
Abstract
Hereditary retinal dystrophy is clinically defined as a broad group of chronic and progressive disorders that affect visual function by causing photoreceptor degeneration. Previously, we identified mutations in the gene encoding receptor expression-enhancing protein 6 (REEP6), in individuals with autosomal recessive retinitis pigmentosa (RP), the most common form of inherited retinal dystrophy. One individual was molecularly diagnosed with biallelic REEP6 mutations, a missense mutation over a frameshift mutation. In this study, we generated Reep6 compound heterozygous mice, Reep6L135P/− , which mimic the patient genotype and recapitulate the early-onset retinal degeneration phenotypes observed in the individual with RP. To determine the feasibility of rescuing the Reep6 mutant phenotype via gene replacement therapy, we delivered Reep6.1, the mouse retina-specific isoform of REEP6, to photoreceptors of Reep6 mutant mice on postnatal day 20. Evaluation of the therapeutic effects 2 months posttreatment showed improvements in the photoresponse as well as preservation of photoreceptor cells. Importantly, guanylyl cyclase 1 (GC1) expression was also restored to the outer segment after treatment. Furthermore, rAAV8-Reep6.1 single treatment in Reep6 mutant mice 1 year postinjection showed significant improvements in retinal function and morphology, suggesting that the treatment is effective even after a prolonged period. Findings from this study show that gene replacement therapy in the retina with rAAV overexpressing Reep6 is effective, preserving photoreceptor function in Reep6 mutant mice. These findings provide evidence that rAAV8-based gene therapy can prolong survival of photoreceptors in vivo and can be potentially used as a therapeutic modality for treatment of patients with RP.
Introduction
Retinitis pigmentosa (RP) is a heterogeneous genetic disease of the eye, which is characterized by retinal degeneration that results in irreversible loss of light-sensing neurons in the retina, called photoreceptors. In RP, the rod photoreceptors, which play essential roles in providing dark and peripheral vision, are affected first, followed by progressive degeneration of the cones, which provide color and fine-detailed vision. 1,2 Ultimately, the progressive nature of the disease leads to loss of visual acuity and can lead to complete blindness. 1 –5 Over the past decades, pathogenic variants in more than 85 genes have been identified that can be transmitted through different modes of inheritance, and at least 60 genes have been identified to be associated with autosomal recessive RP (arRP). 6,7 However, mutations in these genes can explain the genetic basis of approximately 60% of RP cases, leaving 40% of the patients without a molecular diagnosis. 4 Thus, in efforts to identify pathogenic variants underlying retinal dystrophy, we performed a large-scale screen, using whole-genome and whole-exome sequencing, which led to the identification of mutations in a gene encoding receptor expression-enhancing protein 6 (REEP6). Specifically, we identified individuals with REEP6 variants in five unrelated families from diverse ethnicities that were clinically diagnosed with nonsyndromic RP. 4 Although the nature of the mutations identified varied, the affected individuals were diagnosed with nyctalopia, visual field constriction and peripheral retinal atrophy, severely impaired or diminished responses to light as measured by eletroretinograms, 8 marked reduction in the thickness of the overall retina and photoreceptor outer nuclear layer, and characteristic deposition of bone spicule-like pigments in their fundus. 4 Interestingly, studies have identified several additional RP families with REEP6 variants that also lead to retinal degeneration, 9,10 further exemplifying the significance of REEP6 in photoreceptor function and survival.
REEP6 belongs to the REEP family of proteins, which have previously been implicated in enhancing expression of cell surface receptors and in endoplasmic reticulum (ER) membrane shaping. 11 –16 REEPs are homologous to the yeast Yop1/Yip proteins, which have been shown to regulate intracellular trafficking and targeting of vesicle cargos, particularly G protein-coupled receptors (α2A and α2C adrenergic receptors), to the plasma membrane via interaction at the carboxyl termini. 15 In addition, they function as membrane-shaping adapter proteins and are described as ER morphogens. 13 When Reep6 was first cloned, it was referred to as deleted in polyposis 1-like 1 (Dp1l1) as it was highly homologous to a mouse homolog (Dp1) of the human gene TB2/DP1, and in situ hybridization showed that Dpl1l mRNA was abundantly expressed in the retinal ganglion cells. 17 Furthermore, gene expression profiling of mice deficient in Nrl, which encodes the Maf-family leucine zipper transcription factor NRL, revealed that Reep6 is directly regulated by NRL, which is specifically expressed in rod photoreceptors in the retina. 18 We and other have shown that REEP6 is predominantly expressed in rod photoreceptors, specifically in the endoplasmic reticulum, and is absent from cone photoreceptors. 3,4,9 Mice carrying mutations in Reep6 show a phenotype similar to that of human patients. Specifically, Reep6 L135P knock-in (KI) mice, which harbor a missense variant seen in a patient, exhibit adult-onset retinal dysfunction and photoreceptor degeneration, demonstrating that the p.Leu135Pro missense variant is indeed pathogenic. Reep6 null mice present with a more severe, early-onset phenotype of photoreceptor dysfunction that precedes photoreceptor degeneration. 3 Therefore, Reep6 mutant mice can serve as a good model to study human disease.
One of the interesting observations in Reep6 mutant mice is that the reduction in electroretinogram (ERG) response occurs before obvious photoreceptor degeneration. One explanation for this phenotype is that it is likely due to altered trafficking and/or stability of specific proteins involved in the phototransduction pathway, including guanylyl cyclase, phosphodiesterase 6 (PDE6), transducin-α, and G protein subunit α transducin 1 (GNAT1), in Reep6 mutant mice. Defects in delivery of proteins to their specific subcellular compartments in the photoreceptors can lead to severely compromised regulation of the phototransduction cascade, evident in Reep6-deficient mice as well as several other retinal disease models. 19 –24 Furthermore, Reep6 knockout (KO) mice exhibited ER stress and induction of the unfolded protein response pathway, as well as abnormal mitochondrial proliferation, likely resulting from protein transportation defects in photoreceptors, leading to a progressive loss of vision.
Given that loss of REEP6 leads to severe perturbation of retinal function in individuals with RP and that the associated disease pathogenesis is progressive, it provides us an opportunity to devise new treatment strategies for REEP6-associated RP. Adeno-associated virus (rAAV)-based gene therapies have been shown to be successful in rescuing disease phenotypes in several animal models of retinal degeneration 25 –38 as well as in patients. 7,39 –42 Indeed, most recently, RPE65 gene therapy became the first directly administered treatment approved by the U.S. Food and Drug Administration for patients with Leber congenital amaurosis 2 (LCA2; OMIM entry MIM204100) and retinitis pigmentosa 20 (RP20) (MIM#613794). rAAV vector plasmids have been studied extensively for delivery of functional genes in various cell types in the retina. Specifically, the rAAV serotype 8 (rAAV8) vector plasmid can provide high transduction efficiency and rapid expression of transgenes in photoreceptor cells under the control of the human rhodopsin kinase (hGRK1) promoter. 31,32,36
In this study, we generated and characterized compound heterozygous mice harboring a combination of the missense p.Leu135Pro allele and a loss-of-function allele, recapitulation the REEP6 variants identified in an individual with simplex RP. 3,4 Reep6L135P/− compound heterozygous mice exhibit photoreceptor degeneration starting at 1 month of age, which worsens in severity over time. Using the Reep6L135P/− mice as an animal model for gene therapy, we evaluated whether delivery of murine Reep6.1, the retina-specific isoform of REEP6, to photoreceptors of postnatal Reep6 mutant mice could preserve photoreceptor function and survival. We used rAAV8 to deliver Reep6.1 in the retina of postnatal day 20 (P20) Reep6L135P/− mice and show for the first time that rAAV8-Reep6.1 gene therapy can rescue REEP6-associated retinal degeneration in mice over a course of more than 1 year. Collectively, these findings unravel the successful targeting and therapeutic effects of rAAV8-Reep6.1-mediated gene therapy in mice and lay a foundation for the potential of gene therapy treatments in patients with REEP6-associated RP.
Materials and Methods
Production of rAAV8-Reep6.1 viral vector
Full-length Reep6.1 cDNA was tagged with a 3 ×FLAG epitope tag to the N terminus of Reep6.1 (GENEWIZ). The 3 × FLAG-tagged mouse Reep6.1 cDNA was digested with NotI and amplified by PCR, and the sequence was verified. rAAV8 vector plasmid containing the G protein-coupled receptor kinase 1 (GRK1) promoter was used to generate the pTR-Grk1-3xFlag-mReep6.1 plasmid. rAAV8 was used to package Grk1-3xFlag-mReep6.1 to achieve robust transduction efficiency and expression in retinal photoreceptors (Gene Vector Core, Baylor College of Medicine). The resulting viral titer for rAAV8-hGrk1-3xFlag-mReep6.1 was 7.1 × 1013 genome copies (GC)/mL. rAAV8-hGrk1(Y733F)-GFP viral vector (2.31 × 1013 GC/mL) was used as a control for subretinal injections.
Generation of Reep6 compound heterozygous mice
Previously, Reep6 L135P KI and Reep6 KO mice were generated by CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 gene editing. 3,4 In this study, Reep6L135P/L135P homozygous male mice were bred with Reep6+/− heterozygous female mice to generate Reep6L135P/− compound heterozygous mutant mice. Genotyping was performed by a genomic PCR assay as described previously, 3,4 followed by Sanger sequencing. Genotyping analysis for Reep6 control and mutant mice is shown in Supplementary Fig. S1. All Reep6L135P/− compound heterozygous mutant mice were grossly normal and viable.
All mice in this study were maintained under 12-h light and 12-h dark cycle conditions. All animal experiments were conducted in agreement with the Association for Research in Vision and Ophthalmology (ARVO) statement for the use of animals in ophthalmic and vision research, and protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at Baylor College of Medicine.
Subretinal injections
P20 mice were anesthetized with a combination drug consisting of ketamine (22 mg/kg), xylazine (4.4 mg/kg), and acepromazine (0.37 mg/kg), which was injected intraperitoneally. Subretinal injections were performed as described previously. 36 A shallow incision was made through the sclera with a beveled 30-gauge needle. After this step, a 35-gauge blunt needle was presented inside the vitreous cavity and pushed forward until the tip of the needle had moved past the retina. Viral solution was injected into the subretinal space, using an Ultra-Micro-Pump II and Micron-4 Controller (World Precision Instruments). Mice were treated once in the right eye, using 1 μl of rAAV8-hGRK1-3xFlag-mReep6.1 (7.1 × 1013 GC/mL), and coinjected with rAAV8-hGRK1(Y733F)-GFP (1.0 × 1012 GC/mL) for examining transduction efficiency posttreatment, whereas the contralateral left eye was uninjected, serving as an internal control. Altogether, 20 Reep6L135P/− mutant mice were injected for assessment 2 months posttreatment and 1 year posttreatment, In addition, eight age-matched Reep6L135P+ control mice in total were injected in the right eye to assess toxicity. No decline in visual function or morphology was observed in the right eye of control mice compared with the left untreated eye (data not shown).
Electroretinography
Before ERG, the fundus of all mice injected was evaluated for damage resulting from injections, including severe retinal detachment, atrophy, scarring using the Micron-IV retinal imaging system (Phoenix Research Laboratories). Mice were dark-adapted overnight (at least 12 h) before ERG experimentation. ERGs were performed as previously described. 3,4,36 The anesthesia cocktail (described in the previous section) was delivered by intraperitoneal injection into the mice. Pupils were dilated under dim red light with tropicamide (1%) and phenylephrine (2.5%) eye drops, and proparacaine (1%) was used to anesthetize the cornea. Goniosoft (hypromellose, 2.5%) was applied to the cornea to avoid dehydration and facilitate contact before placing ERG electrodes on each eye. For reference, a platinum subdermal needle electrode was placed into the forehead of the mouse. Scotopic ERG was performed at six varying flash intensities (−24, −14, −4, 0, and 10 dB) on Reep6 mutant mice and isogenic control mice at various time intervals (2, 8, and 54 wk postinjection) after subretinal injection treatments. Age-matched Reep6+/− untreated mice Reep6+/− left eye-injected mice were recorded alongside mutant Reep6L135P/− left eye-injected mice at every time point. The LKC UTAS visual diagnostic system and EMWIN software (LKC Technologies) were used to obtain and compile the data. ERG data analysis was performed on GraphPad Prism5 software (GraphPad Software). ERG responses for each flash intensity were averaged from the left-eye (untreated) and right-eye (treated) ERG recordings for each genotype group, and standard t-test/one-way ANOVA was performed for statistical analysis as appropriate.
Histology and immunostaining
Individual mice were placed into a small container with isoflurane-infused paper towels and were cervically dislocated after they were anesthetized. Eyes were enucleated and processed in fresh modified Davidson's fixative 43 for 18 to 24 h at 4°C. After fixation, eyes were dehydrated in ethanol and embedded in paraffin wax according to standard protocol. 3,4,44 Serial paraffin sections (7 μm) were sectioned on a microtome (Leica), and hematoxylin and eosin (H&E) staining was performed. After H&E staining, slides were cleaned, mounted with coverslips, dried at room temperature, and imaged with a light microscope (ApoTome; Zeiss). For immunostaining, tissue sections were processed in xylene for 1 h and rehydrated in ethanol gradients. For antigen retrieval, slides were boiled in containers containing 0.01 M citrate buffer in a rice cooker for 30 min, cooled for 30 min on ice, and rinsed in phosphate-buffered saline (PBS) three times. Hybridization was performed using 10% normal goat serum with 0.1% Triton-X100 in PBS for 1 h in a humidifying chamber at room temperature, after incubation with primary antibodies. Antibodies used in this article include REEP6 (kind gift from A. Swaroop, National Eye Institute), M2 FLAG (F3165; Sigma), guanylyl cyclase 1 (Ret GC1 CAT rabbit polyclonal; a kind gift from A. Dizhoor, Salus University), and caspase-12 (ab3612; EMD Millipore). After overnight incubation at 4°C, slides were rinsed in PBS and stained with 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies), and Prolong Gold antifade reagent (Life Technologies) was used to mount the coverslips on the slides. An Axio Imager.M2m (Zeiss) was used to obtain immunofluorescence images at × 10, × 20, and × 40 zoom.
Results
Reep6 L135P/− compound heterozygous mice exhibit photoreceptor degeneration
Reep6 homozygous null mice exhibit early-onset photoreceptor dysfunction, which is evident starting as early as postnatal day 15 (P15). By P20, photoreceptor degeneration can be observed by histological analysis. 3 In comparison, Reep6L135P/L135P homozygous mutant mice exhibit delayed-onset retinal degeneration that is not evident until 2 months or later. 4 To establish a Reep6 mouse model that has a proper window of treatability, we generated compound heterozygous mice harboring both a null allele and a missense (L135P) mutant allele, termed Reep6L135P/−. Histological analysis of compound heterozygous Reep6L135P/− mouse retina revealed defects in retinal morphology compared with the control Reep6+/− mouse retina, as expected (Fig. 1). Reep6L135P/− mice exhibited mild thinning of the outer nuclear layer (ONL) on P22 (Fig. 1A), which became more severe over time (Fig. 1C–E). By 3 months of age, the retina of Reep6L135P/− mice had lost 40% of the photoreceptor cells as indicated by the reduced thickness of the ONL (Fig. 1C). In 7-month-old Reep6L135P/− mice, the retina is considerably degenerated with more than 65% of photoreceptor cells lost, and a thin ONL with only a few photoreceptor rows remaining (Fig. 1D). In comparison, the other cell layers in the retina, including the inner nuclear layer and ganglion cell layer (GCL), did not exhibit any obvious defects.
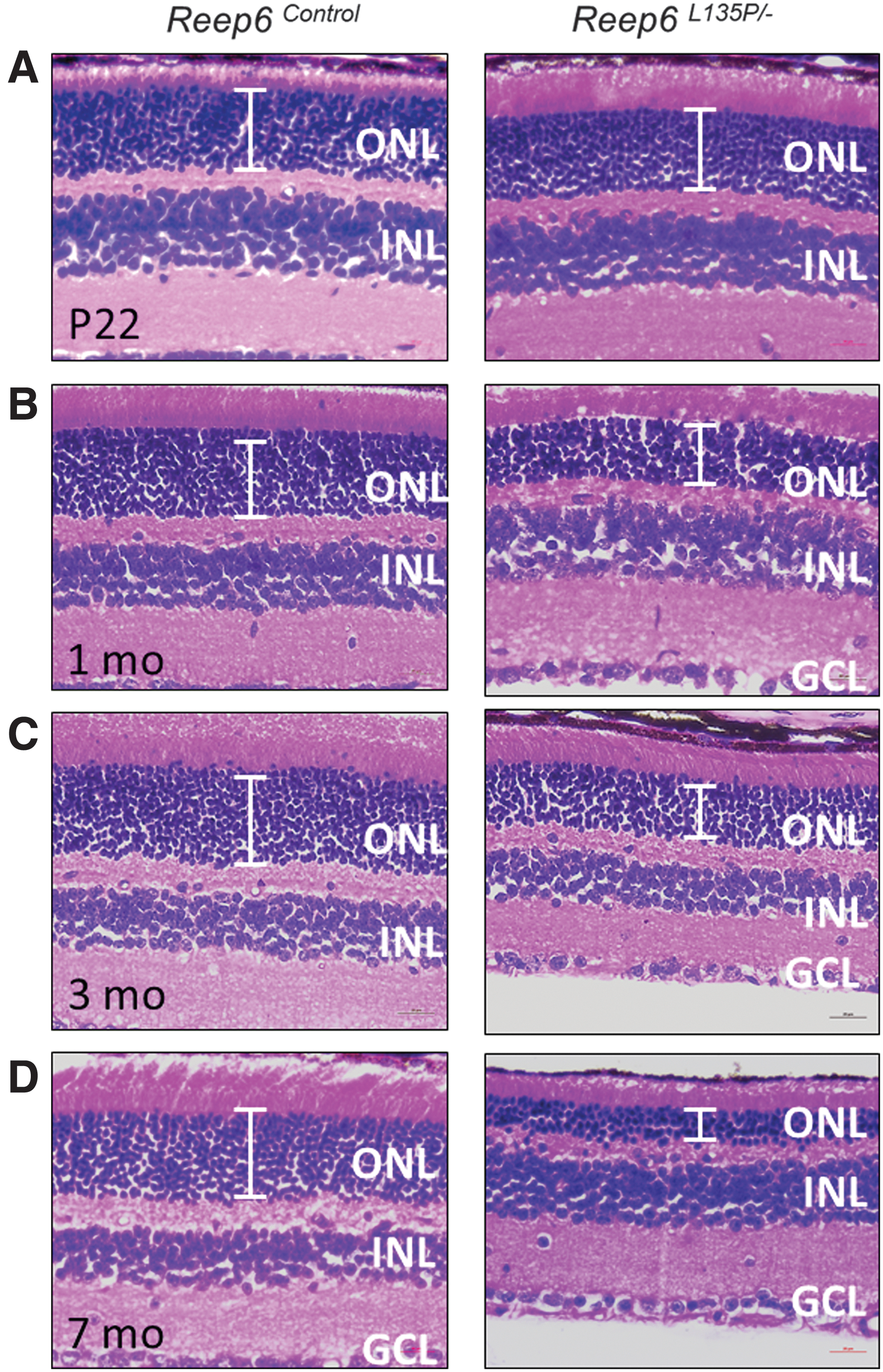
Reep6L135P/−
compound heterozygous mice exhibit photoreceptor degeneration. Histological analysis of retinal sections from Reep6L135P/−
and Reep6+/−
mice was performed on
Reep6 mutant mice exhibit reduced electrophysiological response to light
Full-field electroretinography (ERG) of dark-adapted Reep6L135P/− mice was performed to evaluate the rod photoreceptor-mediated responses at various stages of degeneration. Reep6L135P/− mice exhibited a significant reduction in the scotopic ERG responses as early as P22 compared with heterozygous Reep6+/− and Reep6+/L135P littermate controls (Fig. 2A). The amplitudes of both the a-wave (measuring responses of photoreceptor cells) and b-wave (measuring the cumulative response of rod and bipolar cells) were considerably decreased in Reep6L135P/− mice, suggesting that the rod photoreception function is significantly compromised in mice devoid of REEP6. Furthermore, we tested the ERG responses at additional time intervals, including 3 months and 7 months of age, and consistently observed a significant reduction in the a-wave and b-wave responses, whereas the responses were normal in age-matched Reep6+/L135P and Reep6+/− control mice (Fig. 2B–D).

Reep6L135P/−
mutant mice exhibit defects in response to light. Quantitative evaluation of scotopic a-wave and b-wave responses for dark-adapted mice on
rAAV-mediated REEP6 transduction in photoreceptor cells
To specifically target REEP6 to the photoreceptors, where it is normally localized in the inner segment of rod cells, we used rAAV8 to package mouse Reep6.1 complementary DNA (cDNA), the retina-specific isoform of Reep6. Expression of the Reep6.1 construct is under the control of the human rhodopsin kinase (hGRK1) promotor, which has been established to be the most effective combination for attaining rapid and high transgene expression in mouse photoreceptors. 45 Furthermore, to distinguish the vectored wild-type REEP6.1 protein from the resident REEP6 L135P protein, an N-terminal FLAG tag was included. Because the onset of photoreceptor degeneration in Reep6L135P/− mice occurs at or before P22, we performed treatments with rAAV8-Reep6.1 in mice as early as P20. To determine whether the rAAV8-Reep6.1 treatment is successful in targeting expression of mouse REEP6 in the photoreceptors, we examined the retina of treated mice (n = 6) by immunofluorescence staining 2 months posttreatment. In this study, all treatments were performed in the right eye (RE) and compared with the contralateral left eye (LE).
In Reep6L135P/− mice, we failed to detect expression of endogenous REEP6 (green) in the LE (Fig. 3A and C). In the treated RE of Reep6L135P/− mice, FLAG-tagged transgene expression was robustly detected by positive FLAG staining (red) in the photoreceptors (Fig. 3B). At higher magnification, the FLAG signal was detected specifically in the inner segment of photoreceptors (Fig. 3D) of treated Reep6L135P/− retinas, where the wild-type REEP6 protein is normally localized (Fig. 3E). Further, we performed coimmunostaining of Reep6L135P/− LE and RE retinal sections with an anti-REEP6 antibody. REEP6 immunolabeling was detected in the inner segment of the photoreceptors in Reep6L135P/− RE treated retinal sections, where it colocalized with FLAG-positive staining (Fig. 3B and D). Furthermore, both FLAG and REEP6 immunoreactivity were strongest at the site of injection (indicated by asterisks; Fig. 3B), and the expression extended throughout most of the retina, covering more than 70% of the dorsal and ventral regions. Some regions of the tissue exhibited weaker immunoreactivity due to the inconsistent nature of subretinal injections. Consistent with the expression pattern of REEP6, the areas that were targeted exhibited preservation of overall retinal morphology compared with areas that remained untreated, where marked thinning of the retina was observed on gross examination. On the basis of the data, it is evident that the rAAV8-Reep6.1 vector results in stable and efficient expression of REEP6 in rod photoreceptors of Reep6L135P/− mice after treatment by subretinal injections.
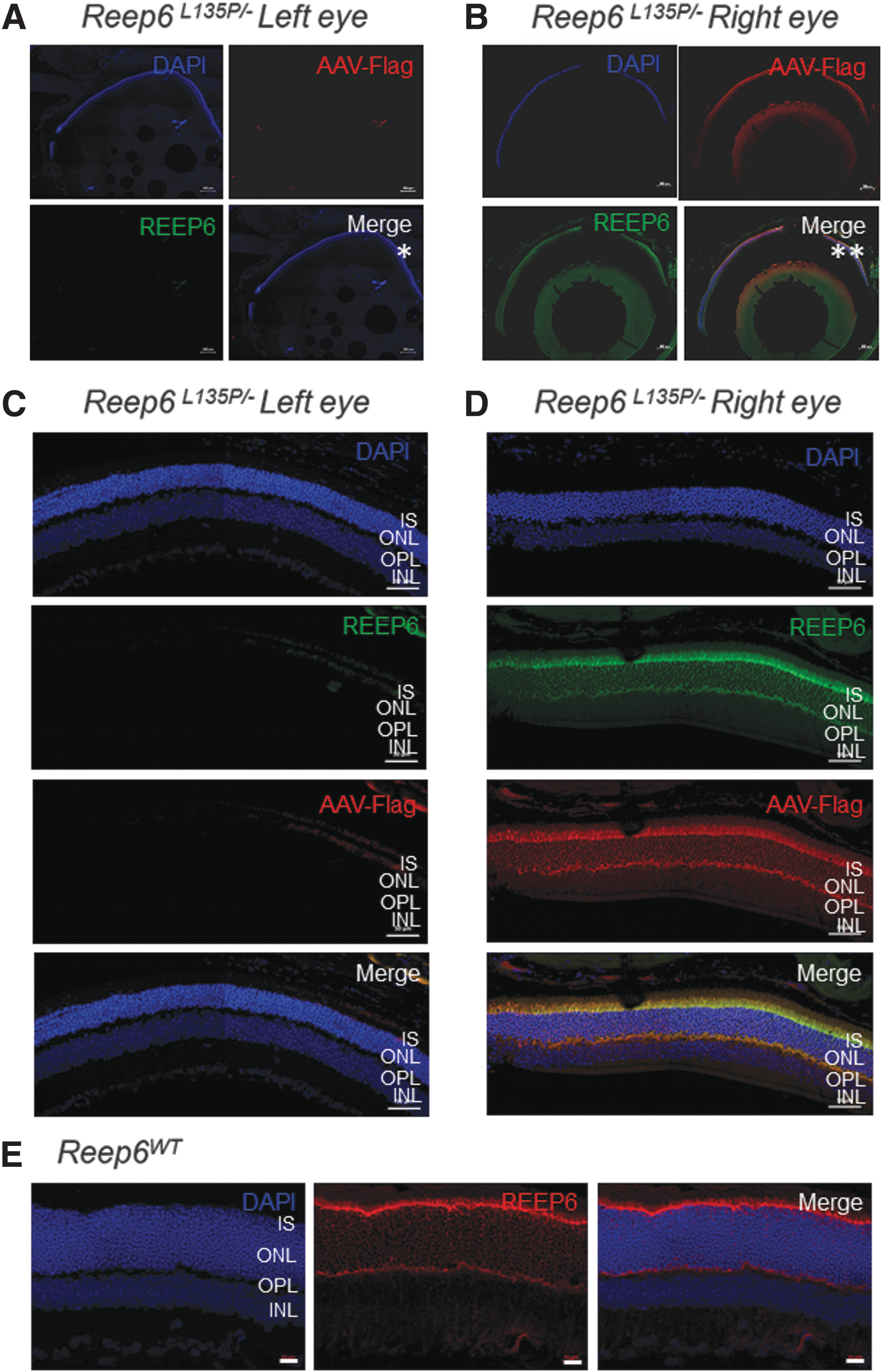
Flag-REEP6.1 and endogenous REEP6 expression and localization in the photoreceptors of rAAV8-Reep6.1-treated and -untreated Reep6L135P/−
mice. Immunofluorescence staining of representative contralateral
Preservation of photoreceptor integrity and function in rAAV8-Reep6.1-treated Reep6 mutant mice
As previously stated, the retina of Reep6L135P/− mice exhibits mild thinning of the ONL starting on P22, which progressively increases in severity. To determine whether the rAAV8-Reep6.1 treatment was successful in restoring retinal morphology in Reep6 mutant mice, we performed histological analysis of both the LE and RE of Reep6L135P/− mice (n = 6) 2 months postinjection (referred to as 11 wk old). H&E staining of the untreated LE in 11-week-old mice shows reduction in overall retinal thickness and marked thinning of the ONL (Fig. 4A and B), in comparison with treated Reep6L135P/− REs and Reep6 wild-type control mice (Fig. 4C and D). On treatment with rAAV8-Reep6.1, Reep6L135P/− mouse REs showed drastic improvements in overall retinal morphology as evident by preservation of photoreceptor cell nuclei in the ONL and increased ONL thickness (Fig. 4C and D) in regions where REEP6 is robustly expressed (Fig. 3B and D). Consequently, in areas of the retina distant from the site of injection and that lack REEP6 expression, the retinal morphology was less intact in the RE, but improved nonetheless compared with the untreated LE. Quantitative morphometric analysis of Reep6 wild-type, Reep6L135P/− LE, and Reep6L135P/− RE shows the ONL thickness averaged from six retinas per group; evidently, there is an improvement in the morphology of the photoreceptors in the treated group.
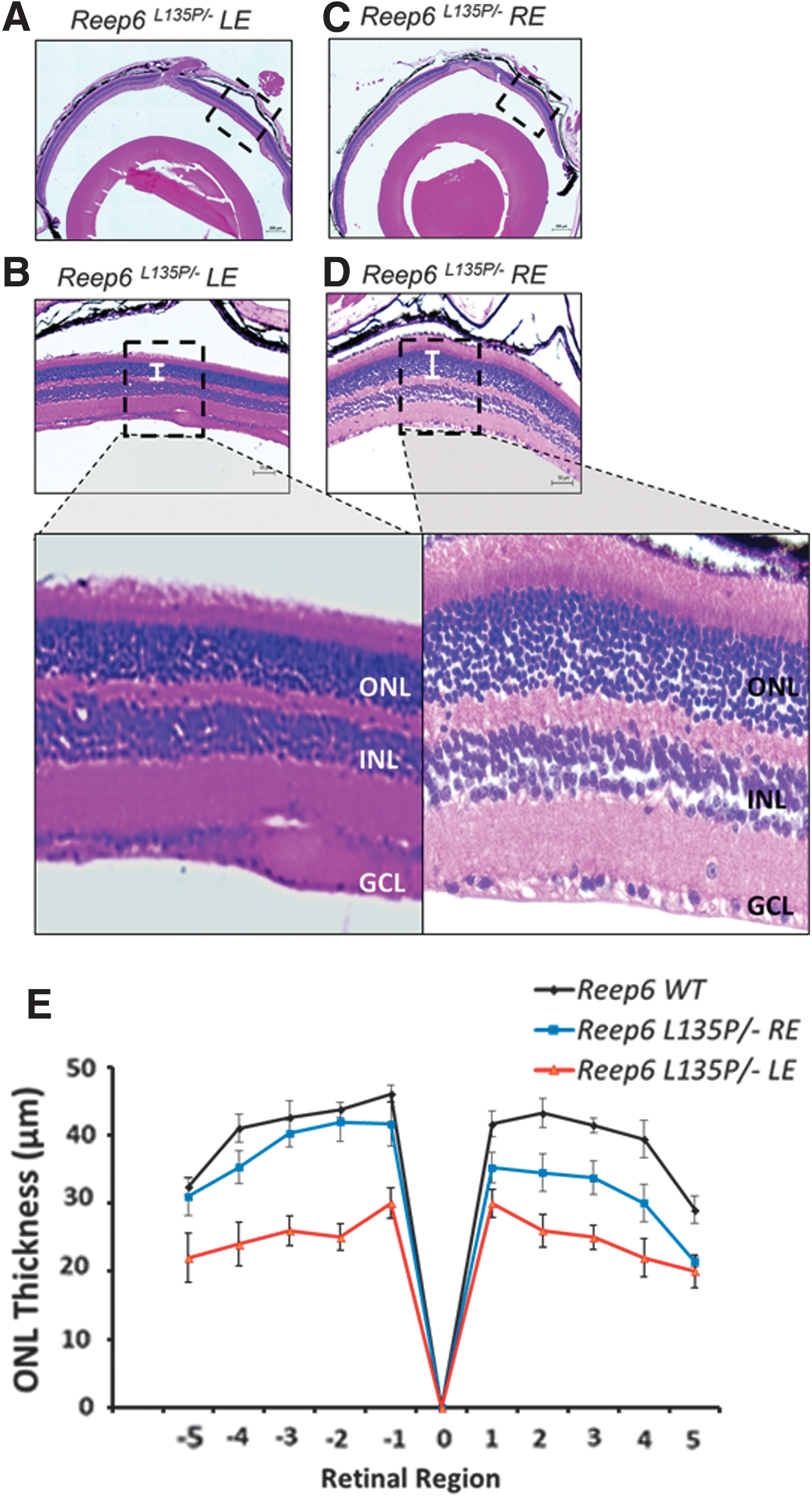
Preservation of photoreceptor morphology in Reep6L135P/−
mice 2 months after treatment. Shown are hematoxylin and eosin staining of paraffin-embedded retinal sections from
To assess whether morphological preservation of photoreceptors in Reep6L135P/− mice posttreatment is concordant with functional rescue of electrophysiological response to light, we examined the ERGs of dark-adapted Reep6L135P/− mice by testing both the LE and RE independently. At 11 weeks of age (2 mo posttreatment on P20), Reep6L135P/− mice exhibited an attenuated scotopic ERG response in the contralateral untreated LE, where both the a-wave amplitude and the b-wave amplitude were significantly decreased (indicated by an asterisk) (Fig. 5A and B, red line) compared with healthy Reep6 wild-type mice (black line). In comparison, the treated RE of Reep6L135P/− mutant mice had improvements in the dark-adapted scotopic response at various flash intensities. There was a statistically significant improvement in the a-wave response (indicated by a hashtag symbol); the b-wave (combined rod photoreceptor and inner retinal neurons response) response was also improved, although not significantly. Furthermore, there was no significant difference between the treated RE and the wild-type scotopic b-wave responses (Fig. 5A and B, blue line), demonstrating functional retention of rod photoreceptors resulting from expression of the Reep6.1 transgene

Functional assessment of scotopic response in rAAV8-Reep6.1-treated mice based on electroretinography (ERG). Before ERG, fundus examination was performed to check for any damage that was persistent after subretinal injections of all mice. Mice that incurred retinal damage were excluded from ERG analysis. ERG was performed in dark-adapted Reep6L135P/−
mouse left eye (LE) and right eye (RE) 2 months after treatment at various flash intensities (x axis).
rAAV8-Reep6.1-mediated gene therapy alleviates ER stress response resulting from REEP6 deficiency and restores guanylyl cyclase 1 localization
Previously, we reported that loss of REEP6 causes induction of the ER stress response pathway and activation of signature ER stress markers in Reep6 homozygous knockout mice on P20, before any major phenotypic defects can be observed. In Reep6L135P/− mice, we observed activation of caspase-12, a marker of ER stress-induced apoptosis, in the untreated LE at 11 weeks of age (Fig. 6B). Close examination of caspase-12-positive staining shows a punctate pattern in the ONL (shown by arrows) at higher magnification (Fig. 6B), suggesting activation of the ER stress/unfolded protein response pathway. In 2-month posttreatment RE of Reep6L135P/− mice (Fig. 6A), we failed to detect strong positive signals for caspase-12, suggesting that rAAV8-Reep6.1 gene therapy can alleviate ER stress response in Reep6 mutant mice and prolong photoreceptor cell survival.

rAAV8-Reep6.1-mediated gene therapy alleviates endoplasmic reticulum (ER) stress response and restores guanylyl cyclase 1 (GC1) expression.
Furthermore, we have previously shown that guanylyl cyclase 1 (GC1) and GC2 expression in Reep6 KO mice is undetectable by immunofluorescence microscopy. 3 To determine whether REEP6 expression can rescue endogenous expression of GC1 to the photoreceptor outer segment (OS), we performed immunostaining on retinal sections from Reep6L135P/− mice 2 months posttreatment. GC1-positive staining was detected in the OS of Reep6L135P/− right eye, and the expression was consistent throughout the retina and coincided with FLAG expression in the posttreated eyes. GC1 localization to the OS was comparable to wild-type GC1 localization. In contrast, GC1 expression was not detected in the untreated eyes of Reep6L135P/− mice, consistent with the findings observed in Reep6 KO mice, as expected.
Long-term rescue efficiency of rAAV8-Reep6.1-mediated gene therapy in Reep6 mutant mice
Retinal sections of both LE and RE from Reep6L135P/− mice (n = 4) treated only once on P20 were examined to determine the long-term potential of rAAV8-Reep6.1 gene therapy treatment. At the late-stage 1-year time point, the ONL of the untreated LE retina was reduced to only two or three layers of nuclei, consistent with the progressive degeneration observed between P22 and 7 months of age (Fig. 7A). In contrast, rAAV8-Reep6.1-treated retina exhibited substantial ONL thickness in the RE of Reep6L135P/− mice (Fig. 7B), indicating significant retention of the photoreceptor integrity after prolonged treatment. In addition to restoration of retinal morphology, Reep6L135P/− mouse RE also retained a functional photoresponse as measured by ERGs in dark-adapted mice (Fig. 7C and D). Whereas Reep6L135P/− mouse LE had a severely diminished a-wave amplitude, the Reep6L135P/− mouse RE had a significantly increased a-wave amplitude (Fig. 7A). Similarly, the treated RE of Reep6L135P/− mice had an elevated b-wave response (Fig. 7B). On the basis of the comparative ERG analysis, it is evident that rod photoreceptor response is significantly preserved in Reep6 mutant mice, even up to 1 year posttreatment.

Long-term rescue efficiency of rAAV8-Reep6.1-mediated gene therapy in Reep6 mutant mice.
Discussion
Ultimately, the goal of identifying the genetic causes underlying RP is to develop therapeutic treatments for patients who have retinal degeneration; however, to date, there are no gene therapy treatments for patients with autosomal recessive RP. With emerging findings of success in clinical trials for gene therapy-based approaches to prevent and/or restore loss of photoreceptor cells in the retina, there is evidence for translation of these treatment strategies in humans. 25 –38 Thus, this study set out to develop rAAV-based gene therapy for REEP6-associated RP to assess its therapeutic efficacy in Reep6 mutant mice as a preclinical model of retinal degeneration. Here, we have shown that compound heterozygous Reep6L135P/− mice exhibit progressive retinal degeneration, similar to the clinical observations of an individual who harbored these variants, and that rAAV8-Reep6.1 regulated by the hGRK1 promoter can effectively drive mouse REEP6 expression in the rod photoreceptors, which results in restoration of retinal morphology and photoreceptor function. This effect of gene replacement therapy can persist up to 1 year postinjection, demonstrating its long-term therapeutic potential in treating retinal degeneration. Importantly, transgene expression of Reep6.1, the retina-specific isoform of Reep6, induced proper GC1 expression and localization to the OS, resulting in alleviation of ER stress and restoration of visual function and photoreceptor survival.
We have established and characterized two animal models of REEP6-associated retinal degeneration. 3,4 Reep6L135P/L135P mice exhibit adult-onset photoreceptor degeneration that is evident at about 4 months of age, whereas Reep6 germline-null mutant mice exhibit a much more severe, rapid onset of retinal dysfunction, which precedes retinal degeneration, as early as P15. In the REEP6-associated RP cohort, two mutations, a missense mutation (c.404T>C [p.Leu135Pro]) and a single-nucleotide deletion also in exon 4 (c.448del [p.Ala150Pfs*2]), were identified in an individual with RP. To recapitulate the genotype of this individual, which we deemed would be an interesting model to assess the efficacy of gene therapy, we generated compound heterozygous Reep6L135P/− mice. Reep6L135P/− mice exhibit photoreceptor dysfunction that is evident by P22, although the morphological defects become evident at about 1 month of age. Furthermore, although Reep6L135P/− mice display the presence of at least three photoreceptor layers by 1 year of age, Reep6−/− mice have merely one photoreceptor layer remaining in the late stages of disease progression. Thus, the severity of degeneration observed in Reep6L135P/− mice is modestly mild compared with the pathogenesis observed in Reep6 KO mice, making it an ideal candidate for gene therapy clinical trials as it provides an optimal window of treatment to evaluate its therapeutic potential.
The retina-specific isoform of Reep6, referred to as Reep6.1, was identified by gene expression profiling of Nrl KO mice, 18 and is expressed specifically in rod photoreceptors. Compared with the canonical transcript, termed REEP6.2, we have shown that REEP6.1 is the predominant isoform in developing human photoreceptors and, indeed, REEP6.1 expression appears to be limited to rod photoreceptors in human 3D optic cups. We performed further analysis to look at the transcripts of both REEP6 isoforms in human tissues and confirmed that REEP6.1 is the predominant isoform expressed in the human retina. Furthermore, we have reported a biallelic single-nucleotide duplication within intron 4 of the REEP6.2 isoform that causes premature termination of the REEP6.1 isoform (c.557dupC [p.Val187Glyfs*13]) in an affected individual, whereas REEP6.2 remains unperturbed. 4 Collectively, these findings suggest that REEP6.1 is a novel transcript within rod photoreceptors, where it likely plays essential roles in retinal function/homeostasis, disruption of which results in the observed retinal dystrophy phenotypes in mice and patients. Thus, in this study, we chose to deliver species-specific Reep6.1 cDNA to photoreceptors packaged in the rAAV8 vector to evaluate the therapeutic potential of gene therapy and to gain further insights into the role of REEP6.1 specifically in rod photoreceptors.
On treatment of Reep6L135P/− mice RE with rAAV8-Reep6.1, we observed rapid and robust expression of both FLAG and REEP6 in the retina, where they colocalized to the inner segment of the photoreceptors. These data provided confirmation that the hGRK1 promoter was successful in restricting expression of REEP6 to photoreceptors, and no off-target expression resulted in the retina because of overexpression of the rAAV8-Reep6.1 viral vector. Furthermore, treatment of the RE of Reep6L135P/− mice enhanced photoreceptor cell survival and preserved the scotopic response to light under dark-adapted conditions. On the basis of analysis of the a-wave (photoreceptor response) in the RE versus the LE of Reep6 mutant mice, there was a significant improvement in the ERGs. However, the improvement in the b-wave (combined photoreceptor and inner retinal neurons response) was not as strong as that of the a-wave, which may reflect an underlying secondary defect in the second-order neurons in the inner retina. In this study, we did not measure the isolated response of bipolar cells, which could provide further insights regarding the ERG b-wave data obtained.
As expected, the improvements in retention of photoreceptor integrity and function led to an alleviated ER stress response in the RE, whereas the untreated LE had strong caspase-12-positive staining present throughout the retina. It is known that induction of the ER stress pathway and the unfolded protein response can lead to ER stress-induced apoptosis, which results in photoreceptor cell death. 46 –51 Here, we have shown that treatment with rAAV8-Reep6.1 is sufficient in rescuing this fate, allowing the photoreceptors to survive and function properly for an extended period of time posttreatment. Indeed, several already established mouse models of retinal degeneration exhibit ER stress, and it would be of interest to determine whether therapeutic approaches such as targeted gene therapy for these other mice can also have a similar effect in alleviating the cell stress response.
On the basis of the positive results obtained from gene therapy trials in Reep6 mutant mice, we also wanted to investigate the molecular mechanisms involved in restoration of visual function and photoreceptor survival. Previously, we have shown that loss of Reep6 leads to misexpression and/or mistrafficking of select proteins involved in the phototransduction pathway. GC1 function is integral in the synthesis of cyclic GMP (cGMP) in the retina, which regulates the opening/closing of cGMP-gated channels, which in turn leads to depolarization/hyperpolarization of the photoreceptors, respectively. 3 In Reep6 KO mice, we hypothesized that when GC1 function is compromised, depletion of intracellular cGMP leads to failure of opening of the cGMP-gated ion channels, resulting in prolonged hyperpolarization of the rod photoreceptors, eventually leading to overexertion and subsequent cell death. The rescue of GC1 localization to the OS by Reep6 transgene expression in the photoreceptors validates our previous hypothesis. It also coincides well with the elevated ERG response posttreatment, indicating that the phototransduction machinery has likely been restored in Reep6L135P/− mutant mice.
In addition, we observed preservation of rod photoreceptor ERG responses as well as increased photoreceptor survival in mice up to 1 year posttreatment after just one round of treatment. This strong effect of rAAV8-Reep6.1 gene therapy can likely be attributed to the high transduction rate of the viral vector in the photoreceptors, as well as proper localization of FLAG-tagged Reep6.1. Indeed, Reep6.1-FLAG-tagged transgene expression sufficiently replenished endogenous REEP6 expression and localization in the retina, illustrating that proper targeting of rAAV-based vectors in the appropriate regions of interest has a significant impact on the overall therapeutic potential. In future, it would be interesting to investigate the effects of restoring the canonical isoform, Reep6.2, using similar approaches utilized in the current study to determine whether it has the potential to rescue photoreceptor degeneration in Reep6 mutant mice.
In conclusion, this work highlights the success of the first clinical rAAV8-Reep6.1-based gene therapy in Reep6 mutant mouse models of retinal degeneration. In particular, we show that 2 months and up to 1 year posttreatment, Reep6 mutant mice showed improvements in the photoresponse as well as preservation of photoreceptor cells, demonstrating that the treatment is effective even after a prolonged period of time. Findings from this study show that gene replacement therapy in the retina with rAAV8-Reep6.1 can improve photoreceptor function in Reep6 mutant mice, extend survival of photoreceptors in vivo, and can be potentially used as a therapeutic target for treatment of patients with RP.
Footnotes
Acknowledgments
The authors thank the Genetically Engineered Mouse Core, which is partially supported by National Institutes of Health (NIH) grant P30CA125123 at the Baylor College of Medicine for generating Reep6 mouse models using CRISPR-gene editing. The authors also thank the Gene Vector Core at the Baylor College of Medicine. This project was funded by the Foundation Fighting Blindness (BR-GE-0613-0618-BCM), the National Eye Institute (R01EY022356, R01EY020540) to R.C., and by 5T32EY007102-23 awarded to S.A. (PI: Dr. Graeme Mardon).
Author Disclosure
There is no conflict of interest and/or competing financial interests to be declared by the authors that contributed to this study.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.