
Abstract
Follistatin-like 1 (Fstl1) protects cardiomyocytes from a broad spectrum of pathologic injuries including myocardial infarction (MI). It is worthy of note that although cardiac Fstl1 is elevated in post-MI microenvironment, its cardioprotective role is still restricted to a limited extent considering the frequency and severity of adverse cardiac remodeling following MI. We therefore propose that intrinsic Fstl1-suppressing microRNA (miRNA) may exist in the heart and its neutralization may further facilitate post-MI recovery. Here, miR-9-5p is predicted as one of the potential Fstl1-targeting miRNAs whose expression is decreased in ischemic myocardium and reversely correlated with Fstl1. Luciferase activity assay further validated Fstl1 as a direct target of miR-9-5p. In addition, forced expression of miR-9-5p in H9c2 cells is concurrent with diminished expression of Fstl1 and vice versa. Importantly, transfection of miR-9-5p mimics in hypoxic H9c2 cells exacerbates cardiac cell death, lactate dehydrogenase release, reactive oxygen species accumulation, and malonyldialdehyde concentration. More importantly, in vivo silencing of miR-9-5p by a specific antagomir in a murine acute MI model effectively preserves post-MI heart function with attenuated fibrosis and inflammatory response. Further studies demonstrated that antagomir treatment stabilizes Fstl1 expression as well as blocks cardiac cell death and reactive oxygen species generation in both ischemia-challenged hearts and hypoxia-treated cardiomyoblasts. Finally, cytoprotection against hypoxic challenge by miR-9-5p inhibitor is partially reversed by knockdown of Fstl1, indicating a novel role of miR-9-5p/Fstl1 axis in survival defense against hypoxic challenge. In summary, these findings identified miR-9-5p as a mediator of hypoxic injury in cardiomyoblasts and miR-9-5p suppression prevents cardiac remodeling after acute MI, providing a potential strategy for early treatment against MI.
Introduction
Myocardial infarction (MI) remains the most common cause of cardiac morbidity and mortality in the world. Following MI, insufficient oxygen/blood supply, as well as oxidative stress accumulation, leads to irreparable loss of cardiomyocytes, and this is further exacerbated by toxic substances released from dead cells. 1,2 In addition, these immediate damages also trigger left ventricular (LV) remodeling, which eventually results in functional decomposition and heart failure. 3 Thus, maintaining sufficient cardiomyocytes and favorable infarct microenvironment is essential for preservation of post-MI heart function and structure.
Follistatin-like 1 (Fstl1), also known as TSC-36, is a secreted glycoprotein induced by transforming growth factor β1. 4 Recently, we reported that Fstl1 protects cardiomyoblasts from cell death through Akt and Smad1/5/9 signaling. 5 Previous research has also demonstrated Fstl1 as a protective cardiokine during post-MI cardiac remodeling 6 –8 and pathological cardiac hypertrophy process. 9 It is worthy of note that although cardiac Fstl1 is obviously elevated in post-MI microenvironment, 6 its cardioprotective role is still restricted to a limited extent considering the frequency and severity of adverse cardiac remodeling following MI. We, therefore, propose that intrinsic Fstl1 suppressor may exist in the ischemic myocardium and its silence may further assist post-MI recovery.
MicroRNAs (miRNAs) are a class of ubiquitously expressed, noncoding RNAs with a length of 17–24 nucleotides. They function as gene silencers by inhibiting translation and/or by promoting degradation of mRNA. 10,11 The miRNAs involved in cardiac remodeling can be broadly categorized as pro-remodeling and anti-remodeling miRNAs. For example, miR-17-3p contributes to exercise-induced cardiac growth and protects against myocardial ischemia-reperfusion injury. 12 In contrast, excessive miR-195 in failing myocardium directly targets mitochondrial Sirt3, resulting in enhanced global protein acetylation and impaired cardiac energy metabolism. 13 We therefore hypothesize that miRNA(s) may represent those intrinsic Fstl1 suppressor(s) and its neutralization may further alter cardiac remodeling process and benefit recovery from MI.
Our present study identified miR-9-5p as an Fstl1 suppressor that directly targets its 3′-untranslated regions (3′-UTRs) and negatively regulates its expression. Excessive miR-9-5p in cardiomyoblasts contributes to multiple hypoxia-initiated cellular injuries. Importantly, myocardial injection of its antagomir stabilizes Fstl1 expression and simultaneously attenuates post-MI cardiac dysfunction and remodeling. Further studies demonstrated that antagomir treatment blocks cardiac cell death and reactive oxygen species (ROS) generation in both ischemia-challenged hearts and hypoxia-treated cardiomyoblasts. Intriguingly, abrogated remodeling in antagomir-treated hearts is also associated with diminished fibrosis and inflammatory response. Finally, knockdown of Fstl1 abrogates cytoprotective role of miR-9-5p inhibitor against hypoxic challenge, indicating Fstl1 as a functional downstream of miR-9-5p in this process. Collectively, our data provide critical information for establishing miRNA-based therapeutic approaches against myocardial infarction.
Materials and Methods
Cell culture and treatment
Rat embryonic cardiomyoblast-derived H9c2 cells and 293T cells were maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum. For hypoxic treatment, cells were cultured in a tri-gas incubator (Thermo, Germany) composed of 94% N2, 5% CO2, and 1% O2 for 48 h. miR-9-5p mimics, inhibitors, siFstl1, and their corresponding negative controls (NC) were purchased from GenePharma (China) and transfected into the cells with lipofectamine 2000 (Invitrogen). Rat siFstl1 sequence was described as previously. 5 The negative control small interfering RNA contains a 19-bp scrambled sequence with 3′dT overhangs.
Upstream miRNA prediction
TargetScan 6.2, miRDB, and miRanda were used to calculate the potential target relationship between miR-9-5p and Fstl1. Subsequent expression regulation and luciferase reporter assay were further established to validate this prediction. All analyzed miRNAs are of mouse (Mus musculus) origin, and therefore, the prefix “mmu” was omitted throughout the text.
Dual-luciferase reporter assay
Luciferase activity assay was performed in 293T cells using the dual-luciferase reporter assay system (Promega) according to the manufacturer's instructions. Briefly, wild-type (WT) or mutant (MUT) 3′-UTRs of Fstl1 were subcloned into the pmiR-RB-REPORT™ vector (Ribobio, China) downstream of the luciferase gene. Fstl1-3′-UTR-WT or Fstl1-3′-UTR-MUT vectors were co-transfected with miR-9-5p mimics or negative control. Renilla luciferase activity (Ruc) was measured 48 h posttransfection and normalized to firefly luciferase activity (Luc).
Detection of intracellular ROS production
The production of ROS in H9c2 cells was fluorometrically monitored using the nonfluorescent probe, 2′,7′-dichlorofluorescein diacetate (DCFH-DA) (Yeasen, China). DCFH-DA passively diffuses into cells and is deacetylated, changing into the fluorescent compound, 2′,7′-dichlorofluorescein (DCFH). DCFH reacts with ROS to form the fluorescent product, DCF, which is trapped inside cells. For detection of intracellular ROS, H9c2 cells were treated with DCFH-DA (10 μmol/L) at 37°C for 30 min. To evaluate tissue production of ROS (O2 − in particular), fresh and frozen myocardium sections were incubated with 10 μmol/L dihydroethidium (DHE; Beyotime, China) for 1 h. All images were monitored using an inverted fluorescence microscope (Olympus, Japan). DHE signals were calculated using Image J software and fluorescence intensity in experimental group was finally normalized to control group.
Measurement of lactate dehydrogenase and malonyldialdehyde levels
The release of lactate dehydrogenase (LDH) indicates loss of membrane integrity in damaged cells and was measured using a commercially available kit (Beyotime, China). Briefly, culture supernatant was collected for reaction and absorbance was measured at 490 and 650 nm on a multifunctional microplate reader (BIO-TEK). Malonyldialdehyde (MDA) levels were measured using a MDA detection kit (Beyotime, China) according to the manufacturers' protocol. 14 LDH release (OD) and MDA concentration (μmol/g protein) in experimental group were finally normalized to control group.
Primary culture of neonatal rat ventricular myocytes and Annexin V apoptotic assay
Primary neonatal rat ventricular myocytes (NRVMs) were isolated and cultured as previously described. 15 NRVMs were transfected with miR-9-5p mimics/inhibitor followed with 24 h of hypoxic treatment. Apoptosis was verified using Annexin V-PE and 7-aminoactinomycin D (7-AAD) double staining as previously described. 16 Early and late apoptotic cells were recognized as Annexin V+7-AAD− and Annexin V+7-AAD+ cells, respectively.
Terminal deoxynucleotidyl transferase dUTP nick-end labeling assay and examination of nuclear condensation by DAPI staining
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay was performed according to the manufacturer's protocol (Roche). Briefly, frozen sections were treated with proteinase K and incubated with terminal deoxynucleotidyl transferase and fluorescein (FITC)-labeled deoxyuridine triphosphate. TUNEL-positive cells were counted in randomly selected fields and expressed as percentage of total nuclei. Nuclear condensation of H9c2 cells was examined by 4′,6-diamidino-2-phenylindole (DAPI) staining as described previously. 5 Briefly, fixed cells were stained with DAPI (Beyotime, China) and examined under a fluorescence microscope (Olympus, Japan). Images were recorded using Image J software and condensed nuclei were expressed as percentage of total nuclei.
Detection of cellular adenosine triphoshate levels
Cellular amount of adenosine triphosphate (ATP) was measured using a firefly luciferase-based ATP assay kit (Beyotime, China) according to the manufacturer's instructions. Briefly, cells were lysed and centrifuged at 12,000 g. The supernatant was then mixed with ATP detection solution for subsequent reaction. Luminance was measured using a multifunctional microplate reader (BIO-TEK).
Animal experiments
Adult male C57BL/6 mice (8–10 weeks old) were obtained from the Experimental Animal Center of the Chinese Academy of Medicine Sciences of Soochow University. All animal protocols were approved based on the local ethics legislation with respect to animal experimentation. Food and water were freely available throughout the experiments.
MI mice model was established as previously described. 17 Briefly, adult mice were given general anesthesia and a rodent ventilator was used to supply oxygen during the surgical procedure. Chests were opened by a horizontal incision between the fourth and fifth intercostal space. MI was achieved through permanent ligation of the left descending coronary artery. After surgery, the thoracic wall was carefully closed. Sham operated animals underwent an identical surgical operation without ligation of the coronary artery.
miRNA antagomir/agomir delivery
miR-9-5p antagomir/agomir (GenePharma, China), the chemically modified mimics/inhibitors with higher stability and affinity for cell membrane, was used for animal studies. For in vivo inhibition or activation of miR-9-5p, antagomir (3 mg/kg BW) or agomir (1 mg/kg BW) mixed with Dharmafect Duo (Thermo) were intramyocardially injected into the border zone region through an insulin syringe with a 29-gauge needle (BD) as described previously. 18 Antagomir NC and agomir NC served as negative controls.
Echocardiography
Echocardiography was performed in lightly anesthetized mice using Vevo 2100 system (VisualSonics, Canada) with an 80 MHz probe as described previously. 18 The following parameters were measured from M-mode images taken from the parasternal short-axis view: ejection fraction (EF), fractional shortening (FS), left ventricular internal diameter in diastole, and left ventricular posterior wall thickness in diastole.
Histology and immunofluorescence staining
Fibrosis was analyzed using a Masson's Trichrome stain kit (Sigma) and quantified with Image J software as described previously. 19 The percentage of infarct size was calculated as (fibrosis area/total LV area) × 100%. The antibodies specific for CD68, tumor necrosis factor alpha (Abcam), and CD206 (Proteintech) were used for immunoflurescence staining as described previously. 20 The nucleus was labeled with DAPI. Fluorescence microscopy images were obtained using an inverted fluorescence microscope (Olympus, Japan) equipped with a digital camera.
Reverse transcription PCR and quantitative real-time PCR
Total RNA was extracted and quantified using ND2000 spectrophotometer (NanoDrop Technologies, USA) as described previously. 21 Quantitative real-time PCR was carried out using Step One-Plus Real-Time PCR System (Applied Biosystems). miRNA and mRNA expression were normalized to U6 and GAPDH, respectively. Primer sequences are provided in Table 1.
Primers used for quantitative real-time PCR
Western blot analysis
Protein lysates were processed for western blot following standard protocol. 22 Primary antibodies against Fstl1 and GAPDH (Santa Cruz) were used. Immunoreactivity was detected by routine enzymatic chemiluminescence.
Enzyme-linked immunosorbent assay
Serum Fstl1 levels were measured using standard quantitative sandwich enzyme-linked immunosorbent assay (R&D) as described previously. 23 Optical densities were determined using a multifunctional microplate reader (BIO-TEK) at 450 nm.
Statistical analysis
Statistical analysis was performed using Graphpad Prism 5 software. The p values were based on the two-tailed Student's t-test, and p < 0.05 was considered significant.
Results
Fstl1 is a putative target of miR-9-5p
miRNAs regulate gene expression by posttranscriptional gene silencing. 24 We and others have previously shown that Fstl1 resists cardiac cell death challenged by a wide range of injuries. 5 –7,9 Nevertheless, the upstream Fstl1-targeting miRNA(s) remain largely unknown. We believe that antagonizing such Fstl1-suppressing miRNA(s) may further assist post-MI recovery. To screen Fstl1-targeting candidates, we set the following standards for bioinformatics prediction: (1) targets 3′UTR of Fstl1; (2) evolutionarily conserved between mouse, rat, and human; and (3) must be predicted by TargetScan, miRanda, and miRDB together. Intriguingly, miR-9-5p is predicted as the only Fstl1-targeting miRNA whose targeting site is evolutionarily conserved (Fig. 1A). Subsequently, a time-course study was conducted at post-MI days 0, 3, 7, and 14 to quantify dynamic expression of Fstl1 and miR-9-5p in ischemic border zone of murine hearts. Consistent with previous reporting, 6 mRNA level of Fstl1 reaches its peak with a 9.84-fold increase as early as post-MI day 3, and remains at similar levels afterwards (Fig. 1B). It is worthy of note that miR-9-5p expression declines to 69.08% on post-MI day 3 and to minimum (10.55%) on post-MI day 7, suggesting reverse correlation with that of Fstl1 (Fig. 1C). Next, we constructed a dual-luciferase reporter vector to perform luciferase reporter assay (Fig. 1D). As shown in Fig. 1E, miR-9-5p remarkably reduces luciferase activity of wild-type Fstl1 3′-UTR (p < 0.001), whereas that of mutant Fstl1 3′-UTR remains unchanged, suggesting direct binding of miR-9-5p to 3′-UTR of Fstl1. Besides, enforced expression of miR-9-5p in H9c2 cells (Fig. 1F) is concurrent with the diminished expression (Fig. 1G) and secretion (Fig. 1H) of Fstl1 and vice versa (Fig. 1I–K). Collectively, miR-9-5p is an Fstl1-targeting miRNA that negatively modulates its expression.
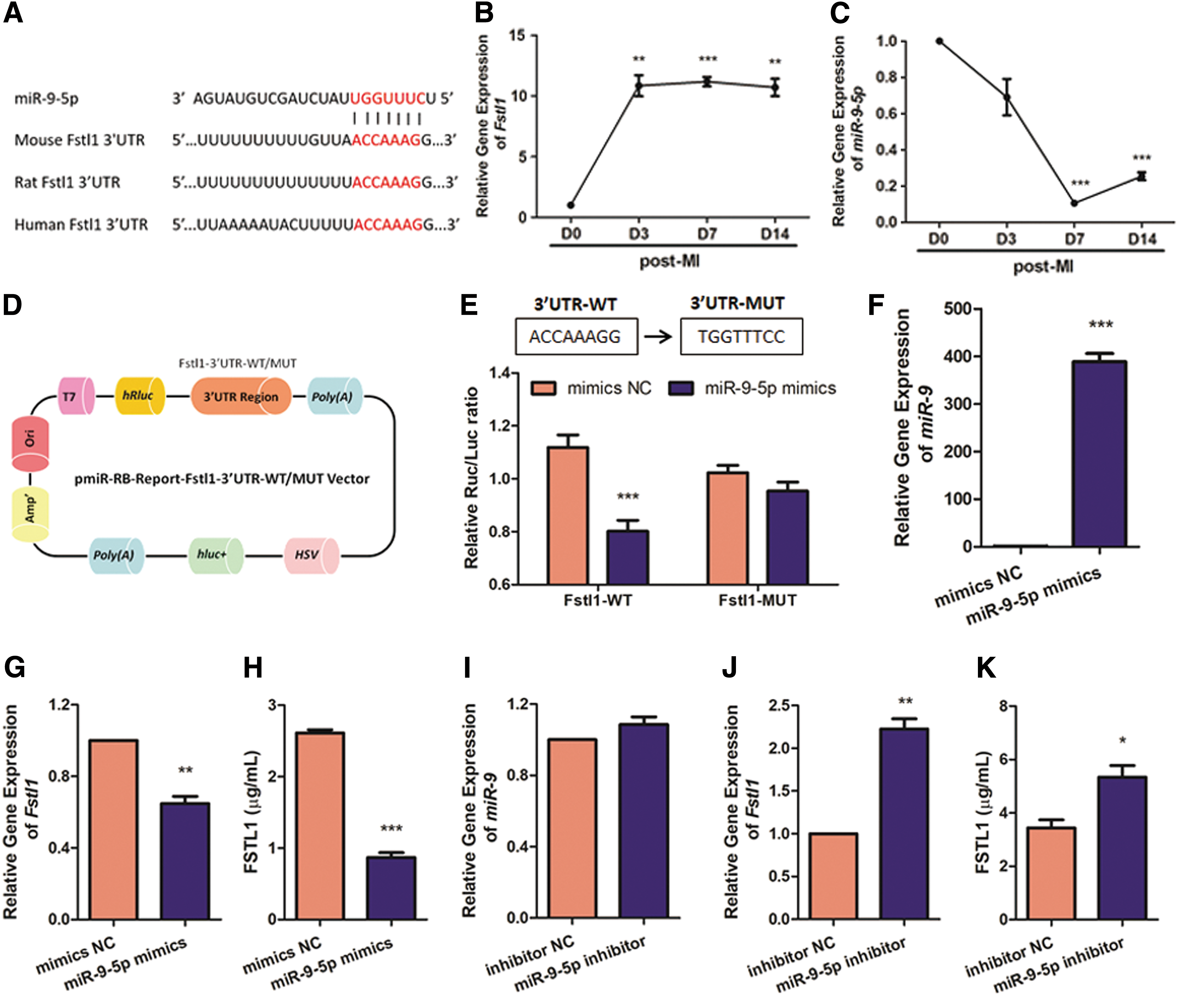
Fstl1 is a putative target of miR-9-5p.
Enforced expression of miR-9-5p aggravates cellular injury after hypoxic treatment
To directly investigate functional role of miR-9-5p in vitro, cardiomyoblast-derived H9c2 cells were transfected with miR-9-5p mimics before subsequent hypoxic treatment. Nuclear condensation, a widely used marker for apoptotic cells, 5,25 were identified by condensed and coalesced nuclei with a brighter blue fluorescence of DAPI staining. Notably, transfection of miR-9-5p mimics further elevates hypoxia-induced nuclear condensation by 30.58% (Fig. 2A and B). We also isolated neonatal rat ventricular myocytes (NRVMs) and evaluated their apoptosis with Annexin V/7–-AAD staining. As illustrated in Fig. 2C and D, both early and late apoptosis in NRVMs are obviously triggered by hypoxic treatment, and this is further exacerbated by miR-9-5p overexpression (+13.76% in early apoptosis and +78.52% in late apoptosis). Consistent with above observations, hypoxia-initiated LDH release, marker for cell injury, 26 is also enhanced notably by miR-9-5p mimics treatment (+26.22%, Fig. 2E). Next, intracellular ROS production was evaluated by DCF fluorescence intensity. As illustrated in Fig. 2F, hypoxic treatment contributes to excessive ROS accumulation and this is further exacerbated by transfection of miR-9-5p mimics, indicating pro-oxidative function of miR-9-5p. Similarly, hypoxic treatment elevates intracellular MDA content by 53.70% and this effect is further aggravated by excessive miR-9-5p (+21.37%, Fig. 2G). Finally, miR-9-5p mimics also exacerbates reduction trend of ATP production following hypoxia, indicating compromised mitochondrial function (−13.10%, Fig. 2H).
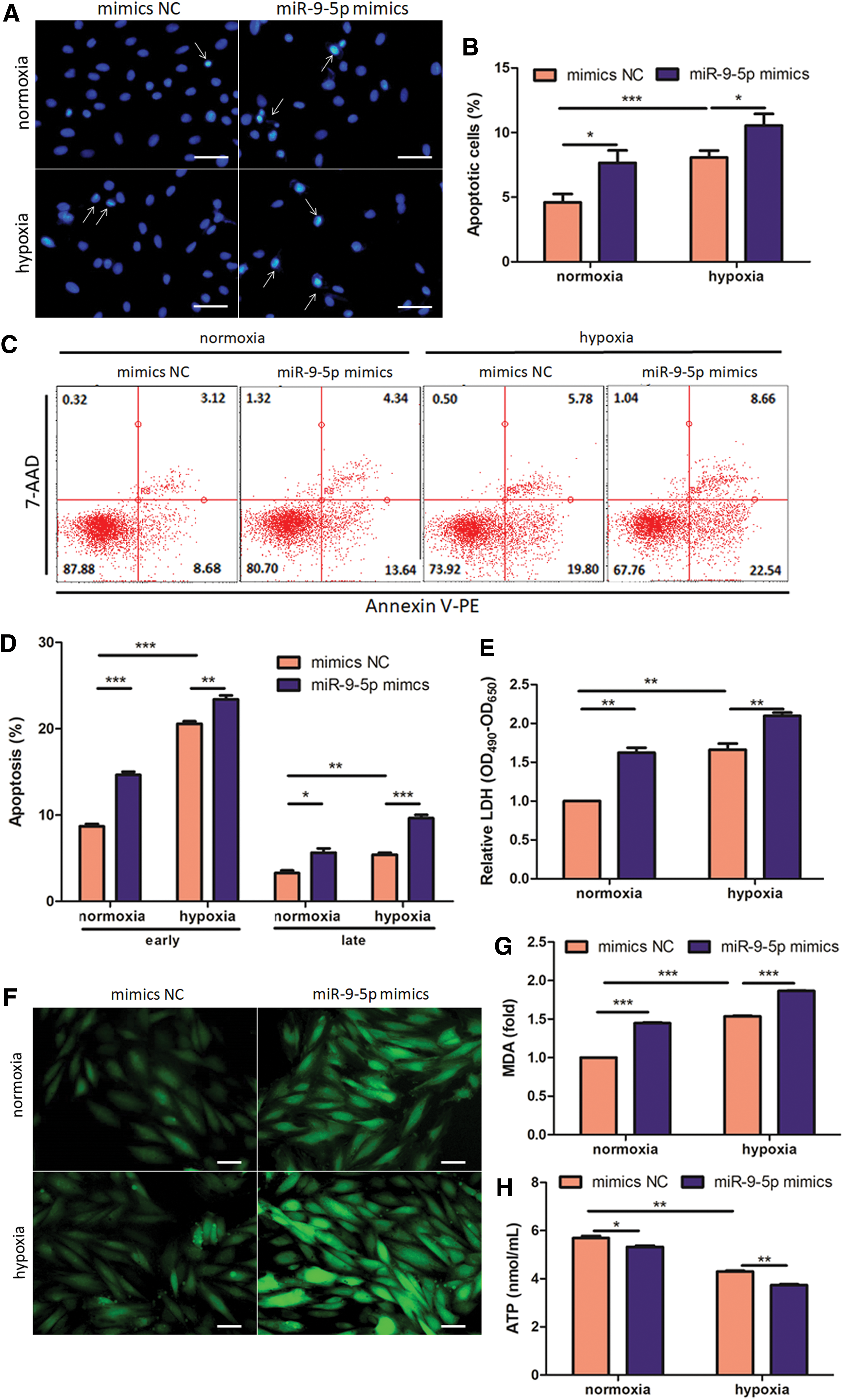
miR-9-5p aggravates hypoxia-induced cardiac injury. H9c2 cells were transfected with miR-9-5p mimics and exposed to hypoxia for 48 h.
Local inhibition of miR-9-5p preserves post-MI cardiac function in adult mice
In order to neutralize endogenous miR-9-5p, the engineered oligonucleotides termed “antagomir” were designed to form complementary base pairs with it and effectively inactivate its function. miR-9-5p antagomir was directly delivered through myocardial injection to study its potential role in antagonizing post-MI cardiac remodeling. As expected, miR-9-5p antagomir effectively depletes its own expression by 63.63% (Fig. 3A) and elevates both mRNA (Fig. 3B) and protein level (Fig. 3C) of myocardial Fstl1 accordingly. Consistently, an increase in serum Fstl1 is also evidenced by ELISA analysis (+110%, Fig. 3D). One week after myocardial infarction, echocardiography was demonstrated to determine cardiac function and volume. As illustrated in Fig. 4A–E, MI-induced reduction in EF (Fig. 4B) and FS (Fig. 4C) is partially reversed by miR-9-5p antagomir (+114% in EF and +128% in FS). Intriguingly, despite reduced LV function, myocardial volume (LV internal diameter in diastole; LV internal diameter in diastole; Fig. 4D) and wall thickness (LV posterior wall thickness in diastole; LV posterior wall thickness in diastole; Fig. 4E) remain unchanged following MI challenge. Consistently, miR-9-5p antagomir treatment also attenuates post-MI cardiac expression of atrial natriuretic factor (ANF) and brain natriuretic peptide (BNP), two remodeling markers, by 29.68% and 67.41%, respectively (Fig. 4F). Importantly, improved post-MI EF by antagomir treatment can be monitored for as long as 4 weeks (Fig. 4G). Conversely, myocardial injection of miR-9-5p agomir, the chemically modified mimics, markedly elevates its own level (Supplementary Fig. S1A), and, consequently, antagonizes Fstl1 level (Supplementary Fig. S1B). In accordance with the above loss-of-function data, miR-9-5p agomir dramatically aggravates post-MI cardiac dysfunction (Supplementary Fig. S1C–G). Collectively, myocardial injection of miR-9-5p antagomir stabilizes Fstl1 expression and impressively maintains post-MI cardiac function in a murine model.
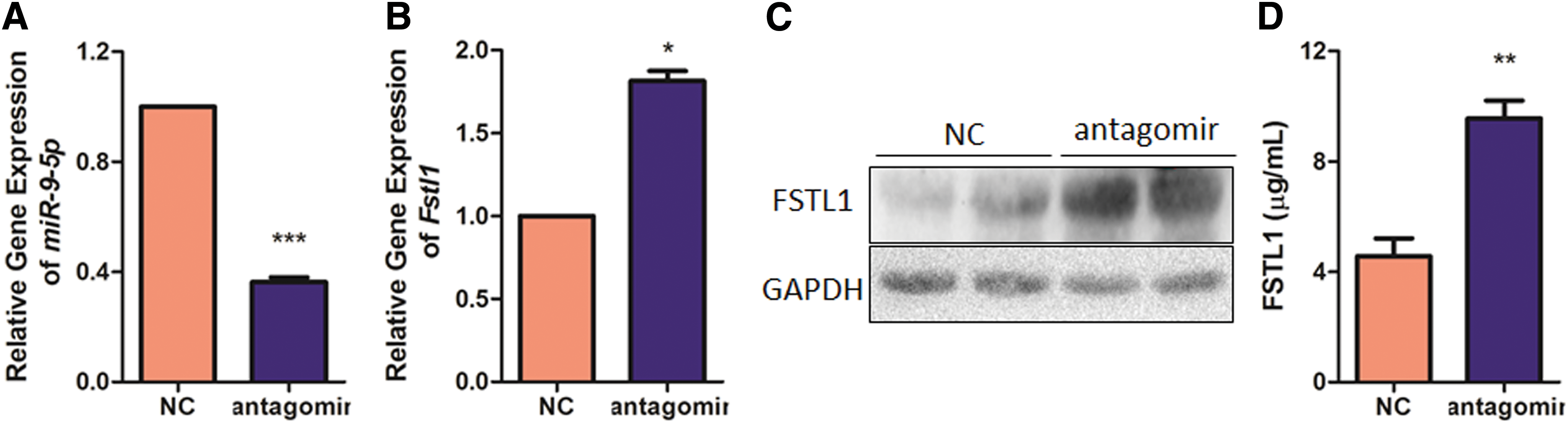
Myocardial silencing of miR-9-5p by local injection of its antagomir in hearts. Murine myocardium proximal to the injection site was collected 7 days after myocardial delivery of miR-9-5p antagomir. Expression of miR-9-5p
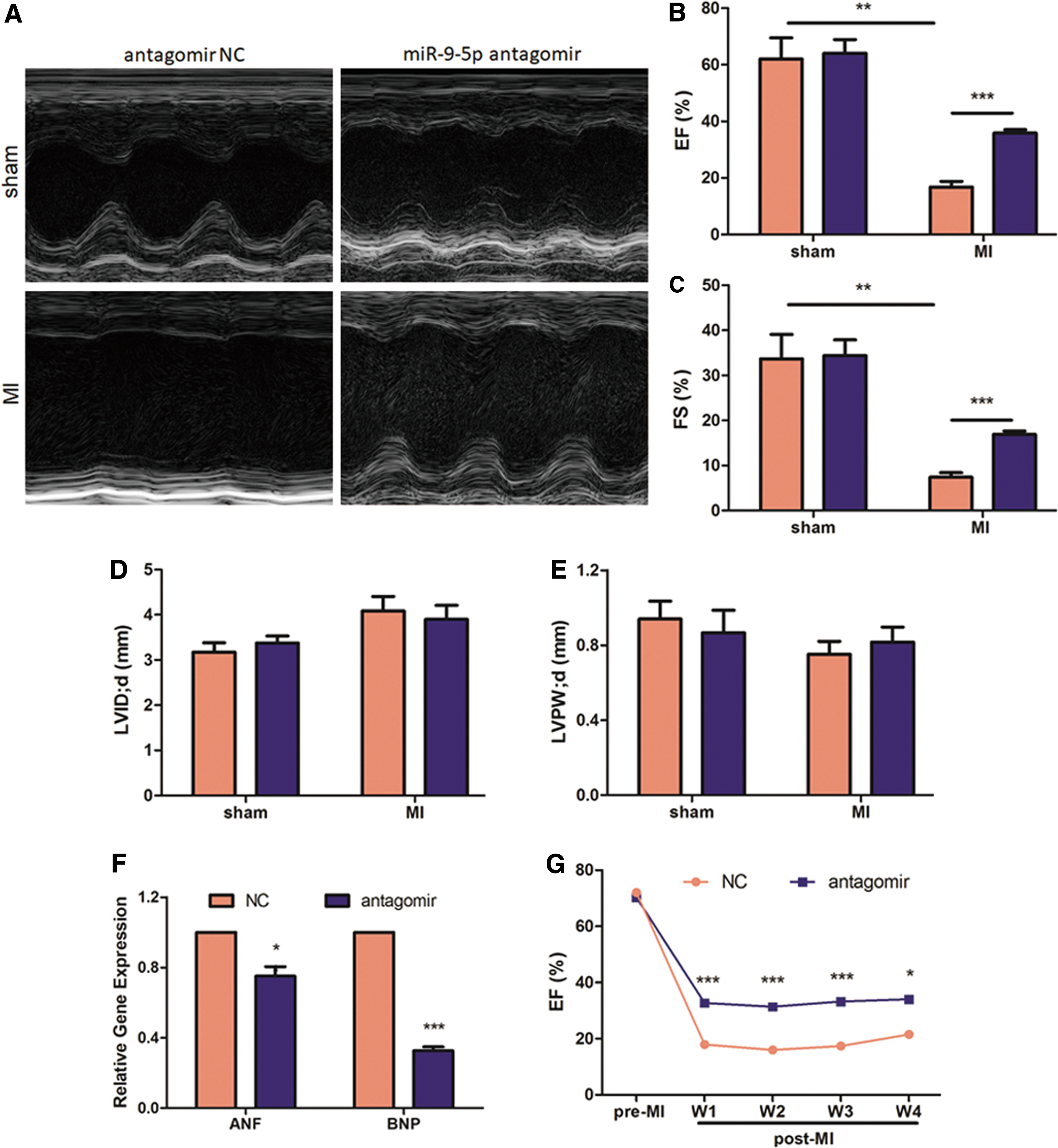
Myocardial inhibition of miR-9-5p alleviates post-MI cardiac dysfunction.
Antagomir-induced blockade of miR-9-5p attenuates cell death in ischemic myocardium
Loss of cardiomyocytes is a hallmark in the early phase of myocardial infarction. As shown in Fig. 5A and B, percentage of TUNEL-positive cells in the two sham groups is similar and very low. Nonetheless, MI triggers cell apoptosis in the ischemic border zone and miR-9-5p neutralization diminishes apoptotic ratio by 78.22% (Fig. 5B). Reversely, miR-9-5p agomir treatment exacerbates percentage of TUNEL-positive cells by 41.35% on post-MI 7 d (Supplementary Fig. S2). To further evaluate the pro-survival role of miR-9-5p inhibition, we transfected H9c2 cells with miR-9-5p inhibitor and subjected them to hypoxic treatment for 48 h in vitro. Notably, inhibition of miR-9-5p ameliorates hypoxia-induced cell death in cardiomyoblasts, as illustrated by a 31.04% reduction in nuclear condensation (Fig. 5C and D). Additionally, Annexin V/7-AAD staining on NRVMs further confirms the above findings (Fig. 5E). Consistently, hypoxia-initiated LDH release is also attenuated by 21.45% after miR-9-5p inhibitor treatment (Fig. 5F). Taken together, these data suggest that inhibition of miR-9-5p attenuates cell death both in ischemic myocardial injury in vivo, and in hypoxic cardiomyoblasts in vitro.
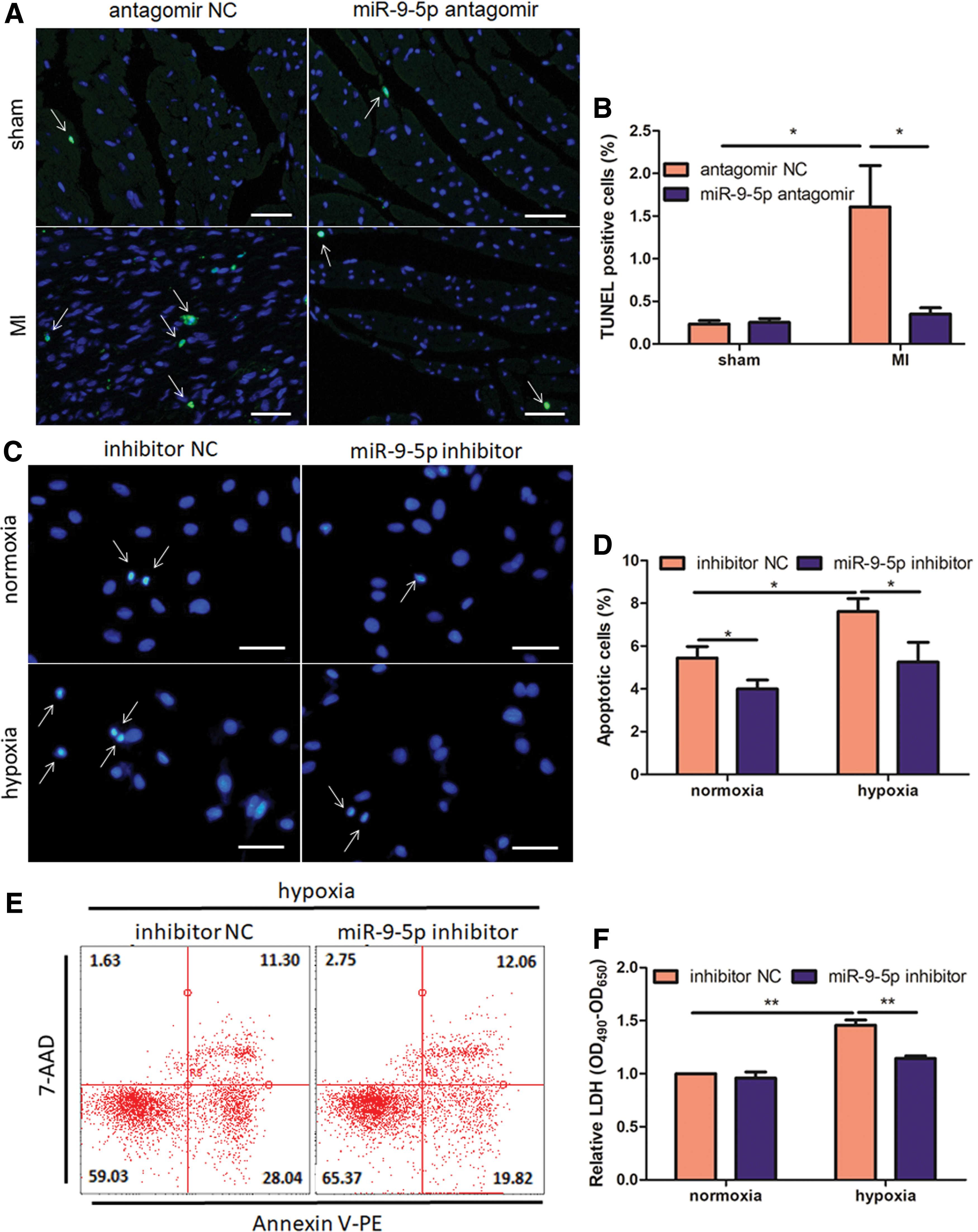
Inhibition of miR-9-5p protects against cardiac cell death in vivo and in vitro.
Pharmacological knockdown of miR-9-5p ameliorates excessive myocardial oxidative stress
ROS overproduction is a critical feature of infarcted myocardium and contributes to subsequent cardiac injury. 27 We therefore quantified myocardial O2 − content using DHE staining in infracted hearts. Consistent with previous report, 28 MI leads to extensive ROS generation in peri-infarct zones (Fig. 6A and B). Importantly, knockdown of miR-9-5p significantly abrogates MI-induced increase in O2 – production (p < 0.05; Fig. 6A and B), indicating a favorable microenvironment for cardiac recovery. We further determined cellular ROS and ATP levels in hypoxic H9c2 cells. As indicated by DCF fluorescence intensity, inhibition of miR-9-5p also reverses hypoxia-induced ROS over-accumulation in vitro (Fig. 6C). Finally, hypoxic ATP production is also partially maintained by transfection of miR-9-5p inhibitor (Fig. 6D). Collectively, these data indicate abrogated ROS generation in both ischemia-challenged hearts and hypoxia-treated cardiomyoblasts.
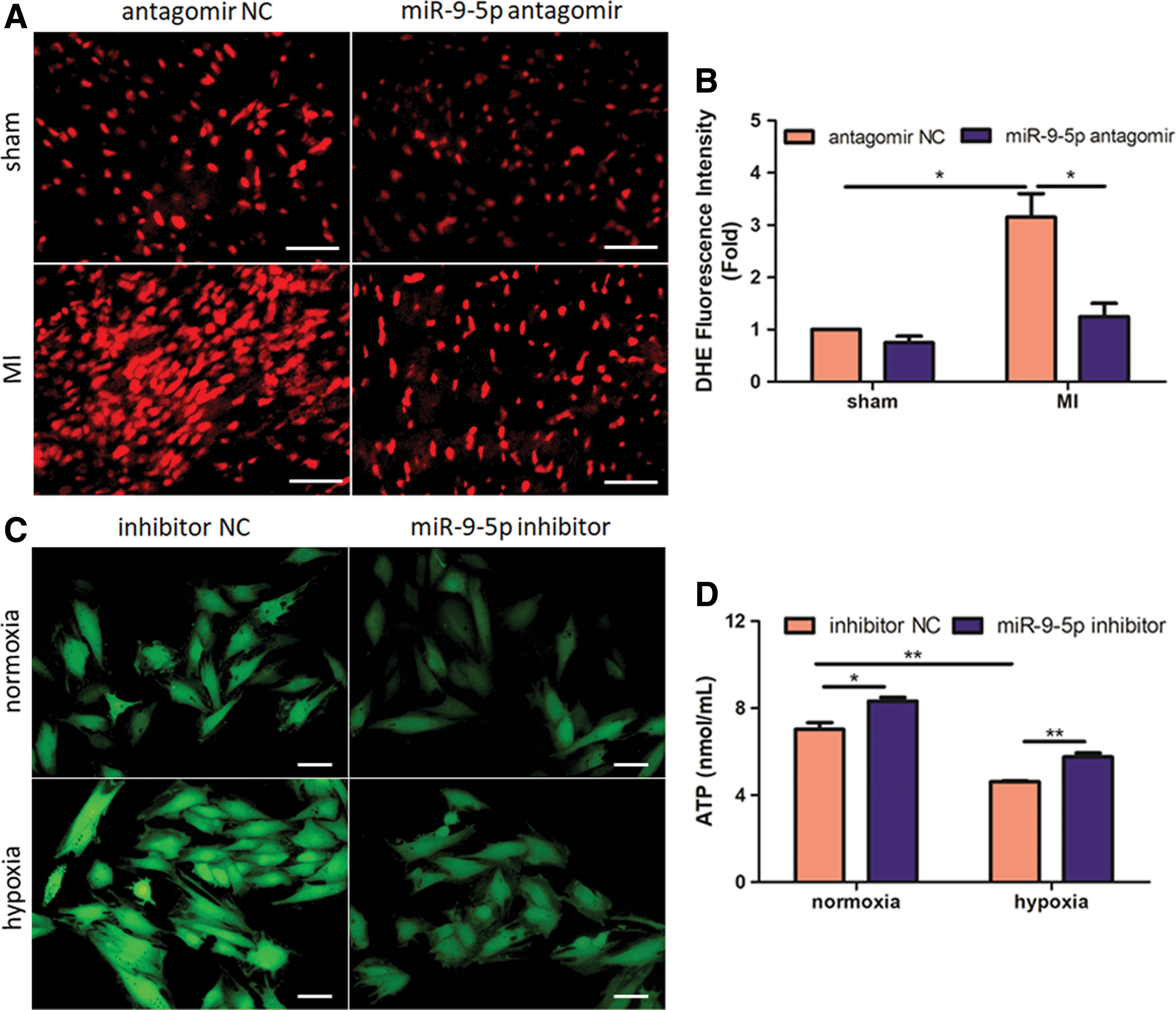
Inhibition of miR-9-5p leads to attenuation of myocardial oxidative stress in vivo and in vitro.
Myocardial fibrosis is ameliorated in miR-9-5p-antagomir-treated hearts
We next evaluated whether the preserved post-MI cardiac function by miR-9-5p antagomir treatment is somehow associated with alleviated fibrosis and scar formation. Masson's trichrome staining was conducted to reveal extracellular matrix deposition in the interstitial regions of peri-infarct zones. Notably, miR-9-5p suppression diminishes both infarct area (−48.66%; Fig. 7A and B) and interstitial fibrosis (Fig. 7C). Moreover, attenuated fibrosis by miR-9-5p antagomir treatment was also confirmed by decreased mRNA for type 1 collagen (−67.14%, Fig. 7D) and fibronectin (−64.59%, Fig. 7E). Importantly, attenuated scar formation by antagomir treatment was also validated on post-MI 4 w (Fig. 7F and G). Consistently, myocardial injection of miR-9-5p agomir abrogates post-MI scar formation by 104% (Supplementary Fig. S3A and B). In summary, we observed impaired fibrosis and collagen deposition in miR-9-5p-antagomir-treated hearts.
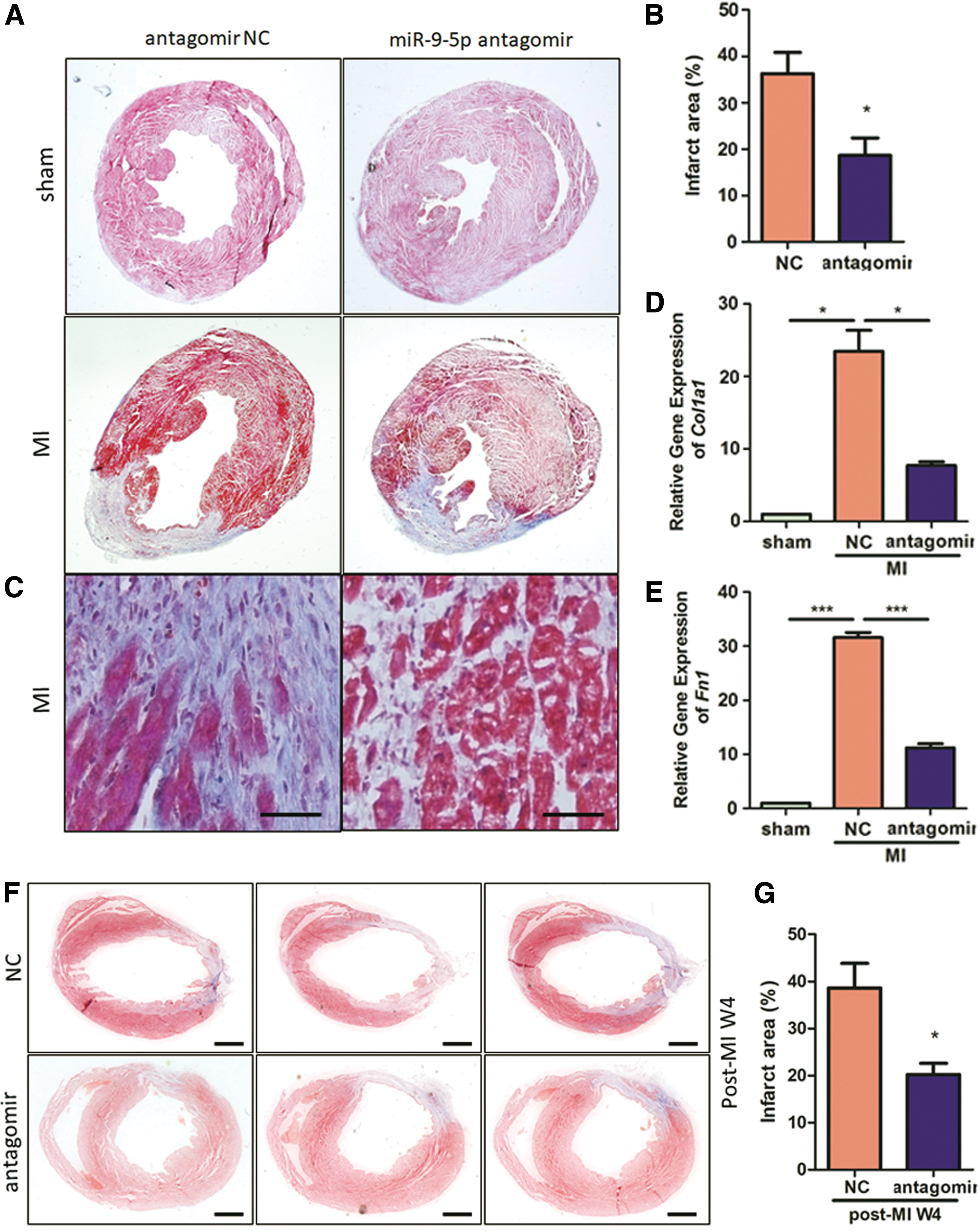
Myocardial fibrosis is ameliorated in miR-9-5p-antagomir-treated hearts. Macroscopic view
Attenuated inflammatory response in miR-9-5p-antagomir-treated hearts
As a central cellular protagonist in innate immunity, excessive inflammation triggered by macrophages can exacerbate post-MI cardiac dysfunction, 29,30 thus heart sections were stained with anti-CD68 antibody to identify infiltrated macrophages in peri-infarct myocardium. Importantly, we noted a reduction in CD68 positive macrophage infiltration into antagomir-treated peri-infarct zones (Fig. 8A). Following MI, classic M1 macrophages dominate in peri-infarct area and produce pro-inflammatory cytokines in the context of innate immune response. 31 –33 Accordingly, we monitored these M1 macrophages and quantified cardiac production of pro-inflammatory interleukin-6 (IL-6) and interleukin-1β (IL-1β). As illustrated in Fig. 8B and C, we observed impaired tumor necrosis factor alpha– positive M1 macrophage infiltration as well as decreased cardiac IL-6 (−45.29%, p < 0.05) and IL-1β levels (−73.47%, p < 0.001). M2 macrophages, which produce anti-inflammatory cytokines, are associated with responses to anti-inflammatory reactions and tissue remodeling. 31 –33 As illustrated in Fig. 8D and E, enhanced CD206 positive M2 macrophage infiltration as well as significantly elevated cardiac interleukin-10 (IL-10) levels (+287.37%, P < 0.05) were observed in antagomir-treated hearts, indicating an infarct environment favorable for tissue repair. In summary, we observed blocked macrophage infiltration and inflammatory response in miR-9-5p-antagomir-treated MI hearts.

Knockdown of miR-9-5p blocks inflammation in peri-infarct myocardium. Representative images of CD68-positive macrophage
miR-9-5p inhibitor protects against hypoxic challenge through Fstl1
To further determine whether Fstl1 is responsible for cytoprotection by miR-9-5p inhibition, we treated H9c2 cells with miR-9-5p inhibitor and/or siFstl1 and subjected them to subsequent hypoxic treatment. As illustrated in Fig. 9A, Fstl1 mRNA is declined by 65.67% after transfection of siFstl1 + miR-9-5p antagomir, compared with that transfected with siNC + miR-9-5p antagomir. Next, we observed that Fstl1 knockdown with siFstl1 deprives the cytoprotective role of miR-9-5p inhibitor against hypoxia-induced apoptosis and ROS over-accumulation. siFstl1 administration increases nuclear condensation in hypoxia + miR-9-5p inhibitor group by 176% compared with that treated with hypoxia + miR-9-5p inhibitor alone (p < 0.01; Fig. 9B and C). Besides, ROS generation after siFstl1 treatment exhibits a similar trend with cell death results (Fig. 9D). Taken together, our data demonstrate a novel role of miR-9-5p/Fstl1 axis in cellular defense against hypoxic injury, consistent with our previous bioinformatics prediction.
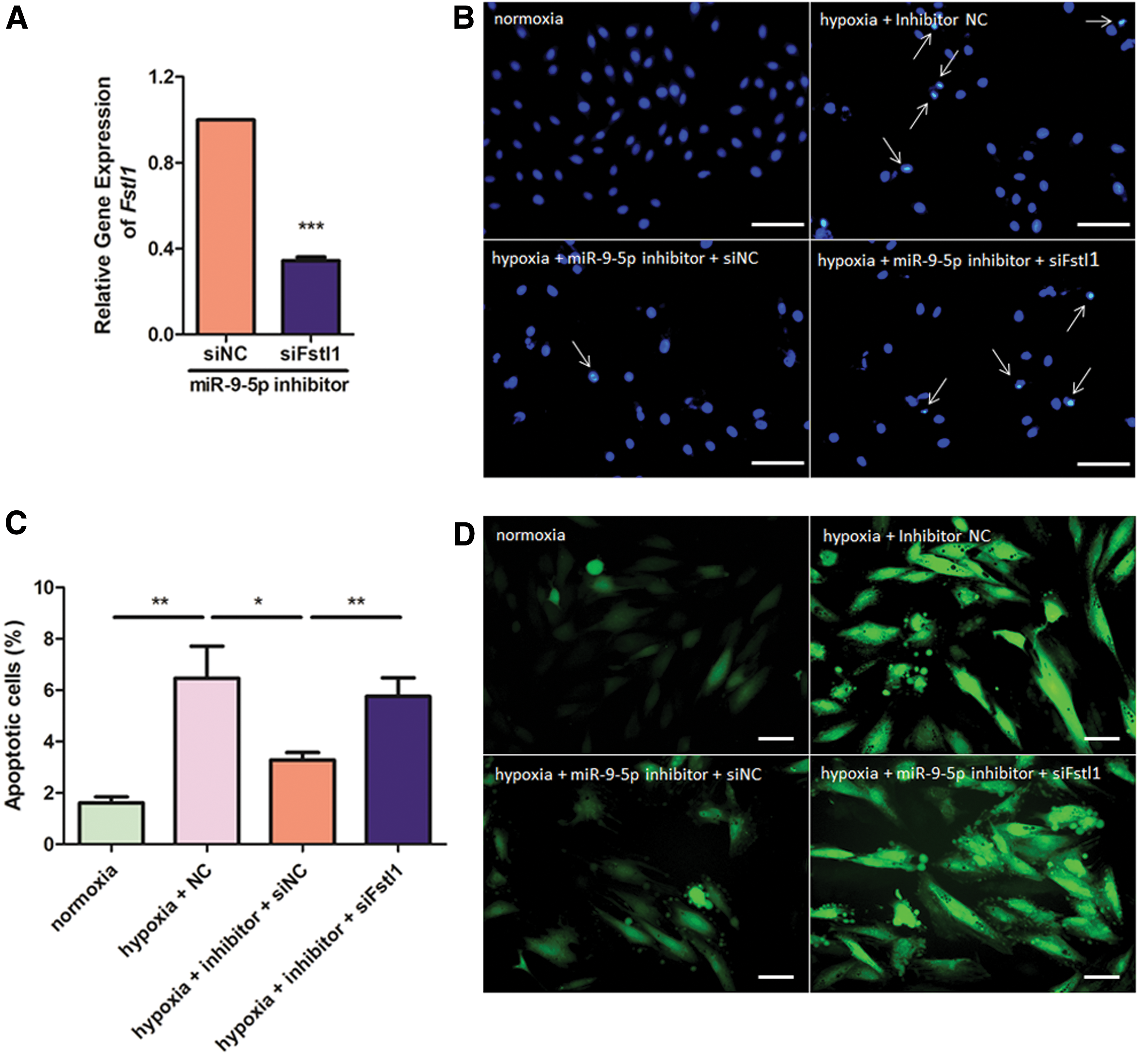
miR-9-5p inhibitor exerts its cytoprotective role through Fstl1. H9c2 cells were transfected with miR-9-5p inhibitor and/or siFstl1 and exposed to hypoxic treatment for 48 h.
Discussion
In our current study, miR-9-5p is identified as an upstream miRNA that directly targets the cardiokine Fstl1. Overexpression of miR-9-5p aggravates cell death, LDH release, and ROS overaccumulation in hypoxic cardiomyoblasts. We therefore neutralized myocardial miR-9-5p to identify a potential strategy modulating post-MI cardiac remodeling. As expected, miR-9-5p antagomir abrogates its own level and stabilizes Fstl1 expression. Furthermore, pharmacological inhibition of miR-9-5p preserves post-MI cardiac function by restraining cell death and oxidation in ischemic myocardium. Besides, diminished fibrosis and inflammation were also observed in antagomir-treated hearts. Finally, the cytoprotective role of miR-9-5p inhibitor against hypoxic challenge was proved to be mediated through Fstl1. These results demonstrated the cardio-protective role of miR-9-5p antagomir against MI, supporting its utility for acute repair of infarcted hearts.
Recent evidences have highlighted the significance of controlling redox status in treatment of MI. 34 Oxidative stress in ischemic heart alters the balance of production and elimination of intracellular oxygen free radicals and contributes to overaccumulation of ROS, which is an essential player in inducing loss of cardiomyocytes. 35 Similarly, our study reveals the anti-oxidative role of miR-9-5p antagomir/inhibitor in both ischemia-challenged hearts and hypoxia-treated cardiomyoblasts, further emphasizing the importance of suppressing oxidative stress for protection against infarct injury. Consistent with our findings, improving ischemic microenvironment by suppressing excessive oxidative stress has also been reported to effectively promotes therapeutic efficacy of transplanted stem cells in ischemic injury. 35
Our present results validated miR-9-5p as an Fstl1-targeting miRNA which negatively inhibits its local expression. As a cardiokine, circulating and/or myocardial Fstl1 levels are elevated in patients with acute coronary syndrome 36,37 and chronic systolic heart failure. 38 Recently, the significance of Fstl1 in cardiac remodeling has been increasingly recognized. It was reported to suppress cardiac dysfunction in both murine 6 and porcine models of ischemia/reperfusion 7 through its anti-apoptotic and anti-inflammatory actions. Besides, restoration of its nonglycosylated epicardium-producing isoform enhances proliferation of immature cardiomyocytes and reverses post-MI remodeling. 8 Furthermore, mice with cardiac-specific Fstl1 knockout (cFstl1-KO) develop cardiac hypertrophy and ventricular dysfunction in response to transverse aortic constriction. 9
Fstl1 mRNA reaches its peak as early as post-MI day 3 and remains at similar levels afterward (Fig. 1B), whereas miR-9-5p expression declines to 69.08% on post-MI day 3 and to minimum (10.55%) on post-MI day 7 (Fig. 1C), suggesting, except miR-9-5p, other Fstl1-targeting molecules may exit in the heart. For example, miR-27a inhibits migration and invasion of fibroblast-like synoviocytes by tmiargeting Fstl1 in rheumatoid arthritis. 39 Besides, miR-32-5p targets Fstl1 and modulates host defense against mycobacterial infection. 40 Actually, we also analyzed expression of both has-miR-27a and has-miR-32-5p in a published gene-profiling dataset of MI patients (GSE76591), 41 and cardiac expression of these two miRNAs remains unchanged compared to control subjects (data not shown). From this point of view, miR-27a and miR-32-5p seem to be uninvolved, at least, in post-MI cardiac remodeling. Finally, Fstl1-targeting molecules include but not restrict to miRNA. Further efforts are still needed to identify these molecules involved in cardiac remodeling process.
Till now, many additional miR-9-5p targets have also been validated. It is therefore possible that other genes may be dysregulated by antagonizing intrinsic miR-9-5p. To the best of our knowledge, miR-9-5p targets platelet derived growth factor receptor-β (PDGFR-β), and, consequently, suppresses paracrine angiogenic capacity of cardiomyocytes. 42 Besides, miR-9-5p inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAV-like protein 1 (ELAVL1). 43 Actually, we have examined dynamic expression of these two genes on post-MI days 0, 3, 7, and 14. Expression of PDGFR-β is slightly elevated to about two-folds after MI (Supplementary Fig. S4A). It is therefore promising that PDGFR-β level may be elevated by silencing miR-9-5p, which is beneficial for cardiac recovery through paracrine angiogenic capacity of cardiomyocytes. On the other hand, post-MI expression of ELAVL1, the hyperglycemia-induced pyroptosis promotor, remains unchanged (Supplementary Fig. S4B), suggesting that miR-9-5p regulate post-MI cardiac remodeling via a mechanism that is distinct from ELAVL1.
In our present study, antagonizing miR-9-5p in ischemic myocardium ameliorates post-MI fibrosis and scar formation. This seems to be contradicting previous reports, as miR-9-5p has been illustrated as an antifibrotic miRNA and Fstl1 as a profibrotic factor. For example, miR-9-5p abrogates hepatic fibrosis in CCl4-induced liver fibrotic mice 44 as well as inhibits neonatal cardiac fibroblasts proliferation and collagen production. 45 Besides, inhibition of Fstl1 impairs bleomycin-induced pulmonary fibrosis by blocking activation of fibroblasts to myofibroblasts. 46 Regarding our findings, we propose that both the attenuated ischemic cell death and less oxidative microenvironment in treated hearts restrains activation and migration of myofibroblasts, leading to diminished scar formation.
miRNA-based therapies are advancing in a rapid speed, with successful completion of phase 1 and phase 2 clinical trials of Santaris Pharma's miravirsen, targeting miR-122 for hepatitis C virus infection treatment. 47 It is therefore promising to develop miRNA-based therapies directing cardiovascular diseases. The major finding of our present study indicates that miR-9-5p exacerbates hypoxic injury in vitro and pharmacological inhibition of miR-9-5p preserves post-MI cardiac function. Previous studies in inflammation revealed a novel role of miR-9-5p in lipopolysaccharide-mediated release of proinflammatory cytokines 48 and in cancer metastasis. 49 However, the function of miR-9-5p in cardiovascular injuries seems to be divergent and controversial depending on different experimental models. In contrast to our results, some studies have identified miR-9-5p as an antihypertrophic miRNA which inhibits cardiac hypertrophy by targeting myocardin. 50 In addition, miR-9-5p attenuates hyperglycemia-induced cardiac pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. 43 It is therefore highly needed to further clarify the specified function of miR-9-5p in cardiac remodeling over a broad spectrum of cardiac injuries.
In conclusion, we validated miR-9-5p as an Fstl1-suppressing miRNA. miR-9-5p mimics significantly aggravates hypoxia-induced injuries to H9c2 cells. Furthermore, myocardial silencing of miR-9-5p is able to attenuate post-MI cardiac remodeling and maintain heart function. Our data support the promise of miR-9-5p antagomir as a novel strategy for treatment of acute MI and shed new light on the understanding of cardiac remodeling machinery regulated by miRNAs.
Footnotes
Acknowledgments
This work was supported by Jiangsu Province's Key Discipline / Laboratory of Medicine (XK201118), National Natural Science Foundation of China (NSFC-81770258), National Key R&D Program of China (2017YFA0103700), Science and Technology Project of Suzhou (SYS201705), National Natural Science Foundation of China (No. 31500944), Natural Science Foundation of Jiangsu Province (BK20150687 and BK20160321), and Taishan Scholar Project of Shandong Province of China (tsqn20161066 to Wencheng Zhang).
Author Disclosure
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.