1 dsDNA both in the cytoplasm and in the nucleus, and impair the late reverse transcription and viral DNA integration.
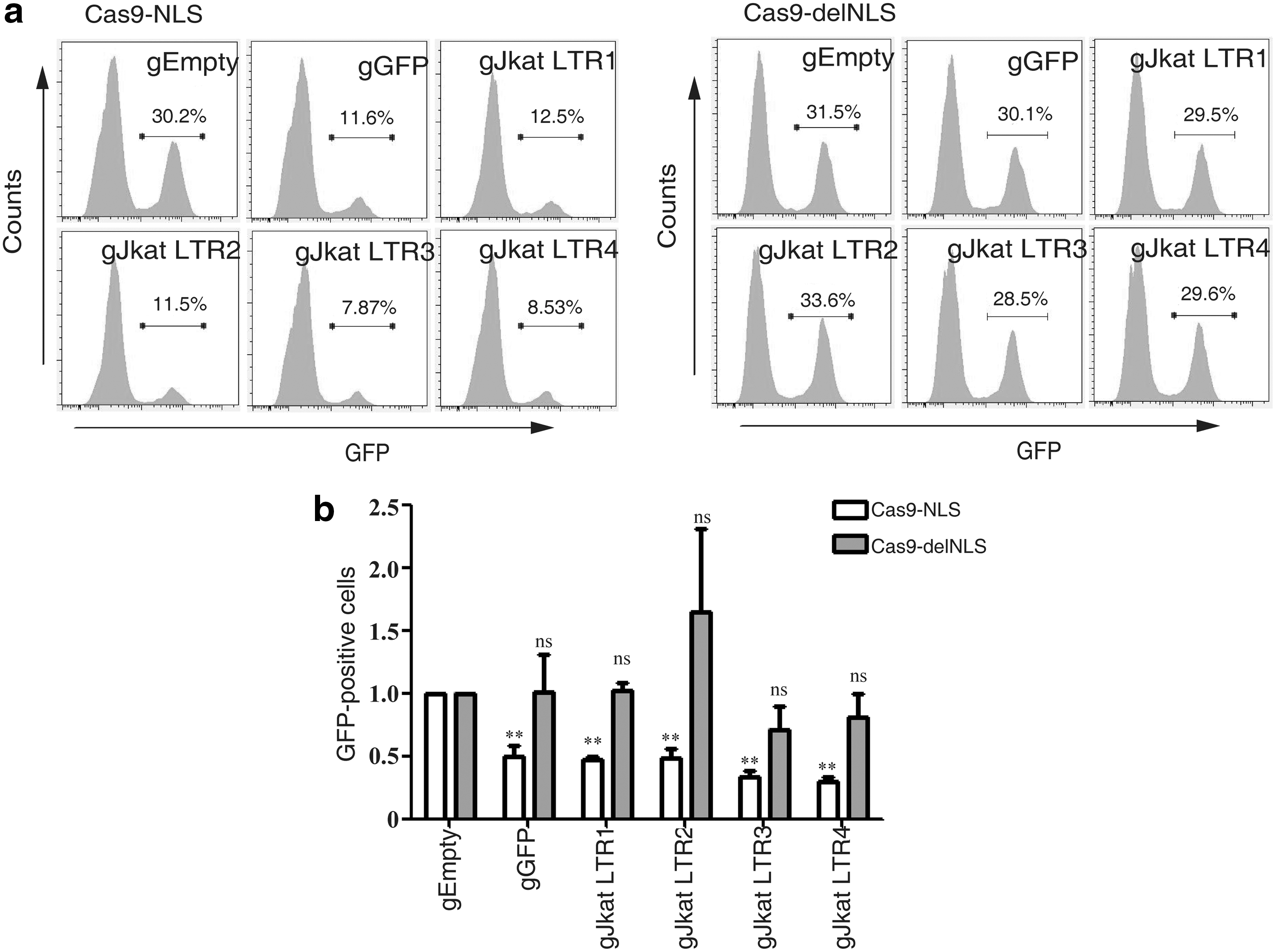
Cas9-delNLS/gRNA does not target the integrated HIV-1 DNA.
Discussion
The CRISPR/Cas9 system has been explored for its potential utility in providing new and unique treatments for HIV. Ebina, Liao, and Zhu designed CRISPR/Cas9 targeting HIV-1 LTR or gene coding region, and observed editing and inactivation of the latently integrated viral genome, suggesting that the CRISPR/Cas9 system may be a useful tool for curing HIV-1 infection. 52,56,75 Kaminski constructed a Tat controlled Cas9 system armed with multiplex gRNA targeting HIV-1 LTR, which successfully excised viral DNA segments. 59 Hu and Kaminski were able to delete the entire integrated proviral DNA completely utilizing CRISPR/Cas9 targeting the U3 region of HIV-1 LTR. 54,58 Considering HIV-1 LTR sequence heterogeneity and patient-derived variants, Dampier designed gRNAs targeting the predominant sequence of the integrated proviral LTR or designed broad-spectrum anti-HIV-1 gRNAs to target patient derived variants. 53,63 CXCR4 and CCR5 are co-receptors for HIV-1 infection. Several studies reported that CRISPR/Cas9 editing CXCR4 or CCR5 confers HIV-1 resistance to human primary CD4(+) T cells or hematopoietic stem cells. 67,71,73,74,98,99 Utilizing the CRISPR/Cas9 system comprising gRNA with a nuclease deficient Cas9 (dCas9) fused to the transactivation domain, the latently proviral DNA was reactivated, providing an alternative approach for the “shock and kill” treatment. 84,85,87 –89,100 AAV vector was explored for gene therapy in vivo. 65,101 –105 Yin effectively excised HIV-1 proviral DNA in solid tissues/organs from HIV-1 Tg26 transgenic mice by AAV-mediated gene delivery of multiplex sgRNAs/saCas9. 65 The efficacy of HIV-1 excision in animals using the AAV delivery vector showed great promise in clinical translation of Cas9 in treating HIV-1 infection in humans.
Nevertheless, few studies have investigated which steps of HIV-1 infection are impaired by CRISPR/Cas9. This study first tested multiple sgRNA target sites in HIV-1 genome, and strong inhibition of HIV-1 infection was observed by CRISPR/Cas9. Among the gRNAs that were tested, targeting the R region of LTR, especially the Tat-responsive stem bulge-loop structure (TAR) region, generated the strongest inhibitory effect on HIV-1 expression (Fig. 1b and c), which agrees with previous reports. 52,75 Furthermore, targeting the primer activation signal and the primer binding site also significantly inhibited HIV-1 replication (Fig. 1b and c), which supports the importance of these functional motifs in HIV-1 replication. Further, real-time PCR was performed to determine the levels of newly synthesized HIV-1 DNA, and it found that Cas9 targets and diminishes the late reverse transcription viral DNA and the integrated viral DNA. Importantly, using a version of Cas9 devoid of nuclear location signal, it was demonstrated that CRISPR/Cas9 targets HIV-1 genome prior to its nuclear entry.
CRISPR/Cas9 editing induces double-strand DNA breaks at the target sites, which often triggers the NHEJ and leads to indels and nucleotide substitutions. 17,22 Previous reports have demonstrated that many of these indels are lethal to the virus, but some lead to the emergence of replication competent viruses that are resistant to Cas9/sgRNA. 61,62,79 –81 Wang and Lebbink employed a combination of two carefully selected gRNAs targeting different regions of the HIV genome to abrogate viral replication and prevent viral escape. 60,77 Zhao et al. combined CRISPR-Cas9 and RNA interference to attack HIV-1 DNA or RNA and observed synergistic inhibition of HIV-1 infection. 106 This study now reports that in addition to causing indels by NHEJ repair, the action of Cas9 also leads to reduction in the levels of viral DNA products, including the late reverse transcription virus DNA and integrated viral DNA. By testing a version of Cas9 that only exists and functions in the cytoplasm, it was found that editing HIV-1 DNA by Cas9 can occur as early as when viral DNA is synthesized in the cytoplasm. However, the cytoplasmic Cas9 is unable to edit and excise the integrated HIV-1 DNA, which highlights the necessity of using the nuclear Cas9 (Cas9-NLS) to inactivate proviral DNA.
In summary, several gRNAs have been identified that are able to guide Cas9 to edit and suppress HIV-1 infection efficiently. The results also reveal a new mechanism of Cas9 inhibition of HIV-1 infection by decreasing the levels of viral DNA products, in addition to causing indels by NHEJ repair. Furthermore, it has been found that the newly synthesized viral DNA is already accessible to editing by Cas9 in the cytoplasm. The findings are expected to aid the design of new Cas9 variants with higher anti-HIV-1 activity.
Footnotes
Acknowledgments
We thank Yan Xiao and Li Li for technical assistance in performing confocal microscopy. This study was supported by funds from the National Key Plan for Scientific Research and Development of China (2016YFD0500307), from the Ministry of Science and Technology of China (2013ZX10001005-001-002), from the CAMS Innovation Fund for Medical Science (CIFMS 2017-I2M-1-014), from the National Natural Science Foundation of China (81371808, 81528012, 81401673, 81601771), from the Canadian Institutes of Health Research (CCI-132561), from the PUMC Youth Fund/Fundamental Research Funds for the Central Universities (3332016083), and from the CAMS general fund (2016ZX310049).
Author Disclosure
The authors declare that there are no competing interests associated with the article information.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.