
Abstract
Over the last decade, the role of the assembly-activating protein (AAP) has begun to be dissected for the formation of adeno-associated virus (AAV) capsids based on different viral serotypes. Recently, the authors' group has specifically studied AAP's relevance during production of AAV gene therapy vectors in mammalian or insect cells, and AAP was found to be essential for capsid protein stabilization and generation of functional vector particles. Here, the lingering question is additionally addressed of whether molecular AAV evolution via DNA family shuffling of viral capsid genes would perturb AAP functionality due to concurrent and inadvertent recombination of the AAP open reading frame. To this end, a battery of complementary experiments was conducted in which: (1) the ability of chimeric AAP from AAVDJ, a hybrid of serotypes 2, 8, and 9, was tested to rescue AAP knockouts in the three parental serotypes; (2) the functionality of 60 chimeric AAPs extracted from five shuffled, unselected capsid libraries was measured; (3) whether production of different shuffled libraries, 10 wild-type serotypes or 25 individual chimeric capsids, can be enhanced by overexpression of AAP cocktails was assessed; and (4) the activity of 12 chimeric AAPs isolated from a shuffled library that was iteratively selected in vivo in mouse livers was studied. Collectively, the data demonstrate a remarkable tolerance of AAP for recombination via DNA family shuffling, evidenced by the findings that (1) all chimeric AAPs studied here retained at least partial activity, even in cases where the cognate hybrid capsid may be non-functional, and that (2) ectopic AAP overexpression did not enhance production of shuffled AAV chimeras or libraries, implying that the inherently encoded hybrid AAP variants are sufficiently active. Together, this work provides compelling evidence that AAP is not rate limiting during AAV capsid shuffling and thereby relieves a major concern in the field of AAV vector evolution.
Introduction
A
In contrast, there is a consensus that another stunning revelation, first reported by the Kleinschmidt lab in 2010, 12 has an equal impact on future work with both wild-type and recombinant AAVs. In this pivotal study, Sonntag et al. identified a 23–26 kDa protein translated from roughly 600 bp that are encoded in the second open reading frame of the 2.2 kb AAV2 capsid gene and that overlap with the N termini of the VP2 and VP3 capsid proteins. 12 As demonstrated in the original work and two follow-up studies, 12 –14 this protein exerts a crucial role in the assembly of intact AAV2 particles, leading to its designation as assembly-activating protein (AAP). A decisive experiment proving this role was to knock out AAP by site-directed mutagenesis, resulting in a loss of assembled AAV2 particle formation that could be rescued by ectopic AAP overexpression.
To date, the exact mode of action of AAP remains elusive though, in spite of extensive additional work from the Nakai laboratory and the authors, confirming the essential role of AAP not only for AAV2, but also for many other AAV serotypes in which the AAP sequence is largely conserved. 15 –18 Possibly, AAP acts as a scaffold or chaperone that directly interacts with free AAV capsid proteins and induces conformational changes that subsequently foster capsid assembly. Alternatively, AAP–VP interaction may lead to translocation of VP proteins to cellular sites or structures, such as the nucleolus, where other factors reside that then trigger VP assembly. Arguing against this, it has recently been shown that AAP is also critically required for recombinant AAV2 assembly in insect cells that lack these putative mammalian factors, and that AAPs from distinct AAV serotypes differ in their intracellular localization pattern, 17 congruent with data from Earley et al. 16 Interestingly, it was also found that AAP knockout results in a loss of free VP proteins (but not capsid mRNAs) and thus also assembled capsids in AAV2-producing cells, and that artificial VP stabilization via the addition of proteasome inhibitors does not rescue particle formation. 17 Likewise, Naumer et al. showed that forced localization of free AAV2 VP proteins to the nucleolus, where AAV2 capsid assembly occurs, 19 is not sufficient to generate intact particles in the absence of AAP. 13
Together, these and other data support the idea that AAP directly interacts with AAV capsid proteins and fosters their assembly into virions, which counteracts the otherwise rapid turnover of free VP proteins in the cell (the latter is perhaps due to ubiquitination and subsequent proteasomal degradation). For a more comprehensive discussion of the possible mechanism(s) of AAP action in the AAV life cycle, the reader is referred to the authors' latest publication, including the model there in fig. 9. 17
This study expanded on the aforementioned recent observations that AAP is critical for AAV vector production in two heterologous production systems, and that all serotypes tested thus far require AAP for efficient assembly of functional particles. This is crucial for the use of AAV vectors in human gene therapy, considering that a major asset of AAV is the ability to engineer designer capsids genetically that potently transfer DNA to specific tissues or cells, ideally in the presence of neutralizing anti-AAV antibodies. To achieve this, a wealth of techniques for capsid gene modification have been devised, comprising site-directed mutagenesis, peptide display, or ancestral reconstruction. 20,21 In addition, the method of DNA family shuffling is used for capsid gene diversification, whose hallmark is the creation of chimeric AAV capsid genes that are composed of fragments from multiple parental isolates. To this end, capsid genes of interest are first fragmented by controlled DNase I digestion, then recombined based on partial sequence homologies in a primer-less PCR, and finally cloned into a replication-competent AAV plasmid for production and selection of a capsid library. 22 –24
In particular, when using this protocol for AAV vector evolution, it is key to be aware of AAP and of its central role, as AAP is largely conserved in all natural serotypes that are routinely used for AAV shuffling. 14 Moreover, the overlap of the AAP open reading frame with that of the VP proteins implies that VP shuffling automatically also yields chimeric AAPs whose function may be perturbed because of recombination. This, in turn, would limit AAV vector evolution through DNA family shuffling—a risk that was originally predicted by Sonntag et al. 12 and that would have to be considered in future AAV evolution schemes. This study thus comprehensively assessed the ability of shuffled AAPs to trans-complement AAV/AAP knockout helper plasmids and to rescue AAV vector production, allowing it to be concluded that this risk is probably negligible.
Materials and Methods
Cloning of AAV, AAP, or cap expression plasmids
Plasmids employing a cytomegalovirus (CMV) promoter for expression of chimeric AAPs, including the one from AAVDJ, were generated essentially as described before.
17
Briefly, they were polymerase chain reaction (PCR) amplified from the respective capsid gene by using primers binding at the start or end of each AAP frame. These primers comprised NotI (forward primer) or EcoRI (reverse primer) restriction sites, allowing for cloning of the AAP sequences into the abovementioned acceptor vector containing a CMV promoter for robust AAP expression (pIRES-Ago2).
25
All primer sequences are listed in Supplementary Table S1 (Supplementary Data are available online at
For use in trans-complementation assays, the cap gene of selected chimeras was transferred from a replication-competent AAV library construct containing AAV2 inverted terminal repeats and rep (Herrmann et al., manuscript in preparation) into an AAV helper context 17 via HindIII and SpeI sites that are present in both donor and acceptor vector.
Cell culture
HEK293T, SF539, and MCF7 cells were cultured under standard sterile growth conditions in Dulbecco's modified Eagle's medium (DMEM) with GlutaMAX (Life Technologies) at 37°C and 5% CO2. The medium was supplemented with 100 IU/mL of penicillin-streptomycin (Life Technologies) and 10% fetal bovine serum (Sigma–Aldrich). Cells were split routinely when they reached around 80% confluency.
AAV crude cell lysate experiments
Production of crude lysates (i.e., non-purified, AAV vector-containing HEK293T cell extracts) was performed as described recently, using polyethylenimine (PEI; linear, MW ∼25,000; Polyscience) for plasmid triple transfection (AAV/green fluorescent protein [GFP] vector, AAV helper, and adenoviral helper plasmid). 17,26 In trans-complementation assays, AAV helper constructs were used that encoded wild-type, chimeric, or AAP-deficient (AAPmut) capsids, and either the indicated AAP expression plasmid or sheared salmon sperm stuffer DNA (Invitrogen) was added to the mix as a fourth component. Per well of a six-well plate, a total of 2.6 μg of DNA was transfected, composed of either 3 × 867 ng (AAV/GFP vector, AAV helper, and adenoviral helper) in the case of a regular vector production without extra AAP, or 3 × 690 ng (same plasmids as above) plus 530 ng of AAP plasmid or stuffer DNA. After 48 h, the medium was removed, and AAV particles were extracted from the cells as described 17 and then stored at −20°C for later use.
For transduction experiments, cells were seeded onto 96-well plates (0.5 × 104 [MCF7, SF539] to 1.5 × 104 [HEK293T] cells per well) 24 h prior to the addition of 5 μL of AAV solution (undiluted crude lysate or diluted 1:10 or 1:100). After 48 h, transduction efficiency was determined by measuring the percentage of GFP-positive cells as a surrogate readout. For this purpose, the cells were washed with 100 μL phosphate-buffered saline (PBS), detached with 30 μL of trypsin (Life Technologies) for 5–10 min at 37°C, and then re-suspended in 170 μL of 1% bovine serum albumin (Roth) in 1 × PBS. Amounts of GFP-positive cells were determined by flow cytometry using a FC500 MPL flow cytometer (Beckmann Coulter) and the MXP software for data analysis. To avoid saturation artifacts, the dilution was further analyzed in which the wild-type control gave <95% positive cells, and all other values were then normalized to this control (set to 1).
For a selected chimeric capsid (clone #18), crude lysates were titrated by droplet digital (dd)PCR, using the QX200 ddPCR system (Bio-Rad). Initially, the crude lysates were diluted 1:100 in benzonase buffer (see next section), before 5 μL of sample was mixed with 5 μL of 1:5 diluted DNaseI (Roche; 27 mg/mL stock solution). The samples were incubated in a PCR cycler for 1 h at 37°C followed by DNaseI inactivation for 30 min at 75°C. Next, 10 μL of proteinase K (recombinant, PCR grade; Roche) was added from a 1:50 dilution (20 mg/mL stock) and incubated at 56°C for 2 h, followed by inactivation at 95°C for 30 min. The ddPCR reaction was set up and processed as previously described. 27 The PCR consisted of an initial heating step of 94°C for 10 min, followed by 40 cycles of heating at 94°C for 30 s and amplification at 60°C for 1 min, as well as a final heating step at 98°C for 10 min. Primers and probe were directed against the eGFP insert (all sequences are listed in Supplementary Table S1).
AAV library production, purification, and titration
Plasmid libraries of chimeric AAV cap genes were generated by DNA family shuffling 22 using an optimized workflow (Herrmann et al., manuscript in preparation), and single plasmid clones were analyzed by Sanger sequencing. Viral libraries were derived by standard transfection of HEK293T cells. Therefore, an adenoviral helper construct, 28 a plasmid pool encoding shuffled AAV capsid libraries based on different serotype combinations, and either sheared salmon sperm stuffer DNA or a mixture of plasmids encoding the respective parental AAPs were transfected in a ratio of 1.3:1.3:1 using the PEI transfection method. Briefly, per 15 cm plate of HEK293T cells, a total of 32.4 μg of DNA was mixed and filled up to 790 μL with water. A second mix containing 352 μL of PEI and 438 μL of H2O was prepared in parallel. After the addition of 790 μL of 300 mM NaCl to both mixes, they were combined, vortexed, and incubated at room temperature for 10 min. Then, the mix was added dropwise to the cells. Three days later, cells were scraped off, transferred to tubes, centrifuged for 10 min at 400 g, washed with 1 × PBS, and re-suspended in 500 μL of benzonase buffer (50 mM Tris-HCl, pH 8.5, 150 mM NaCl, and 2 mM MgCl2) per plate. After five freeze/thaw cycles, 75 IU of benzonase (Merck) per milliliter were added, and the lysate was incubated at 37°C for 1 h while inverting the tube every 10 min. Subsequently, cell debris was pelleted twice at 4000 g for 15 min at 4°C, and AAV particles were purified through iodixanol density gradient centrifugation. 29 Genome-containing virus particles were determined via quantitative PCR using a primer-probe set specific for rep2 (Supplementary Table S1). 13,29
Immunostaining and microscopy analysis
HEK293T cells were seeded into polylysine (Sigma–Aldrich) coated eight-chambered LabTeks (Thermo Fisher Scientific) and the next day transfected with 1 μg of total DNA using TurboFect transfection reagent (0.8 μL/well; Thermo Fisher Scientific). This DNA comprised equal amounts of (1) an AAV helper encoding Rep of AAV2 and the indicated AAP-deficient capsid, (2) an AAP expression vector or stuffer DNA, and (3) an AAV vector plasmid encoding Renilla and Firefly luciferase. Twenty hours later, the cells were fixed with 4% paraformaldehyde in PBS for 30 min and permeabilized by incubation for 10 min with 0.1% Triton. Next, the samples were blocked for 60 min in blocking buffer (5% fetal calf serum [Sigma–Aldrich] in PBS), before they were stained with 120 μL of primary antibody (undiluted hybridoma supernatants) detecting assembled AAV capsids (A20, ADK8, and ADK9; kind gift from J. Kleinschmidt, German Cancer Research Center, Heidelberg, Germany) or free capsid proteins (B1). 30 An HA-tag was fused to AAP in the AAP expression vectors and detected using an anti-HA antibody (GTX29110; GeneTex). Following 1–2 h of incubation with the antibodies at room temperature, the samples were washed twice with blocking buffer for 10 min each and then incubated with secondary antibodies (1:700 diluted in blocking buffer) for 1 h at 37°C. B1, A20, or ADK were detected using an anti-mouse AF488-labeled secondary antibody, while an anti-rabbit AF647-labeled secondary antibody (both Life Technologies) was used to detect HA-tagged AAP. In addition, Hoechst (Molecular Probes, Thermo Fisher Scientific) was added in a 1:3,000 dilution to stain nuclei. Finally, the samples were washed with 1 × PBS and stored in the dark at 4°C in 200 μL of PBS per well.
Microscopy images were acquired with an Olympus ScanR IX-81 inverted fluorescence screening microscope and processed with the Konstanz Information Miner (KNIME;
Statistics and sequence analyses
Trans-complementation assays were analyzed by comparing the samples relative to the cognate wild-type controls. Usually, at least three independent experiments were performed, and the means are shown with standard deviation (SD). Statistical significance required p-values <0.05, determined by a one-way analysis of variance with Bonferroni's multiple comparisons test. In selected figures, transduction efficiencies are depicted as heatmaps, also showing the means of three independent experiments.
For sequence analyses, complete chimeric cap genes were sequenced and aligned to the respective viral parents using VectorNTI's AlignX software tool (Life Technologies). Using an in-house online tool, Salanto, 17,25 chimeric sequence stretches were then assigned to the best-matching parental sequence and colored accordingly. Details of this tool and the assignment workflow will be reported elsewhere (Herrmann et al., manuscript in preparation). Chimeric AAP sequences were extracted from the corresponding cap sequences.
Results and Discussion
AAP from the prototype shuffled variant AAVDJ can broadly rescue AAP-deficient AAV serotypes
In a first experiment, the ability of AAP from AAVDJ (from hereon called AAPDJ), a chimeric capsid previously created by the authors through DNA family shuffling of eight different AAV serotypes and primarily composed of fragments from AAV2, 8 and 9, 22 to rescue an AAP knockout in its three parental AAV isolates was evaluated. Therefore, the AAP open reading frame from AAVDJ (Fig. 1A and Supplementary Table S2) was cloned into the recently reported AAP expression plasmid, 17 and then the resulting construct was used to trans-complement AAP-deficient helper plasmids (called AAVXmut; with X denoting the serotype) of serotypes 2, 8, or 9 in small-scale AAV-GFP vector productions (workflow in Fig. 1B). Next, fresh cells were transduced with the different vector particle–containing cell culture supernatants (“crude lysates”), and expression of vector-encoded GFP was measured by fluorescent-activated cell sorting as surrogate readout for successful trans-complementation, akin to a recent study by the authors. 17 It was found that AAPDJ can efficiently rescue all three AAP knockout mutants to a similar degree as each cognate AAP (Fig. 1C, compare the brown bars [AAPDJ] to the others), whereas AAP8 or AAP9 only partially trans-complemented AAV2mut (AAP knockout AAV2 construct), independently confirming the prior results. 17

Trans-complementation abilities of chimeric assembly-activating protein (AAP) from AAVDJ (AAPDJ).
Next, this experiment was expanded to helper plasmids from seven other serotypes likewise carrying an AAP knockout, and it was found that AAPDJ could also efficiently rescue these (Fig. 1D). The only exceptions were AAV4 and AAV5 whose rescue was impaired, congruent with other data by others and us showing that these two serotypes prefer their own AAP. 16,17 Reminiscent of earlier findings with AAP2, 17 AAPDJ also enhanced AAV3 vector production from the AAP-deficient AAV3mut helper, even beyond 100% (i.e., the value obtained with a wild-type AAV3 helper), while AAP8 and AAP9 did not.
Interestingly, despite their differing abilities to rescue the AAP2 knockout (Fig. 1C and D), all four AAP variants, including the one from AAVDJ, frequently co-localized with AAV2 capsids in cells transfected with the AAV2mut helper and an AAP expression construct, as detected by specific antibody staining (Fig. 1E, left and middle columns, and Supplementary Fig. S1). In contrast, AAPDJ showed less co-staining with AAV8 or AAV9 capsids (Fig. 1E, right column, and Supplementary Fig. S1), although it rescued AAV8mut and AAV9mut very effectively (Fig. 1C and D). Only limited AAP/capsid co-staining was observed for the other combinations of the AAV8 or AAV9 capsids with AAP from serotypes 2, 8, or 9. A notable exception was AAP2, which was frequently co-detected with AAV9 capsids in the same cell (Supplementary Fig. S1, combination AAP2 with AAV9mut).
Taken together, these data obtained with a specific chimeric AAP imply that AAP recombination, which inevitably occurs during DNA family shuffling of full AAV capsid genes, is not necessarily detrimental. Instead, it can even breed AAP chimeras that exhibit novel and interesting properties compared to the natural AAPs from the input viruses, as evidenced here by the distinct co-staining patterns and rescue efficiencies of AAPDJ. Concurrently, these results support and expand the recent conclusions that AAP functionality is uncoupled from its extent of co-localization with free VP proteins or assembled capsids. 16,17
AAP shuffling during molecular AAV capsid evolution generally results in functional variants
Next, the study aimed to solidify these results further with a larger cohort of chimeric AAPs. To this end, 60 new expression plasmids encoding hybrid AAPs were cloned that were isolated from five shuffled AAV capsid libraries with different input serotypes and complexities (12 arbitrarily selected AAP clones per library; Fig. 2A and B). The 1–10O library contained variants of the AAV4 and AAV5 capsid genes that were recently sequence-optimized (“O”) to increase the homology with AAV2, to in turn facilitate DNA family shuffling (Herrmann et al., manuscript in preparation). The composition of all 60 clones is schematically depicted in Supplementary Fig. S2, and full DNA sequences are shown in Supplementary Table S2. First, these clones were tested in trans-complementation assays with the AAP-deficient AAV2mut construct (same workflow as in Fig. 1B). Strikingly, of the 60 chimeric AAPs, 54 resulted in infectious, GFP-expressing vector particles and were thus able to rescue the AAV2/AAP mutant, albeit to varying extents (Fig. 2C; as cutoff successful rescue was defined as at least 5% of GFP-expressing cells relative to the wild-type AAV2 helper [lower dotted line]). This includes intermediate performers (5–50% rescue, gray area in Fig. 2C) and high performers (>50% rescue). The remaining six AAP chimeras with <5% rescue efficiency in this assay (triangles in Fig. 2C) were initially classified as non-functional.
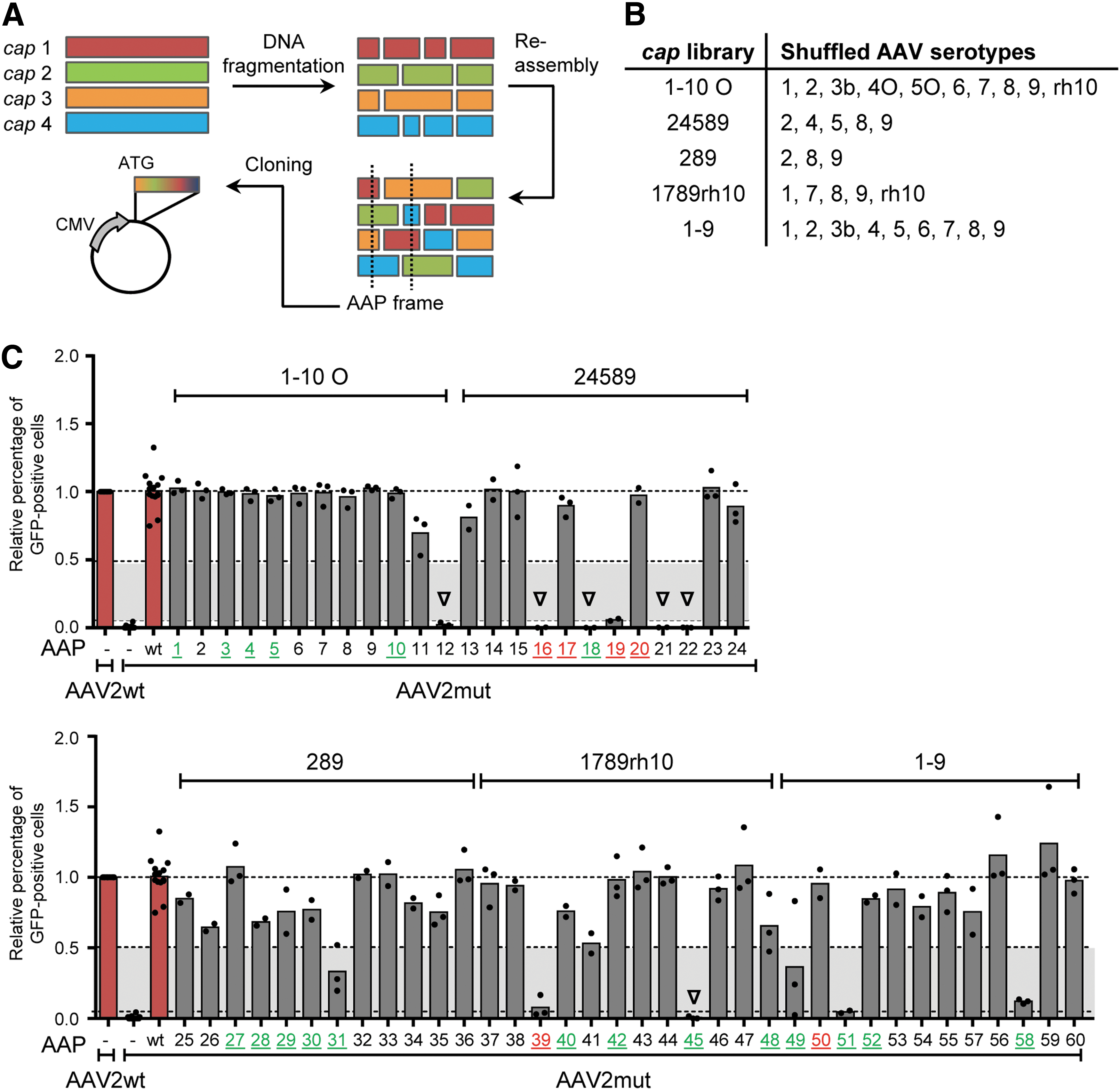
Analysis of 60 chimeric AAP variants randomly selected from five shuffled AAV capsid libraries (12 AAPs per library).
Of note, the data in Fig. 1C and D as well as those in the recent study 17 show that AAPs from different wild-type AAV serotypes vary in their ability to rescue an AAP knockout in AAV2. Thus, all six presumably non-functional variants from Fig. 2C were retested with further AAP-deficient AAV helper constructs, selected based on the composition of the library from which they originated (e.g., AAP#16 from the AAV24589 library was tested with AAV4, 5, 8, and 9). Indeed, all six chimeric AAPs initially classified as defective turned out to be functional with AAV/AAP mutants other than AAV2mut because they consistently rescued at least one parental serotype by >5% (Table 1). Furthermore, three AAP chimeras that showed around 5% activity with the AAP-deficient AAV2 helper (AAP#19, 39, and 51 in Fig. 2C) were able to rescue other parental AAV serotypes by up to 40.8% (AAP#39 and AAV8) or even 87.7% (AAP#19 and AAV9). In total, including this expanded analysis, it was found that 60 of the tested 60 AAPs (i.e., 100%) are functional in combination with at least one AAP-depleted parental serotype.
Results of trans-complementation assays using non-functional (with AAV2mut) AAPs and intermediate performers to rescue other AAP-depleted helper plasmids
Crude cell lysates were tested in MCF7 (AAV4) or SF539 cells (AAV1, 3, 5–9, and rh10; n ≥ 2). Positive cells were determined by flow cytometry and values (shown plus SD) were normalized to the respective AAV wild-type construct (always set to 100%).
Numbers in this column refer to the labels of the chimeric AAP clones, not to AAV serotypes.
These three clones were included, since they barely crossed the 5% assay cutoff in Fig. 2C.
High performers (≥50% activity).
Intermediate performers (5–50%).
Non-functional AAPs (<5%).
AAV, adeno-associated virus; AAP, assembly-activating protein; mut, mutation; SD, standard deviation.
Intriguingly, most AAP chimeras (five out of nine; 55.5%) with reduced or no functionality in Fig. 2C came from libraries containing the evolutionarily distinct capsid genes of AAV4 and AAV5, whereas the remaining four were distributed across the other four libraries. Also interesting is that in five of these AAP chimeras (#12, 16, 18, 21, 22), the N terminus was derived from AAV4, AAV5, or AAV5O (Supplementary Fig. S2). This implies that the N-terminal region of AAP may be especially relevant for its function and thus determine its interchangeability, in line with a previous hypothesis by the Nakai laboratory and the authors, 16,17 and further supported by most recent data from the Asokan and Vandenberghe labs. 31,32
AAP neither rate limits nor enhances the production of individual AAV capsids or capsid libraries
The data thus far suggested that chimeric AAPs retain the capability to rescue AAP knockouts, to extents depending on the AAP composition and the capsid serotype. The latter relationship prompted the question whether overexpression of certain AAP variants or combinations thereof during production could even improve yields of recombinant AAV particles or capsid libraries. To address this question, the following three complementary experiments were performed.
First, wild-type AAV helpers of 10 different serotypes (AAV1–9, rh10) were supplemented with a cocktail of all 10 corresponding AAP variants during GFP vector production, and then the resulting crude lysates were titrated on different cell lines (Fig. 3A–C and Supplementary Fig. S3). These included MCF7 cells, which had previously been found to be permissive for AAV4 transduction. 17 Moreover, different vector dilutions were tested for each cell type and for each AAV-AAP combination in order to avoid saturation of the GFP signals. As exemplified in Fig. 3B and C, the ratios of transduction efficiencies (i.e., percentages of GFP-expressing cells) obtained with stocks produced in the presence versus the absence of the AAP cocktail always approximated 1 (between 0.75 and 1.25). These consistent results suggest neither that AAP is rate limiting during AAV vector production, nor that its overexpression increases the yields of functional particles, at least for the 10 tested wild-type serotypes.

Analysis of the functionality of 25 chimeric capsids randomly selected from five shuffled AAV capsid libraries (five capsids per library).
The second experiment asked whether the outcome would be different for chimeric capsids. Therefore, again, GFP-expressing vectors were produced, but now 25 capsid clones were used from the collection of 60 chimeras in Fig. 2C (five clones were arbitrarily selected per library; sequences are found in Supplementary Table S3). During vector production, a cocktail of AAPs was additionally overexpressed from the parental serotypes that were present in the original libraries. As seen in Fig. 3D, most of the 25 capsid variants reached infectivities >5% (19/25; 76%) in one or more of the three cell lines. An exception were four out of five clones originating from the AAV24589 library (#16, 17, 19, and 20), which failed to yield detectable transduction in any of the three cell lines, although in silico translation revealed no abnormalities or stop codons in the corresponding cap open reading frames. Curiously, among the six chimeric capsids that gave no transduction (#16, 17, 19, 20, 39, and 50; all marked in red in Fig. 2C), three of the cognate AAPs (#17, 20, and 50) were capable of fully rescuing the AAP-deficient AAV2 mutant in the prior assay (Fig. 2C).
Of the 19 functional capsids, most yielded very comparable GFP transduction when produced with or without ectopic AAP overexpression (Fig. 3D and E), congruent with the data for the wild-type capsids (see above, Fig. 3B and C). Still, a striking exception was again noted, namely transduction with capsid chimera #18, which was boosted up to 10-fold when the AAP24589 cocktail was added during its production. Intriguingly, the corresponding AAP clone #18 had failed to rescue AAP-deficient AAV2 (Fig. 2C), as well as its other parents AAV5, AAV8, and AAV9, while it partially rescued AAV4 (22%; Table 1). It was thus interesting to determine how much each of the five AAPs (AAP2, 4, 5, 8, and 9) in the cocktail contributed to the observed improvement for this capsid chimera. Hence, the trans-complementation experiment was repeated, but now the single AAPs were added instead of a mixture during production. Surprisingly, this showed that AAP2 and especially AAP5 support this capsid clone, whereas AAP4 and AAP8 had a minor effect, and AAP9 had no effect at all. This was consistently noted on the levels of cell transduction (Supplementary Fig. S4A), assembly, and DNA encapsidation (Supplementary Fig. S4B). Taken together, and also combined with the fact that capsid #18 showed a marked tropism for SF539 human glioblastoma cells (Fig. 3D and Supplementary Fig. S4), this makes this peculiar chimeric capsid/AAP pair highly interesting for further investigations into AAV/AAP biology.
From all these data, it is concluded that although not all capsids in unselected shuffled AAV capsid libraries (especially those with a high proportion of AAV4 or AAV5) produce functional particles, this defect is not necessarily linked to a malfunction of the cognate chimeric AAP. Instead, it may rather reflect inherent deficiencies of the affected capsid to assemble into transduction-competent virions that cannot be overcome by AAP. In fact, this is the most likely explanation for clones #17 and 20 based on results from independent large-scale production and titration experiments (data not shown), implying a failure of these two capsids to assemble and package DNA, regardless of AAP. Of course, the possibility can also not be excluded that the putatively non-functional capsids have evolved entirely novel tropisms not present in any of the parental serotypes and thus perform well in cell lines other than the three that were tested here. One example is capsid #19, which produced and packaged well in the aforementioned large-scale experiment (data not shown), implying that it may transduce cell types different from the present selection.
Third, to confirm even further the findings that (1) natural or chimeric AAPs are largely interchangeable and (2) AAP does not limit the yields of AAP-expressing AAV variants, a set of shuffled libraries were produced in the presence or absence of a co-transfected mixture of all parental wild-type AAPs. Titration of iodixanol gradient-purified viral library stocks by quantitative PCR revealed that AAP overexpression during library production had no impact on particle titers (Table 2). This implies that either the proportion of low- or non-functional AAPs in typical shuffled libraries is too small to impair overall virus yields, and/or that the remaining functional AAP chimeras can efficiently substitute for the defective counterparts. Regardless of exact mechanism, it is concluded that the production of complex AAV capsid libraries is probably not restricted by the sporadic generation of impaired AAP chimeras in the shuffled DNA pools. In turn, this alleviates the need to overexpress AAP ectopically during library production as a potential countermeasure (see below for a more detailed model and discussion).
Genomic titers of three different AAV cap libraries produced with or without supplementation of an AAP mixture containing the respective parental serotypes
Further evidence that selection of shuffled AAV capsid libraries, while primarily acting on the capsid level, may also breed unique AAPs
Thus far, all the assays in this study and the prior study 17 had employed either naturally occurring AAP variants or chimeras that had been isolated from unselected shuffled libraries, with the exception of AAP from AAVDJ (i.e., a capsid that had been selected under stringent conditions). 22 Notably, as shown above, AAPDJ differs from the AAPs from two of its three parental AAV serotypes in that it effectively rescued AAP knockouts in all of the wild types from which it was derived (like AAP2, but unlike AAP8 and AAP9). This raised the question whether AAP from AAVDJ is a unique case of a broadly active chimeric AAP, or whether AAPs frequently acquire such properties in the process of directed molecular evolution by co-evolving with their cognate capsids during iterative AAV library selection.
To begin to address this complex question, 12 chimeric AAPs (called S1–S12) were subcloned from a library composed of 10 AAV serotypes (AAV1–3, 4O, 5O, 6–9, and rh10) that had been iteratively selected four times in vivo in mouse livers as part of a different project (Herrmann et al., manuscript in preparation). This selection had resulted in an accumulation of sequences from 4 of the 10 parental serotypes—AAV1, 6, 7, and rh10—in the capsid and AAP open reading frames (Supplementary Fig. S5; the full 12 sequences are shown in Supplementary Table S2). Therefore, the focus was on this serotype subset, and the ability of the 12 chimeric AAPs to rescue the four corresponding AAP knockouts was studied (same workflow as in Fig. 1B).
Remarkably, as shown in Fig. 4, all AAPs were indeed able to trans-complement the AAP-deficient helper plasmids effectively by at least 5% (lower dotted line). With the exception of AAP S2, the remaining 11 chimeric AAPs fully rescued the AAP1mut and AAP6mut helpers, akin to wild-type AAP1 and AAP6, respectively. Particularly notable is that 6 of the 12 AAPs (S1, S3, S4, S7, S8, and S10) were also capable of potently rescuing the AAV7 and AAVrh10 mutants by at least 50% and up to 100% (white area between the two upper dotted lines in Fig. 4). Considering the prior findings that these two serotypes—AAV7 and especially AAVrh10—exhibit selectivity for or against specific wild-type AAP variants in trans-complementation assays, 17 this result implies that library selection had indeed enriched AAPs with distinct and enhanced properties. No chimeric AAP outperformed the corresponding four wild-type AAPs that were tested in parallel (left bars in both panels), further supporting the model that AAP is essential but not rate limiting. Hence, one may also not expect the evolution of a “super-AAP.”
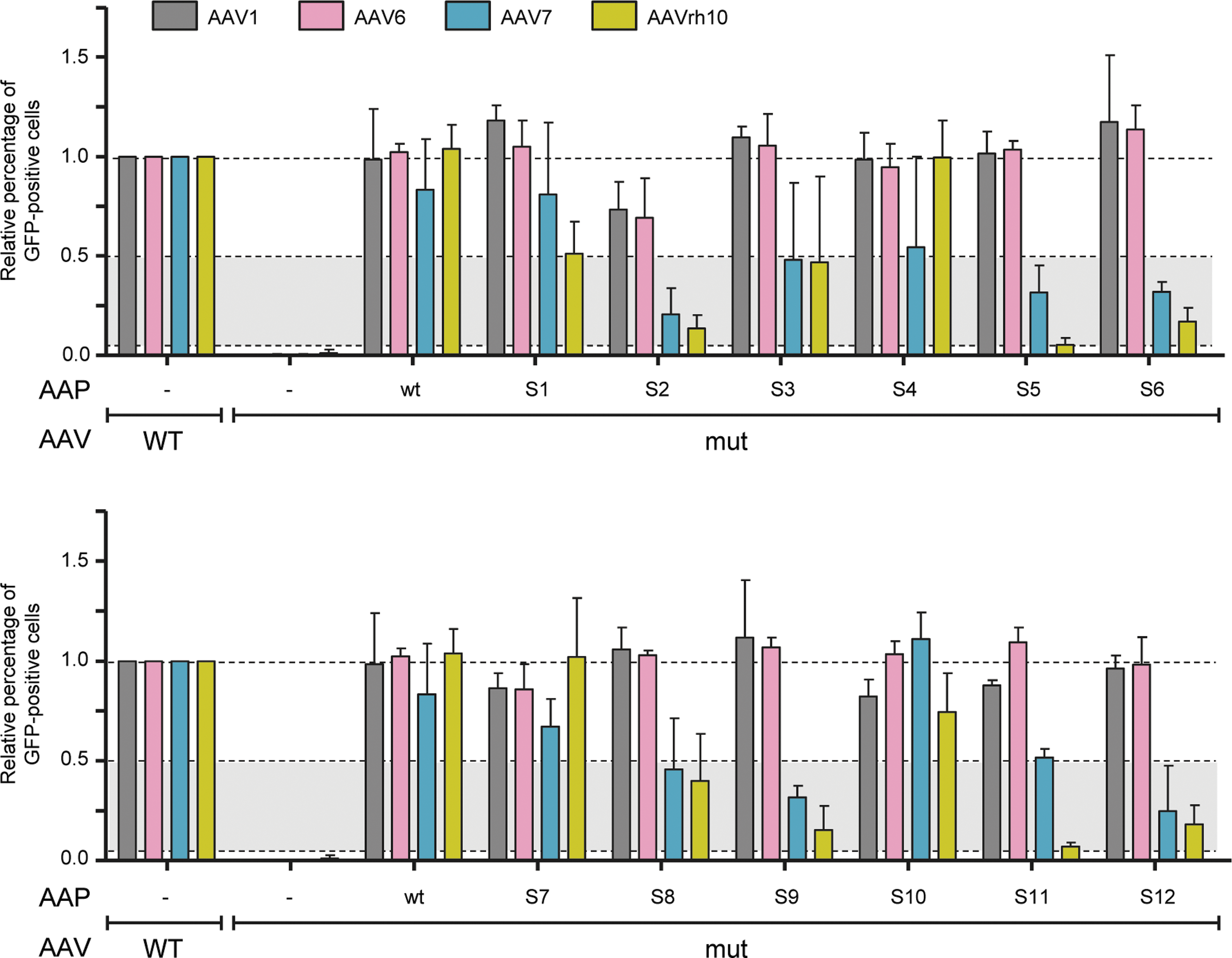
Analysis of the functionality of 12 chimeric AAPs randomly isolated from an iteratively selected AAV1-rh10O library (comprising the sequence-optimized AAV4O and AAV5O cap genes). The four AAV serotypes depicted on top were chosen for this analysis, as they displayed the highest homology to the 12 chimeric AAPs. Accordingly, AAP knockout mutants of these four serotypes were separately trans-complemented with the 12 AAPs, using the workflow in Fig. 1B. Shown are transduction efficiencies (means with SD of at least three independent transfections) of the resulting crude lysates determined in HEK293T cells as amounts of GFP-positive cells. A dilution of the crude lysates yielding transduction rates between 5% and 95% was chosen to illustrate the effect of AAP addition during production relative to the stuffer control. This ratio was set to 1.0 for each of the AAP-competent wt AAVs that were produced as positive controls (left four bars in each graph). As further controls, the four AAP knockout mutants were trans-complemented with the cognate wt AAPs.
Collectively, from these results and the data with AAPDJ (see above), it is cautiously concluded that directed molecular evolution of AAV acts not only on the capsid level, but also on AAP. In other words, positive and/or negative pressure forces the accumulation of specific capsids that fulfill the selection criteria, but concurrently also enriches unique AAP variants that have adapted to their capsid (while showing cross-reactivity), akin to the situation with wild-type AAV capsids and AAPs. Albeit this conclusion that AAP co-evolves together with AAV cap would be congruent with that in an intriguing study from the Nakai laboratory, 18 it is acknowledged that it will require additional experimental validation with more AAPs from different libraries and further selection schemes. This should ultimately help to answer an intriguing chicken or egg question, that is, whether AAV evolution (natural or directed) predominantly acts on the capsid with AAP tagging along, or whether AAP is a key driver itself that partially governs and restricts capsid evolution.
Implications for the future use of DNA family shuffling technology for AAV capsid diversification
The 2010 discovery of the alternative AAP open reading frame sparked concern that the use of DNA family shuffling technology for AAV diversification may come at a hefty price, namely, an inadvertent impact on AAP sequence, structure, and functionality. 12 While these concerns have triggered the present study, it is a relief that the sum of the data allows it to be concluded that AAP is not rate limiting for AAV capsid evolution, at least not during the first step of library production. Most likely, this is due to a combination of two effects, as illustrated in the model in Fig. 5A and B: (1) the majority of AAP chimeras that are inevitably produced by cap shuffling are functional to begin with (AAVC3 in Fig. 5B), and (2) capsids whose AAP is disrupted are efficiently trans-complemented by others in the pool (AAVC1/C2 in Fig. 5B). A good example illustrating the latter scenario is capsid/AAP #18 from Figs. 3 and 4 because (1) its own AAP seems incapable of supporting assembly of the cognate capsid, (2) this assembly is, however, rescued by various heterologous AAPs supplied in trans, and (3) once rescued and assembled, this capsid robustly transduces specific cells.

Model for the role and potential rate-limiting character of AAP during AAV capsid shuffling.
In contrast, the situation may become more complex during subsequent selection of shuffled libraries in cells, when numbers of capsids and hence AAP variants decrease. In extreme cases, the proportion of functional AAP chimeras that can rescue other capsids with a defunct AAP may fall below a critical threshold. It is thus conceivable that a specific capsid fulfills all exogenously applied positive and/or negative selection criteria, but will be eliminated from the pool due to depletion of “helper” AAPs from other capsids and due to the resulting assembly deficiency (AAVC1/C2 in Fig. 5B). Again, the example here is capsid/AAP #18, which would ultimately be lost from a library as soon as all supporting heterologous AAPs would be eliminated too, regardless of the effectivity of capsid #18 in the given target cell. Under such circumstances, the need for capsid/AAP compatibility would turn into an inherent counter-selection pressure and increase the stringency of molecular evolution. Thereby, as already indicated above, AAP itself would become a direct target and concurrently a driver of molecular AAV evolution, as its functionality would supersede that of the corresponding capsid.
In this context, an intriguing very recent study by the Vandenberghe laboratory is noted that examined the natural co-evolution of AAP and AAV capsid proteins, and that perfectly complements the present work. 31 In this study, Maurer et al. postulated that AAP may have evolved in order to grant AAV a higher degree of freedom to mutate the capsid and thus to increase its fitness in a competitive environment. This conclusion—that AAP relaxes structural constraints on the AAV assembly process while enhancing the capsid's freedom to evolve—further fuels the present model that directed AAV evolution is prone to act concurrently on the levels of both AAP and capsid.
Furthermore, capsids may emerge during library production that encode functional AAPs, but whose AAP interaction site is perturbed because of unfavorable VP domain combinations (AAVC4 in Fig. 5B). This site is probably located in the VP C terminus, which forms the capsid's twofold symmetry axis, as mutations in this region abolish VP–AAP interaction and capsid formation. 13 Besides, non-functional particles may also emerge independent of AAP due to frame shifts (especially when libraries include capsids such as AAV4 and AAV5, which are rather divergent compared to most other AAV serotypes), sterical incompatibilities of shuffled domains, or other reasons.
While these AAP-independent adverse events cannot be avoided, it should be rewarding to contemplate solutions to AAP-dependent side effects. For instance, it may pay off to supply different AAP variants during production of secondary capsid libraries (by co-transfection of respective AAP expression plasmids), especially in later stages of selection, to counterbalance depletion of functional AAPs in the sequence pool. Of note, there is ample literature evidencing that stringent selection of shuffled AAV capsid libraries can succeed without such measures, including AAVDJ, which encodes an active AAP, or AAVLK03 whose AAP is derived from AAV3. 22,33 Moreover, the present data with the 12 chimeric AAPs in Fig. 4 additionally show that natural sifting during virus production and proper selection are capable of enriching functional AAV particles with intact AAPs from shuffled capsid libraries. Of course, it is not possible to know whether potent capsids have been lost during iterative library selection in the past for the reason illustrated with clones C1/C2 (and C4) in Fig. 5B and exemplified with capsid/AAP #18 in Figs. 3 and 4. In this respect, selection strategies that depend on helpervirus-mediated amplification of synthetic AAV capsids in the target cell or tissue may be unfavorable, as they increase the risk of losing interesting capsids in case these encode a weak AAP and thus cannot assemble and propagate efficiently. In direct comparison, this risk is much smaller with strategies that rely on PCR-mediated rescue of AAV capsid and hence AAP genes from transduced cells, followed by their re-cloning and production of a secondary library, as these offer the aforementioned option to spike in a cocktail of heterologous “helper” AAPs to foster assembly of all capsid variants in the pool.
Finally, it is noted that DNA family shuffling is also in general a powerful technology to study fundamental structure–function relationships in proteins. For instance, the Asokan laboratory has recently used this approach to dissect determinants in AAVrh10 that govern its trafficking across the blood–brain barrier. 34 Moreover, this technique has previously been exploited by the authors to shuffle the human RNAi proteins Argonaute 1–4, and to identify motifs in Argonaute 2/3 that are critical for Slicer function. 25 Similarly, studies have now started in the authors' laboratory to harness AAP shuffling to unravel domains that are necessary for functionality and for interaction with VP proteins or capsids. Such attempts should largely benefit from juxtaposition with other directed molecular evolution strategies, such as the one reported by the Nakai group that permits separate diversification and analysis of VP and AAP sequence and function. 18 Also, recent data from the Asokan and Vandenberghe laboratories imply that other technologies, such as ancestral capsid reconstruction or AAP mutation and domain swapping, are equally powerful and informative tools to dissect AAP biology further in the future. 31, 32 Until then, the present findings, including the observation that AAP chimeras with an N terminus from AAV4 or AAV5 failed to trans-complement AAV2mut effectively, already exemplify the great power and promise of DNA shuffling for AAV research beyond molecular evolution of new gene therapy vectors.
Footnotes
Acknowledgments
A.-K.H. and D.G. gratefully acknowledge funding by the German Research Foundation (DFG; Collaborative Research Center SFB1129, project TP2) and the Cluster of Excellence CellNetworks (DFG; EXC81). D.G. is thankful for further support by the Transregional DFG Collaborative Research Center TRR179 (project TP18). D.G. and K.B. are grateful for additional funding from the German Center for Infection Research (DZIF; TTU-HIV 04.803). Finally, we acknowledge excellent support by Micha Rosenkranz during the cloning of selected AAP and capsid variants.
Author Disclosure
No competing financial interests exist for any of the authors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.