
Abstract
In recent years, new immunotherapies have greatly contributed to the success of cancer treatment. However, cancer cell–specific antigens limit the effectiveness of these immunotherapies. Identification of neoantigens through tumor sequencing and bioinformatics may help advance research into personalized cancer vaccines. This study evaluated a new immunotherapy by developing a cancer cell–specific gene expression technique that uses a nuclear factor-kappa B (NF-κB)-activating gene expression vector to express a protein or peptide on the surface of cancer cells. These proteins or peptides function as an artificial neoantigen to stimulate the immune system to kill cancer cells. The study demonstrated that NF-κB RelA was widely over-activated only in cancer cells. It also showed that a NF-κB-activating gene expression (Nage) vector, which consisted of a NF-κB-specific promoter (DMP) prepared by fusing a NF-κB decoy sequence to a minimal promoter and a downstream effector gene, could specifically express a protein or peptide on the surface of various cancer cells. By packaging the Nage vector in an adeno-associated virus, in vivo tumors can be significantly inhibited or even eradicated via intravenously injected recombinant adeno-associated viruses.
Introduction
C
Immunotherapies for cancer have developed rapidly in recent years. In fact, the U.S. Food and Drug Administration (FDA) has approved six immunological checkpoint inhibitors (CTLA-4, PD-1, and PD-L1 antibodies) and two chimeric antigen receptor T-cell (CAR-T) immunotherapies for various cancers. 1 –3 However, these immunotherapies are limited by their intrinsic shortcomings. Immunological checkpoint inhibitors have low response rates (average 20–30%) across tumor types. In addition, CAR-Ts are associated with serious side effects, including cytokine-release syndrome, neurotoxicity, and hydrocephalus. 4,5 These immunotherapies may also cause organ inflammation and fatal myocarditis. 2,6,7
CAR-T therapies are restricted to hematological malignancies, including leukemia and lymphoma, and cannot be used to treat solid tumors due to their limited utilizable targets. 8 The most widely used target in CAR-T therapies is CD19, which is highly restricted to the B-cell lineage and can therefore lead to profound B-cell aplasia. 9 Although CD19 displays frequent and high-level expression in B-cell malignancies, it is also required for normal B-cell development in humans and therefore may cause side effects due to off-target effects on healthy B cells. 4,10 CAR-T therapy use is also prohibited by its high cost.
The limitations of current immunotherapies can be overcome by identifying neoantigens through tumor sequencing and bioinformatics to be develop personalized cancer vaccines or to produce therapies without side effects that can be used to treat solid tumors. 11,12 Despite scarce targets and neoantigen identification challenges, it was proposed that an artificial neoantigen could be created on the cancer cell surface to induce the immune system to attack cancer cells in vivo.
Nuclear factor-kappa B (NF-κB) is a sequence-specific DNA-binding eukaryotic transcription factor that plays a critical role in the immune response. 13 –15 The NF-κB family includes five members that can form heterodimers and homodimers to bind the consensus DNA sequences known as κB in genomes. 16 NF-κB is an inducible transcription factor that plays multiple roles in various biological processes upon activation via the NF-κB signaling pathway. 13 NF-κB realizes its physiological and pathological functions by binding and regulating expression of its target genes. 17,18 NF-κB is an important inflammation transcription factor because it regulates the expression of inflammatory mediators, and important inflammatory mediators, such as tumor necrosis factor alpha and interleukin 6, are the direct target genes of NF-κB. 19
NF-κB is widely activated in nearly all types of tumor cells, and it mainly plays an anti-apoptotic role in cancers. 20 –22 Bcl-2 and GADDβ4 are anti-apoptotic proteins, which are overexpressed by a variety of tumors. 23 The genes coding these two proteins are the direct target genes of NF-κB, and over-activation of NF-κB in cancer cells results in their upregulation. 24 NF-κB is constitutively activated in inflammation, immune diseases, and cancers, and the NF-κB pathway is involved in the pathogenesis and proliferation of tumors via the regulation of cell proliferation and apoptosis, promotion of angiogenesis, stimulation of invasion/metastasis, and sustainment of cancer stemness. 25 –34 NF-κB is therefore a key pharmaceutical target, and countless NF-κB inhibitors have been developed as potential candidate drugs. 18,35,36 However, these inhibitors have rarely made it to the clinical stage due to their side effects, as uncontrolled over-inhibition of its activity damages normal physiological functions. 37 –39
To overcome the limitations of current NF-κB inhibitors, a new type of NF-κB inhibitor was recently developed by combining a NF-κB-targeting decoy and microRNA, which can sense the intracellular NF-κB activity and express an artificially designed microRNA that targets NF-κB RelA. 40 In this NF-κB inhibitor, a NF-κB decoy sequence fused with a minimal promoter is used as a NF-κB responsive promoter (DMP) to control the expression of microRNA. This new design can inhibit NF-κB activity in a NF-κB self-controlling manner, which results in apoptosis of cancer cell HepG2 but not normal liver cell HL7702. 40 Therefore, DMP may be a NF-κB-specific promoter that could be employed to develop a cancer cell–specific gene expression tool to express an artificial neoantigen on the cancer cell surface, which could lead to a new gene therapy–based immunotherapy.
This study first detected the expression of NF-κB RelA in various cancer cells from different cancers and normal cells. It was found that NF-κB RelA was specifically expressed in all detected cancer cells but not in normal cells. Based on this observation, a Nage vector was constructed that expresses a reporter gene enhanced green fluorescent protein (eGFP) under the control of DMP. Transfection of this Nage vector demonstrated that the reporter gene was activated in all cancer cells but not normal cells. Then, a new Nage vector was constructed that expresses a streptavidin-binding peptide (SBP) under the control of DMP. The transfection of this Nage vector demonstrated that the SBP was displayed on the surface of all cancer cells but not normal cells. Finally, two new Nage vectors were constructed that express the surface antigen of the hepatitis B virus (HBsAg) and calreticulin (CRT) under the control of DMP. The three Nage vectors were packaged into adeno-associated virus (AAV), and xenographed tumors in mice were treated with three recombinant AAVs (rAAVs). It was found that rAAV administration could significantly inhibit or even eradicate tumors in vivo.
Methods
Cells and culture
Cells used in this study included HEK-293T (human fetal kidney cells), HepG2 (human liver cancer cells), A549 (human lung cancer cells), HT-29 (human colon cancer cells), HeLa (human cervical cancer cells), SKOV3 (human ovarian cancer cells), PANC-1 (human pancreatic cancer cells), MDA-MB-453 (human breast cancer cells), Hepa1-6 (mouse hepatoma cells), RAW264.7 (mouse macrophages), B16F10 (mouse melanoma cells), HL7702 (human normal hepatocytes), and MRC5 (human embryonic fibroblasts). All cells were purchased from the China Center for Type Culture Collection (Shanghai, China). Hepa1-6, HEK-293T, HepG2, HeLa, PANC-1, MDA-MB-453, RAW264.7, B16F10, and MRC-5 cells were cultured with DEME. A549, HT-29, SKOV-3, MRC-5, and HL7702 cells were cultured with RPMI 1640 medium. Both media were supplemented with 10% fetal bovine serum (HyClone), 100 IU/mL penicillin, and 100 μg/mL streptomycin. Cells were incubated at 37°C in a humidified incubator containing 5% CO2.
Plasmid construction
The plasmid vector pDMP-Display-SBP was constructed based on pDisplay (Invitrogen). The expression cassette of pDisplay can display a target protein or peptide on the cell surface using the N-terminus of a rat kappa-chain leader sequence and the transmembrane domain of platelet-derived growth factor receptor (PDGFR). In pDMP-Display-SBP, a DMP promoter replaced the cytomegalovirus (CMV) promoter in pDisplay. The plasmid pDMP-Display-SBP contains a DMP sequence and an SBP coding sequence (Supplementary Data; Supplementary Data are available online at
To construct the pAAV vectors, the pAAV-DMP plasmid was first constructed by replacing the CMV promoter in pAAV-MCS (Stratagene) with the DMP promoter. The pAAV-DMP-Display-HBsAg/CRT/SBP vectors were then constructed by cloning the Display-HBsAg/CRT/SBP coding sequences into pAAV-DMP by using BglII (upstream) and PstI (downstream) restriction sites (Supplementary Data). The Display-HBsAg/CRT/SBP coding sequences were amplified by PCR from pDMP-Display-HBsAg/CRT/SBP. The pAAV-eGFP was constructed by cloning the eGFP coding sequence into pAAV-DMP.
Analysis of NF-κB RelA/p65 expression in cells
Total RNA was extracted from cells using Trizol (Invitrogen). Complementary DNA (cDNA) was prepared from 50 ng of total RNA using 5 × PrimeScript RT Master Mix (Takara) according to the manufacturer's instructions. NF-κB RelA/p65 and GAPDH expression was detected by quantitative PCR (qPCR) using the primers RelA-F/R and GAPDH-F1/R1 (Supplementary Table S3). The 10 μL qPCR reaction contained 1 × Fast SYBR Green Master Mix (ABI), 0.2 μM forward and reverse primers, and 1 μL cDNA. The qPCR program was performed at 95°C for 10 min with 45 cycles of 95°C for 15 s and 60°C for 1 min. A melting curve was then constructed to monitor PCR amplification specificity. PCR programs were run with a real-time PCR machine, StepOne plus (ABI). Data analysis was performed using Applied Biosystems StepOne v2.3, and Ct values were normalized with that of GAPDH. Relative mRNA expression levels were calculated as relative quantity (RQ) according to the following equation: RQ = 2–ΔΔCt.
Evaluation of DMP promoter with reporting eGFP
Cells were seeded onto 24-well plates (1 × 105 cells/well) and cultivated for 12 h. Cells were then transfected with 500 ng of pDMP-eGFP or pCMV-eGFP using Lipofectamine 2000 according to the manufacturer's instructions. Following transfection, cells were cultivated for 20 h and then imaged using a fluorescence microscope (Olympus IX51) equipped with a cold CCD (DP71; Olympus). Fluorescence of cells was also quantified using flow cytometry (Accuri; BD Biosciences).
Evaluation of DMP promoter with displayed SBP
For FITC-streptavidin staining, cells were seeded onto 12-well plates (2 × 105 cells/well) and cultivated for 12 h. Cells were then transfected with pDisplay-DMP-SBP by using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer's instructions. Following transfection, cells were cultivated in fresh medium containing 1 μg/mL FITC-streptavidin (Sigma–Aldrich) for 20 h and then were washed twice with phosphate-buffered saline (PBS) and imaged with a laser scanning confocal microscope (Leica SP8).
To stain cells with streptavidin-IRDye 800CW, cells were seeded onto 24-well plates (1 × 105 cells/well) and cultivated for 12 h. Cells were then transfected with pDisplay-DMP-SBP using Lipofectamine 2000. Following transfection, cells were cultivated in fresh medium containing 1 μg/mL streptavidin-IRDye 800CW (Licor) for 24 h. Cells were washed twice with PBS and imaged using a near-infrared fluorescein (NIRF) imager of the Odyssey Infrared Fluorescence Imaging System (Licor). Cells were then pelleted by trypsinization and imaged again using the Odyssey Infrared Fluorescence Imaging System.
Preparation of rAAV
HEK293T cells were plated in a 75 cm2 flask (5 × 106 cells/flask) and cultivated for 12–20 h. Cells were then co-transfected with two helper plasmids (pHelper and pAAV-RC; Stratagene) and one of several pAAV plasmids, such as pAAV-MCS, pAAV-eGFP, pAAV-DMP-Display-SBP, pAAV-DMP-Display-HBsAg, or pAAV-DMP-Display-CRT. Cells were co-transfected with plasmids using Lipofectamine 2000 according to the manufacturer's instructions. Following transfection, cells were cultured with fresh medium for 72 h. Viruses were then collected and purified as previously described. 41
Titers of AAVs were analyzed by qPCR. A 20 μL qPCR reaction contained 1 × SYBR Green Real-time PCR Master Mix (Roche), 0.25 μM primers (CHS-F1/R1 and GAPDH-F1/R1; Supplementary Table S4), and 2 μL virus or twofold dilutions of a standard DNA (a 212 bp GAPDH fragment). The qPCR program included 95°C for 10 s, 45 cycles of 95°C for 15 s, and 60°C for 1 min. A melting curve was then constructed to monitor PCR amplification specificity. Data analysis was performed using Applied Biosystems StepOne v2.3. The concentration of the virus genome (vg) was calculated according to the standard curve. Quantified viruses were aliquoted and kept at −35°C for later use. The viruses obtained were referred to as AAV-SBP, AAV-HBsAg, AAV-CRT, AAV-eGFP, and AAV-MCS.
Evaluation of the virus by transfecting cells with AAV-SBP
Cells were seeded onto a 24-well plate (1 × 105 cells/well) and cultivated for 12 h. Cells were then transfected with the virus through cultivation in a medium containing AAV-SBP (5 × 1010 vg/well) and 1 μg/mL streptavidin-800CW for 24 h. Cells were washed with PBS and imaged using the Odyssey Infrared fluorescence imaging system. Cells were then pelleted by trypsinization and imaged again using the Odyssey Infrared Odyssey Infrared Fluorescence Imaging System.
Evaluation of rAAV antitumor activity
Four-week-old female BALB/c-Foxn1nu mice with an average body weight of 20 g were purchased from the Changzhou Cavens Laboratory Animal Co. Ltd. All animal experiments followed the guidelines and ethics of the Animal Care and Use Committee of Southeast University (Nanjing, China).
In the first animal experiment, the mouse hepatoma cell Hepa1-6 was seeded onto 24-well plates (1 × 105 cells/well) and cultured for 12 h. Cells were transfected with variant viruses (5 × 1010 vg/well) and cultured for 24 h. Cells were then harvested by trypsinization, re-suspended in PBS, and subcutaneously transplanted to mice on two spots at 1 × 107 cells/spot. The mice were bred for 2 weeks, sacrificed, and photographed. Tumor size was measured with a precision caliper, and tumor volume was calculated as follows: V = (Dd 2)/2, where D is the major tumor axis and d is the minor tumor axis.
In the second animal experiment, mice were subcutaneously transplanted with Hepa1-6 cells on two spots at 1 × 107 cells/place. After breeding for 7–13 days, the mice were divided into different groups and intravenously injected with variant AAVs at 1 × 10 9 vg/mouse. The mice were bred for 7 days, sacrificed, and photographed. Tumor size was measured with a Vernier caliper, and tumor size was measured and calculated as described above.
Detection of AAV virus distribution and effector gene expression by qPCR
Tissues were collected from the mice intravenously injected with AAVs, and total RNA was extracted from tissues using Trizol (Invitrogen). Genomic DNA (gDNA) were extracted by using the TIANamp Genomic DNA Kit (TIANGEN). In a 10 μL reaction, 500 ng of total RNA was reversely transcribed into cDNA using the PrimeScript RT Master Mix (Takara) according to the manufacturer's instructions. Gene expression was detected using a 20 μL qPCR reaction containing 1 × SYBR Green Real-Time PCR Master Mix (Roche), 0.25 μM primers (CHS-F2/R2, eGFP-F/R, and GAPDH-F2/R2; Supplementary Table S5), and 2 μL cDNA. Virus DNA were detected using a 20 μL qPCR reaction containing 1 × SYBR Green Real-time PCR Master Mix (Roche), 0.25 μM primers (AAV-F/R and GAPDH-F2/R2; Supplementary Table S5), and 2 μL gDNA. All qPCR programs were performed at 95°C for 10 min at 45 cycles of 95°C for 15 s and 60°C for 1 min. A melting curve was constructed to monitor PCR amplification specificity. All PCR programs were run with StepOne plus (ABI). qPCR data analysis was performed using Applied Biosystems StepOne v2.3. Ct values were normalized against GAPDH. Relative virus DNA abundance was calculated as relative quantification according to the following equation: RQ = 2–ΔΔCt.
Statistical analyses
Data are expressed as the mean ± standard deviation. Statistical significance was analyzed using t-tests. p-Values of ≤0.05 and ≤0.01 were considered as significant and highly significant differences, respectively.
Results
NF-κB-dependent cancer immunotherapy strategy
A NF-κB-activating gene expression (Nage) technique was developed by utilizing the intracellular activity of transcription factor NF-κB, which is over-activated in inflammation and cancers (Fig. 1). The Nage vector contains a promoter sequence, DMP, which consists of a NF-κB decoy sequence and a minimal promoter sequence. DMP was used to drive expression of the downstream effector gene in the Nage vector. DMP is a NF-κB-specific promoter, which was expected to drive effector gene expression in NF-κB over-activated cancer cells. Since the effector gene can express cell surface–displayed proteins or peptides, it was expected to function as an artificial neoantigen to induce attack to cancer cells.
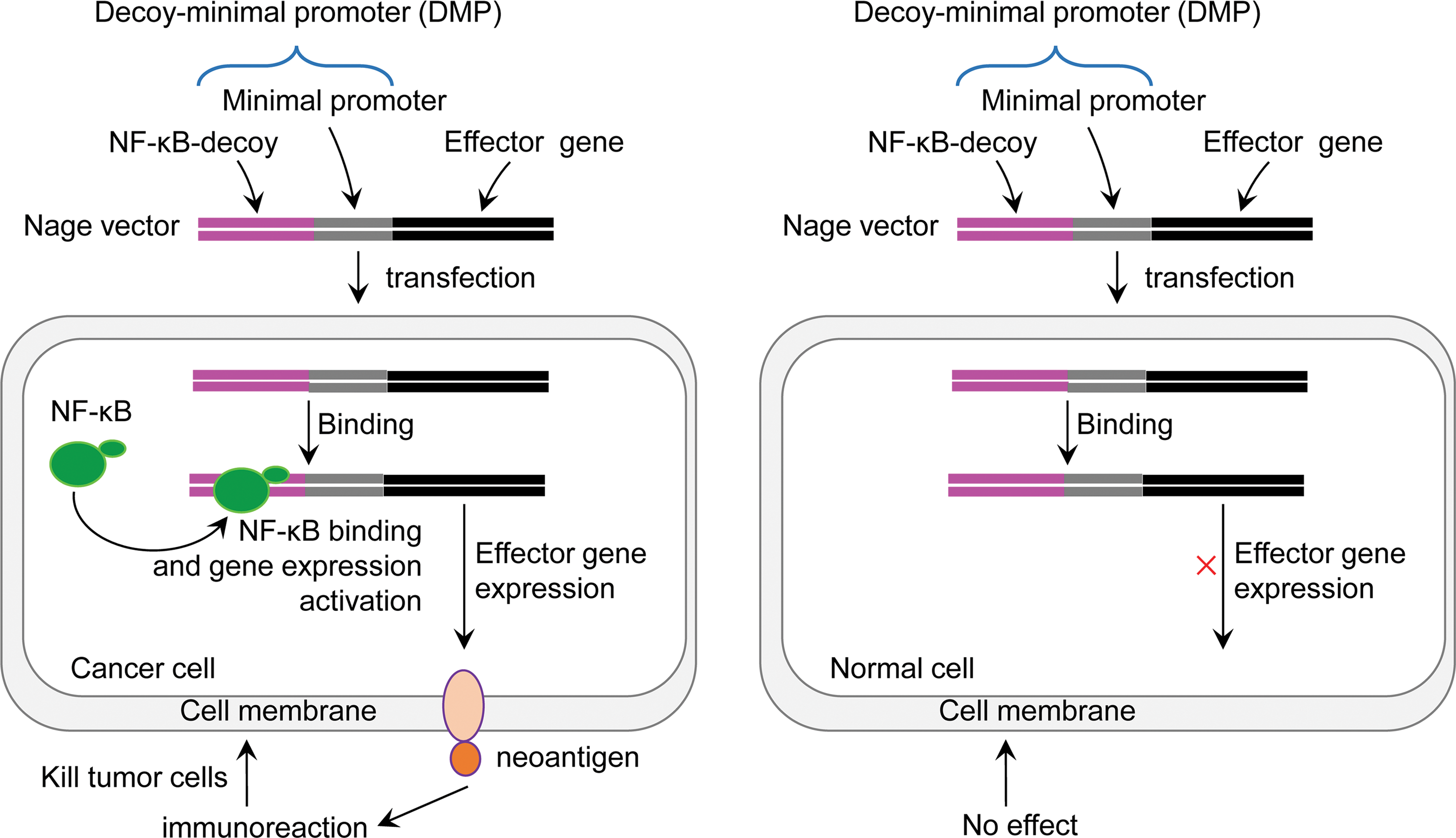
Schematic diagram of cancer immunotherapy with nuclear factor-kappa B (NF-κB)-activating gene expression vector. Color images available online at
Expression of NF-κB RelA in cancer and normal cells
To evaluate NF-κB expression in various cancer and normal cells, NF-κB RelA/p65 expression was measured in eight human cancer cell lines (HepG2, A549, PANC-1, MDA-MB-453, HT-29, HeLa, SKOV-3, and B16F10), two mouse cancer cell lines (Hepa1-6 and RAW264.7), and two human normal cell lines (HL7702 and MRC5) using qPCR (Fig. 2A). It was found that NF-κB RelA/p65 was expressed in all detected cancer cell lines but was not expressed in normal cell lines. Therefore, a NF-κB-specific promoter should drive cancer cell–specific gene expression.
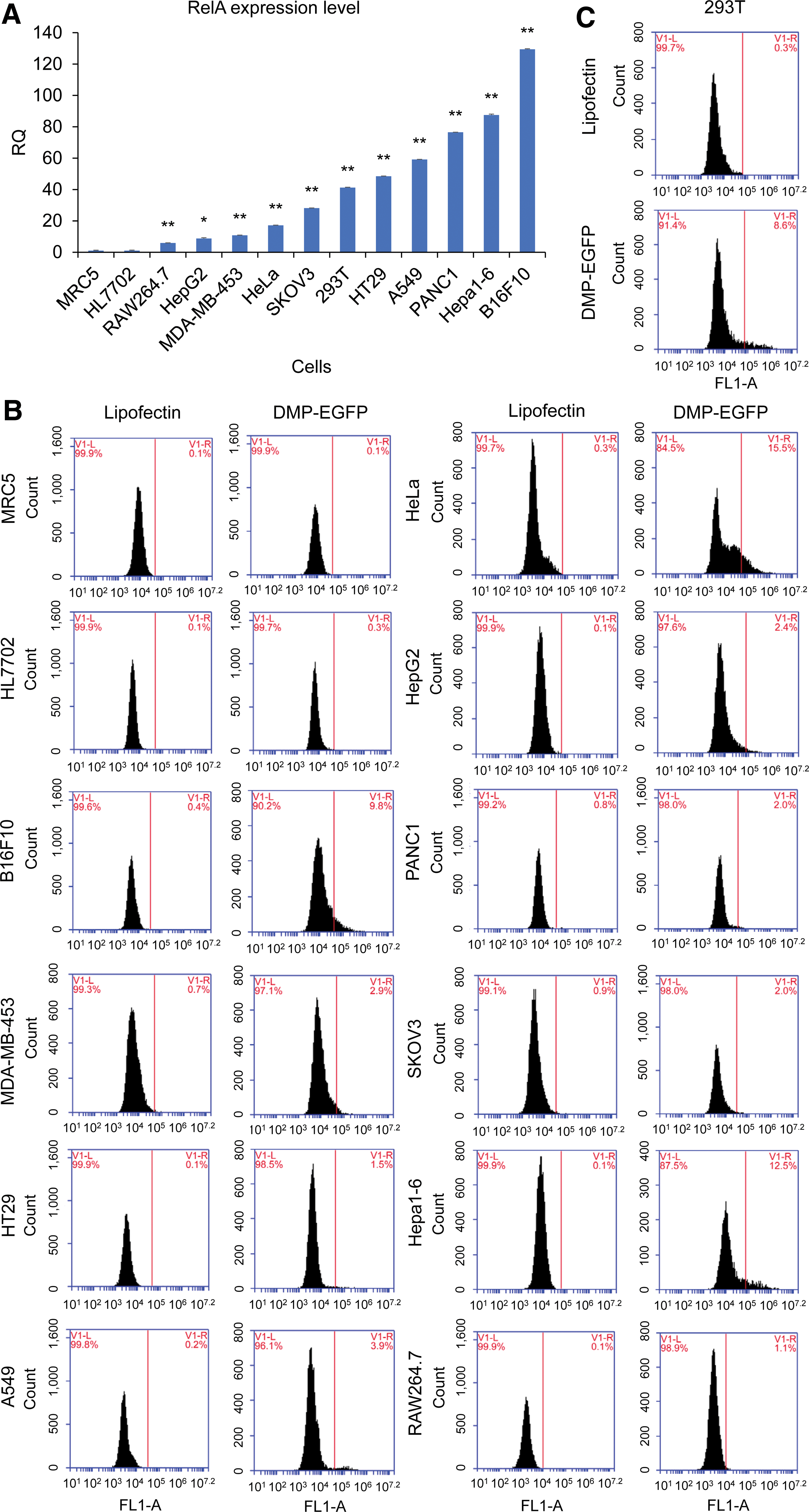
Detection of NF-κB RelA/p65 expression in variant cells.
RelA is a member of the NF-κB family that forms a heterodimer with another member, p50. RelA has both DNA-binding and transcription activation domains, while p50 has only a DNA-binding domain. Therefore, the transcriptional activation function of NF-κB mainly comes from RelA. RelA expression was detected in the 293T cell line, a widely used human cell line with high transfection efficiency. Thus, RelA was also expressed in this cell line.
Evaluation of the DMP promoter with eGFP
To evaluate cancer cell–specific expression of the Nage vector, a plasmid DMP-eGFP (pDMP-eGFP) was constructed by fusing DMP with the eGFP gene, and 13 different cell lines were transfected with this plasmid. It was found that eGFP was expressed in all transfected cancer cell lines, including HepG2, HeLa, PANC-1, MDA-MB-453, A549, HT-29, SKOV-3, Hepa1-6, B16F10, and RAW264.7 (Fig. 2B and Supplementary Fig. S1). However, eGFP was not expressed in two normal cells (MRC-5 and HL7702; Fig. 2B and Supplementary Fig. S1). These results indicate that the Nage vector can be used to realize cancer cell–specific gene expression that is dependent upon NF-κB over-activity in cancer cells. It was also found that eGFP was expressed at the highest level in the 293T cells (Fig. 2C and Supplementary Fig. S1). Therefore, the Nage vector can also express its effector gene in this human cell line in response to its NF-κB over-activity, even though this is not a cancer cell line.
Cells were also transfected with a control plasmid, pCMV-eGFP, in which a widely used promoter, CMV, controlled the expression of eGFP. It was found that eGFP was expressed in all transfected cell lines, including two normal cell lines, indicating that this strong promoter has no cancer cell specificity. The results also revealed that the CMV promoter had higher transcriptional activation activity than the DMP promoter in all transfected cell lines. In comparison, DMP was a NF-κB-specific weaker promoter, which was revealed by the relative low mean fluorescence intensity of all pDMP-eGFP-transfected cell lines (Fig. 2B and C and Supplementary Fig. S1). These results indicate that the Nage vector can only be activated by NF-κB over-activity.
Evaluation of the DMP promoter with displayed SBP
To investigate if a protein or peptide could be expressed on the cell surface in a cancer cell–specific manner, next a new Nage vector, pDMP-Display-SBP, was constructed that could express a streptavidin-binding peptide on the cell surface using a gene display expression cassette under the control of DMP. Five cell lines were transfected with this plasmid, and the displayed SBP was detected by staining cells with FITC-labeled streptavidin. SBP was displayed successfully on the surface of cancer cells Hepa1-6 and HepG2 but not on normal cells MRC-5 and HL7702. SBP was also successfully displayed on the surface of 293T cells due to NF-κB over-activity (Fig. 3).

Evaluation of cancer cell specificity of NF-κB-activating gene expression vector. Variant cells, including cancer (HepG2 and Hepa1-6) and normal (MRC-5 and HL7702) cells and 293T cell, were transfected by pDMP-Display-SBP and then stained with FITC-labeled streptavidin. Cells were imaged with a fluorescent microscope. Color images available online at
To investigate further the cancer cell–specific expression of a protein or peptide on the cell surface, HepG2, 293T, HeLa, PANC-1, MDA-MB-453, HT-29, A549, SKOV-3, Hepa1-6, RAW264.7, B16F10, MRC-5, and HL7702 cells were transfected with pDMP-Display-SBP. The displayed SBP was detected by staining cells with IRDye800CW-streptavidin and imaging them with a NIRF imager. SBP was displayed on the surface of all cancer cells (HepG2, HeLa, PANC-1, MDA-MB-453, HT-29, A549, SKOV-3, Hepa1-6, B16F10, and RAW264.7), but there was no detectable SBP on the surface of two normal cells (MRC-5 and HL7702; Fig. 4A).
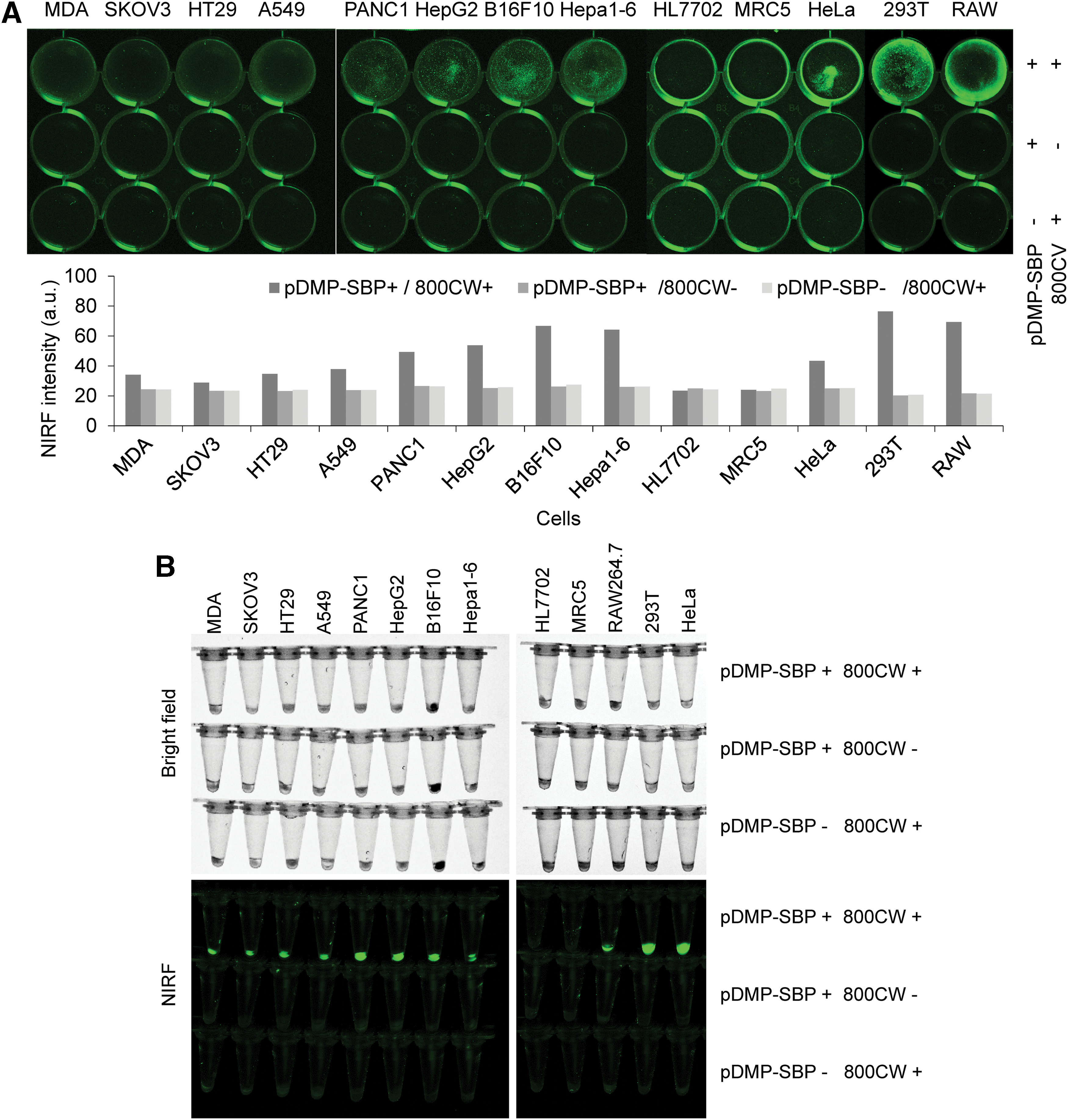
Evaluation of cancer cell specificity of NF-κB-activating gene expression vector. Variant cells, including cancer cells HepG2, HeLa, PANC-1, MDA-MB-453, HT-29, A549, SKOV-3, Hepa1-6, RAW264.7, and B16F10 and normal cells MRC-5 and HL7702, were transfected by pDMP-Display-SBP and then stained with streptavidin labeled by IRDye800CW, a near infrared fluorescence (NIRF) dye.
The stained adherent cells were also pelleted by trypsinization and re-imaged with a NIRF imager, revealing that all cancer cells significantly displayed SBP but the two normal cells did not (Fig. 4B). These results further verify that the Nage vector can specifically display a protein or peptide on cancer cells. In this NIRF assay, SBP was also displayed on the surface of 293T cells due to NF-κB over-activity.
Evaluation of virus vector by transfecting cells with AAV-SBP
The DMP-Display-SBP was packaged into AAV (AAV-SBP) as the in vivo vehicle for the Nage vector. To evaluate AAV-SBP, 293T, HepG2, Hepa1-6, MRC-5, and HL7702 cells were transfected with AAV-SBP, and SBP expression was detected by staining cells with IRDye800CW-streptavidin. SBP was displayed on two cancer cells (HepG2 and Hepa1-6) but not on two normal cells (MRC-5 and HL7702), indicating successful cell infection (Fig. 5A). The stained adherent cells were similarly pelleted by trypsinization and re-imaged with a NIRF imager. These results more clearly showed the cancer cell–specific expression of SBP by AAV-loaded Nage vector (Fig. 5B). In the same way, two other Nage AAVs, AAV-HBsAg and AAV-CRT, were also prepared, which also displayed HBsAg and CRT proteins on the cell surface, and two control AAVs, AAV-MCS and AAV-eGFP, were prepared. Then, the titers of all AAVs were determined using qPCR for in vivo application.
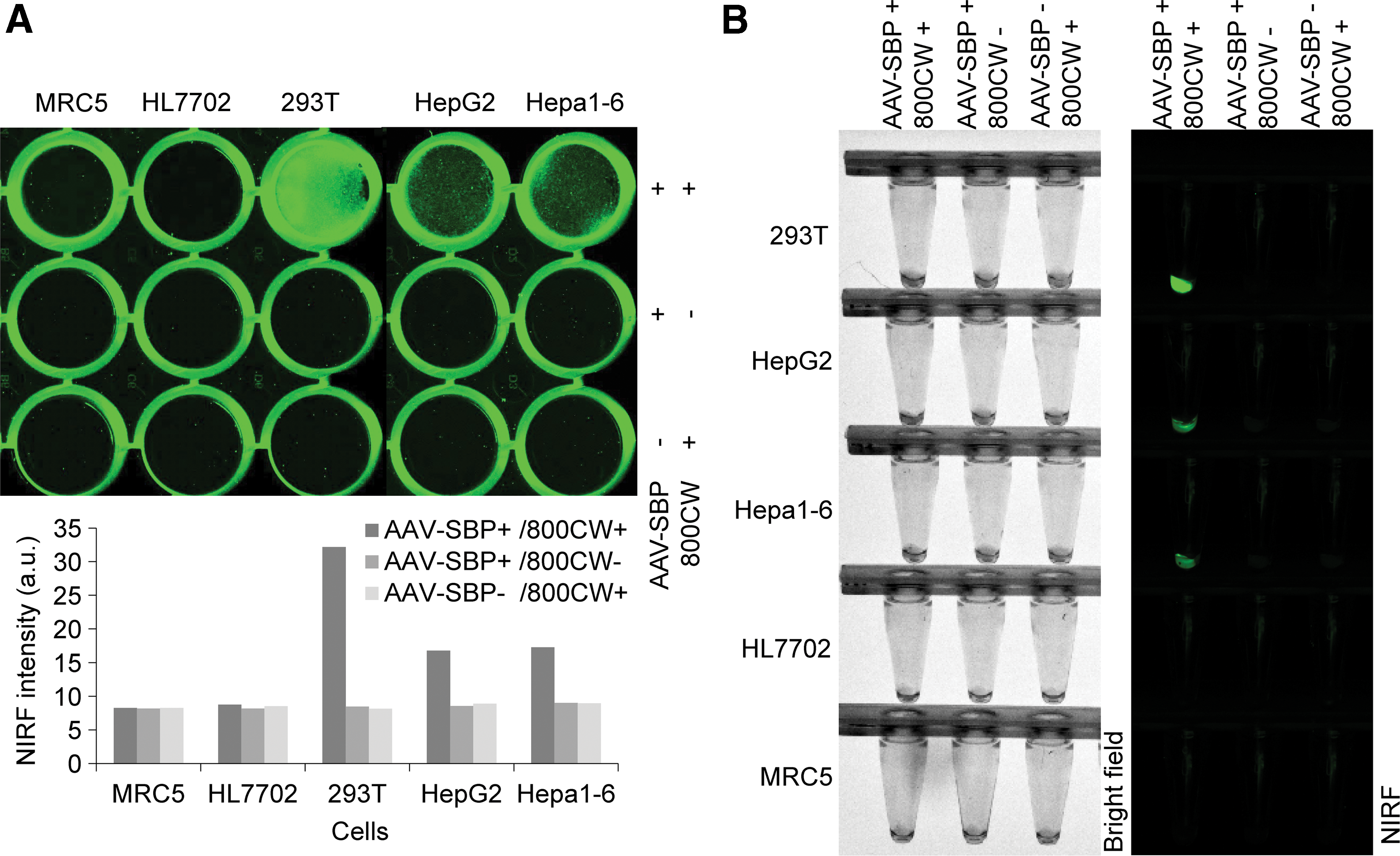
Evaluation of recombinant adeno-associated viruses (rAAVs) with in vitro cell transfection. Cells were transfected with AAV-SBP and detected by staining with IRDye800CW-labeled streptavidin.
Using Nage AAVs as cancer therapies
To evaluate whether the Nage AAVs could be applied to treat cancers in vivo, next, two animal experiments were performed. In the first animal experiment, the in vitro cultured mouse hepatoma Hepa1-6 cells were transfected with AAV-HBsAg, AAV-SBP, AAV-CRT, and AAV-MCS, respectively. The transfected cells subcutaneously transplanted to mice significantly inhibited tumor growth (Fig. 6A). In fact, spots transplanted with the Hepa1-6 cells transfected by experimental AAVs prevented tumor growth on one side in several animals (AAV-HBsAg group: n = 9; AAV-SBP group: n = 7; AAV-CRT group: n = 6). In some cases, treatment prevented tumor growth on both sides (AAV-HBsAg group: n = 4; AAV-SBP group: n = 3; AAV-CRT group: n = 2; Fig. 6A). The measurement of tumor size revealed that the tumor growth was significantly inhibited by the transfection of three experimental AAVs (Fig. 6B).

Treatment of cancer with Nage AAVs (the first animal experiment).
In the second animal experiment, 40 mice were transplanted with the in vitro cultured mouse hepatoma Hepa1-6 cells. After seven days, mice were divided into four groups of 10 each and intravenously injected with four variant viruses (AAV-HBsAg, AAV-SBP, AAV-CRT, and AAV-MCS, respectively). Treatment significantly inhibited tumor growth. Tumors were completely resolved on one side (AAV-HBsAg group: n = 9; AAV-SBP group: n = 8; AAV-CRT group: n = 8) or both sides (AAV-HBsAg group: n = 2; AAV-SBP group: n = 4; AAV-CRT group: n = 7; Fig. 7A). The measurement of tumor size revealed that the tumor growth was significantly inhibited by the intravenously injected AAV-SBP, AAV-HBsAg and AAV-CRT (Fig. 7B).
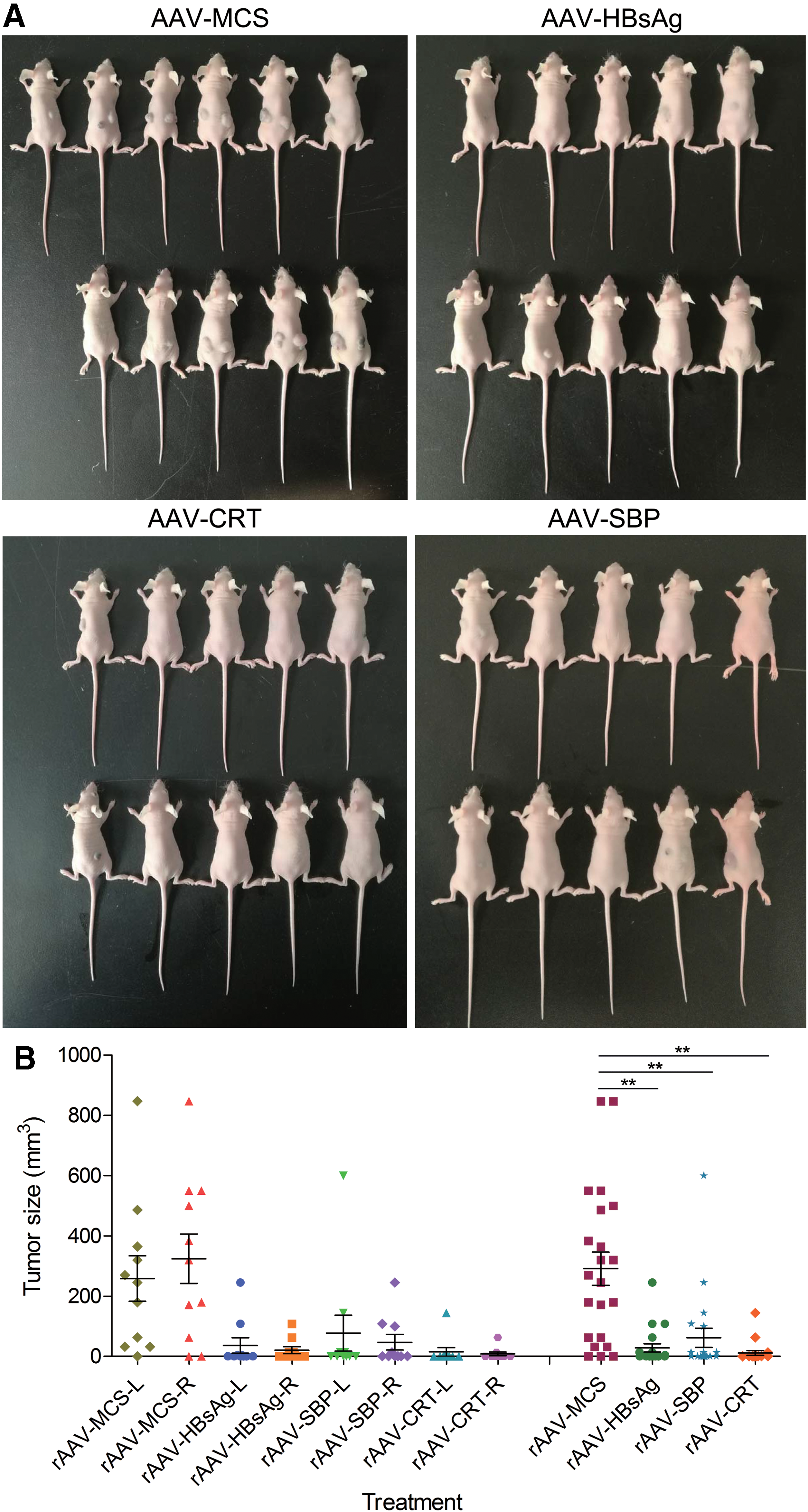
Treatment of cancer with Nage AAV (the second animal experiment: biological replicate 1).
To confirm the effects of treatment further, a replicate of the second experiment was performed. In this experiment, a new control rAAV, AAV-eGFP, was included, which intracellularly expresses eGFP under the control of DMP. Fifty mice were subcutaneously transplanted with Hepa1-6 cells, and after 13 days, tumor size was measured. The tumor-bearing mice were then divided into five groups: AAV-HBsAg (n = 10), AAV-SBP (n = 10), AAV-CRT (n = 10), AAV-eGFP (n = 7), and AAV-MCS (n = 7). The mice were intravenously injected with variant viruses, bred for 7 days, and then sacrificed and photographed (Supplementary Fig. S2). Treatment significantly inhibited tumor growth, and in this animal experiment, tumors were completely resolved in as many as 27 mice. Only three mice showed one or two small tumors (Supplementary Fig. S2). All mice treated with AAV-eGFP and AAV-MCS grew large tumors (Supplementary Fig. S2).
AAV distribution and effector gene tissue expression
To demonstrate the in vivo tumor-specific expression of the Nage vector further, the presence of AAV viral DNA and expression of effector gene were detected in various tissues. Heart, liver, spleen, lung, kidney, and tumor tissues were collected from the mice in the second animal experiment. qPCR revealed that AAV viral genomic DNA (marked by the bGH polyA signal region) was distributed in all tissues (Fig. 8A and Supplementary Fig. S3A). However, the exogenous gene (marked by PDGFR transmembrane domain region) was only expressed in tumor tissues (Fig. 8B and Supplementary Fig. S3B). Detection of eGFP expression in various tissues of mice treated by AAV-eGFP also demonstrated that eGFP was only expressed in tumor tissues (Supplementary Fig. S3B). These data indicate the cancer cell–specific expression of Nage effector genes in vivo.
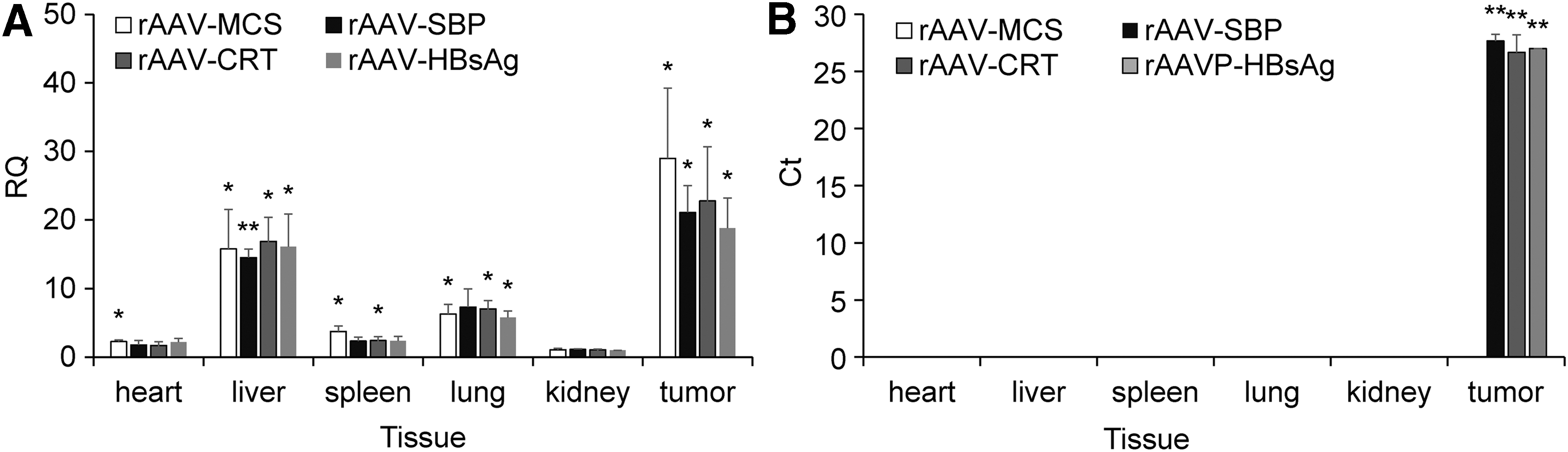
Detection of AAV DNA and effector gene expression in variant tissues. The gDNA and total RNA were extracted from variant tissues, including the heart, liver, spleen, lung, kidney, and tumor tissues. The abundance of virus DNA in gDNAs and the expression of artificial neoantigen genes in total RNA were detected by quantitative polymerase chain reaction.
Discussion
NF-κB is constitutively over-activated in a wide range of cancer tissues. 20,21 However, prior to this study, there were no reports that fully characterized NF-κB activity in various cancer and normal cells, which is critical for the design and development of Nage vectors. Highly sensitive fluorescence characterizations, including visible fluorescence characterization with microscopic imaging, flow cytometry quantification, and near infrared fluorescence characterization with NIRF imaging, demonstrated the cancer cell–specific expression of the Nage vector. Cancer cell–specific expression of Nage vector was also demonstrated via qPCR detection of effector gene expression in various tissues of rAAV-treated tumor-bearing mice. Based on these characterizations, the cancer-cell specificity of the Nage vector was verified, and a feasible NF-κB-utilizing cancer therapy strategy has therefore been proposed.
This strategy uses a safe viral vector, AAV, which is one of the most promising gene therapy vehicles identified in the past 30 years, as it can transduce a wide range of species in vivo with no evidence of toxicity. 42 In the past 2 years, the FDA has approved several human gene therapies that use AAV due to its long-term safety. 43 –45 This study packaged three Nage vectors into AAV and treated tumors in vivo via intravenous administration of rAAVs. This design delivered Nage vectors into cancer cells and resulted in tumor inhibition or eradication, indicating the effectiveness of this gene therapy. Importantly, there were no adverse effects when rAAV was administered by subcutaneous or intravenous injection, indicating its safety.
Nage gene therapy is simple and easy to use compared to CAR-T-based gene therapy, which requires T-cell isolation from patients, T-cell genetic transformation with a lentivirus, and CAR-T cell separation, propagation, and transfusion. Nage-based gene therapy would eliminate the cost and complications associated with CAR-T and creation of personalized vaccines. In addition, Nage-based gene therapy may represent a potential broad-spectrum anticancer drug.
This study displayed three different proteins or peptides on the cancer cell surface: HBsAg, CRT, and SBP. HBsAg is the surface antigen of the hepatitis B virus, which has been used to vaccinate against the hepatitis B virus. 46 HBsAg was used as a typical artificial neoantigen because people in China have been widely immunized by HBsAg, which produces immunological memory in cancer patients. 47,48 Therefore, when HBsAg is displayed on the surface of in vivo cancer cells, it activates immune system memory and induces a prompt and strong immune attack on cancer cells. CRT, an endoplasmic reticulum luminal resident protein, mediates the macrophage immunotherapy of cancers. 49,50 Upon its translocation on the cell surface, it functions as an “eat-me” signal and immediately induces phagocytic uptake in the cell. 50 CRT is expressed in many cancer cells and promotes macrophages to engulf hazardous cancerous cells. 51 In this study, SBP, a short peptide (40 amino acids in length) with high binding affinity to streptavidin, was used as a typical peptide neoantigen. 52,53 However, it can also be used to image tumors using streptavidin-labeled contrast agents for magnetic resonance or positron emission tomography. 54 Since SBP is a bi-functional molecule, an artificial neoantigen for cancer immunotherapy, and a cancer-specific target for tumor imaging, it may be applied to tumor diagnosis and real-time monitoring of cancer immunotherapy.
The Nage vector is a useful technical platform that is adaptable for different applications through different effector genes in the Nage vector, such as molecules for tumor imaging and cancer therapy. For example, suicide genes such as the herpes simplex virus thymidine kinase (HSV-TK) can be used in the HSV-TK/ganciclovir system to kill cancer cells. In addition, some membrane or secretory proteins can be expressed by the Nage vector to induce immunotherapy to cancers. 55 –57
This study separately displayed three proteins or peptides (HBsAg, CRT, and SBP) on the cancer cell surface. However, multiple proteins or peptides can be co-expressed by one Nage vector as long as they can be packaged in one AAV particle. These co-expressed proteins or peptides can play different functions, as recently demonstrated, in which a displayed immunogenic cell-surface protein (e.g., a surface T-cell engager) and three secreted proteins (cytokine, chemokine, and checkpoint inhibitor antibody) were co-expressed to trigger selective T cell–mediated killing of cancer cells synergistically. 57
Initially, this study tried to develop a cancer animal model with immune-competent mice but instead utilized a cancer-bearing nude mouse that is athymus and lacks T cells. Therefore, the exact mechanism underlying cancer clearance by artificial neoantigens was not clarified in this model. It was deduced that this cancer therapy effect may have resulted from the immune cells of innate immunity and B cell–based humoral immunity. The nude mouse has functional B cells, natural killer cells (NKs), and complement. The lymphokine-activated killer cells NKs are more prevalent in nude mice than in normal mice, and their activity seems to be enhanced compared to euthymic mice. 58,59 Mononuclear cells are also present in nude mice, and macrophage function seems to be enhanced. 60 –63 Stimuli in the conventional environment are capable of activating the macrophages of thymic-deficient mice. 64 Nude mice respond poorly to thymus-dependent antigens due to a defect in helper T-cell activity. However, nude mice showed a relatively normal immunoglobulin M response to thymus-independent antigens, and responses to thymus-independent antigens in nude mice are normal. The immunity mechanism underlying cancer clearance by artificial neoantigens in nude mice and in immune-competent mice should be addressed in future studies.
Footnotes
Acknowledgments
The National Natural Science Foundation of China (61571119) and the National Key Research and Development Program of China (2017YFA0205502) supported this work. We thank Professor Guangwen Cao of the Second Military Medical University for gifting us the plasmid with the HBsAg coding sequence.
Author Disclosure
No conflicts of interest declared.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.