
Abstract
Interleukin (IL)-17 and the cells that produce it within the tumor microenvironment appear to promote tumor development and are associated with survival in cancer patients. Here we investigated the role of the IL-17/IL-17 receptor A (IL-17RA) axis in regulating melanoma progression and evaluated the therapeutic potential of blocking the IL-17/IL-17RA pathway. First, recombinant mouse IL-17 (γmIL-17) treatment significantly increased proliferation of mouse B16F10 cells and human A375 and A2058 cells. Silencing IL-17RA by small hairpin RNA (shRNA) in B16F10 cells reduced the γmIL-17–elicited cell proliferation, migration, and invasion, and significantly reduced vascular endothelial growth factor and matrix metalloproteinase production. Remarkably, knockdown of IL-17RA led to a significantly decreased capability of B16F10 cells to form tumors in vivo, similar to that in IL-17-deficient mice. Finally, local application of an adenovirus delivering a shRNA against IL-17RA mRNA not only significantly suppressed tumor development, but also enhanced antitumor immunity by increasing the interferon γ–expressing T cells and not T regulatory cells. Our results highlight the critical role of the IL-17/IL-17RA pathway in tumor progression and imply that targeting IL-17RA represents a promising therapeutic strategy.
Introduction
Chronic inflammation in the tumor microenvironment contributes to the promotion of proliferation, migration, and survival of cancer cells, leading to tumor growth, invasion, and metastasis. 1,2 Such effects are mediated by stromal cells, tumor-associated macrophages, T regulatory cells (Tregs), and tumor cells. 3 –6 Tumor cells themselves generate a number of growth factors and cytokines, such as transforming growth factor beta (TGFβ), which inhibits the functions of immune effectors and directly or indirectly regulates angiogenesis, thereby facilitating cancer development. 7,8 Recent studies showed that a proinflammatory cytokine, interleukin (IL)-17 (also called IL-17A), and the cells that produce it, Th17 and Tc17 cells, are elicited by certain tumors. 9 –12 Although some IL-17–producing cells are present in peripheral blood, 9,13 –15 tumor sites have significantly higher levels of IL-17. 9,11,16,17 For example, while the levels of IL-17–producing cells in tumor-draining lymph nodes and peripheral blood were similar between patients with ovarian cancer and healthy donors, the percentage of IL-17-producing cells was higher in tumor tissues than at other sites in the patients. 9,18 The accumulation of IL-17–expressing cells appeared to reflect promoted tumor development and was correlated with an advanced stage and a poor survival rate. 9,11,13 Chen et al. 11 reported that the level of IL-17 was higher in lung cancer patients and was associated with TNM stage and overall and disease-free survival. Cancer patients with higher levels of IL-17 were more likely than those with lower levels if IL-17 to experience relapse. 13,15,16
IL-17–producing cells play an important role in increasing inflammatory responses during viral infection 19 by promoting host defenses against infectious pathogens. 20,21 They also contribute to the pathogenesis underlying autoimmune diseases. 22,23 Increasing evidence indicates that IL-17 induces many other inflammatory mediators 24 and thereby contributes to the promotion and regulation of allergic responses and leukocyte migration. Although tumor growth was reduced in IL-17 knockout mice and this reduction was recovered by intratumoral supplementation with IL-17, 25,26 the mechanism by which IL-17 mediates tumor promotion remains unclear. It has also been hypothesized that IL-17 might induce tumor angiogenesis by promoting vascular endothelial growth factor (VEGF) to support tumor progression. Indeed, in colon cancer, patients with high levels of IL-17 expression also had high levels of VEGF, and increased VEGF levels were positively correlated with IL-17 expression, 16 indicating that IL-17 mediates VEGF secretion. IL-17 promoted angiogenesis by directly stimulating the proliferation and migration of vascular endothelial cell as well as the release of proangiogenic factors, including VEGF and hepatocyte growth factor (HGF), 27,28 thereby resulting in tumor progression. 15,29,30 The response of cancer patients treated with anti-VEGF therapy appeared to be associated with the signaling of the IL-17/IL-17R axis. 30,31 Alternatively, the loss of IL-17 reduced the expression of Stat3-regulated chemokines, which decreased infiltration by myeloid cells in skin cancer. 32 Moreover, Wang et al. 26 found that IL-17 directly induced IL-6 to activate Stat3 and subsequently enhanced tumor growth. Cytokine signaling is well known to be controlled via the binding of corresponding receptors, which affects subsequent responses. Therefore, enhancement or inhibition of cytokine signaling could be crucial in disease development. In the current study, we investigated the role of IL-17 and IL-17/IL-17RA signaling in tumor growth and tumor behavior by knocking down IL-17RA in tumors. In addition, we explored the therapeutic potential of locally targeting tumor cells with a small hairpin RNA (shRNA) against IL-17RA mRNA.
Materials and Methods
Cell lines
The murine melanoma cell line B16F10 was obtained from Dr. Hsin-Wei Chen (National Health Research Institutes, Taiwan) and maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco, Gaithersburg, MD) supplemented with 10% fetal bovine serum (FBS; HyClone, South Logan, UT) at 37°C with 5% CO2. The B16F10-luc-containing luciferase was cultured in DMEM supplemented with 1 mg/mL G418 (Invitrogen, Carlsbad, CA) under G418 selection pressure. The B16F10-luc was infected with shIL-17RA or shCTRL in a lentiviral system to obtain shIL-17RA-B16F10-luc and shCTRL-B16F10-luc. After cells were infected with lentivirus expressing RFP, they were sorted by FACSAria (BD, San Jose, CA) to create stable shRNA lines. The human melanoma cell lines A375, A2058, and MeWo were obtained from Dr. Hsi-Hsien Lin (Chang Gung University, Taiwan). The A375 and A2058 cells were maintained in DMEM (Gibco) supplemented with 10% FBS (HyClone) at 37°C with 5% CO2. The MeWo cells were maintained in minimum essential medium (Gibco) supplemented with 10% FBS (HyClone) at 37°C with 5% CO2. The 293 cell line was obtained from Dr. Ming-Ling Kuo (Chang Gung University, Taiwan) and maintained in DMEM (Gibco) supplemented with 10% FBS (HyClone) at 37°C with 5% CO2.
Cell proliferation and cell cycle
B16F10-luc cells (1 × 104) were seeded in 96-well flat-bottom plates and then treated with targeted mouse recombinant cytokines or VEGF at optimal concentration for 24 h in quadruplicate. The concentration of recombinant mouse IL-17 (γmIL-17), VEGF, TGFβ, IL-6, IL-10, and interferon γ (IFNγ) was 10 ng/mL. Cell proliferation was assessed by MTT assay according to the manufacturer's recommendations (Sigma-Aldrich, St. Louis, MO). The shIL-17RA-B16F10-luc and shCTRL-B16F10-luc cells were also treated with γmIL-17 (100 ng/mL) for 24 h, and the number of viable cells were counted for assessment of cell proliferation. The human melanoma cell lines A375, A2058, and MeWo were treated with γmIL-17 (100 ng/mL) for 24 h and the number of viable cells were counted for assessment of cell proliferation.
For cell cycle analysis, B16F10-luc cells (5 × 104) were seeded in 24-well flat-bottom plates and treated with γmIL-17 (100 ng/mL) for 6 h. The cells were fixed with 70% ethanol at −20°C and then resuspended in working buffer (Ribonuclease A 0.1%, Triton 0.05%, and propidium iodide 50 μg/mL) and analyzed with a FACSCalibur flow cytometer (BD).
Cell migration and invasion assay
The cell migration assay was performed by using a transwell chamber, and a Matrigel-coated chamber (BD Biosciences, San Jose, CA) was used for the cell invasion assay. The shIL-17RA-B16F10-luc and shCTRL-B16F10-luc cells (5 × 105) treated with γmIL-17 (100 ng/mL) were seeded respectively into the upper chamber in 1% FBS. The lower chamber contained complete culture medium supplemented with 20% FBS. The cells in the lower chamber, which had passed through the membrane (migration, incubated for 48 h) or Matrigel-coated membrane (invasion, incubated for 72 h), were stained with crystal violet. Migration/invasion was then observed and photographed under a microscope (Nikon, Tokyo, Japan). These experiments were repeated in triplicate, the numbers of migrated/invaded cells were quantified per field of view, and the results were statistically analyzed.
Animals
C57BL/6 mice were obtained from the National Laboratory Animal Centre, Taipei, Taiwan. The IL-17 knockout mice were generously gifted by Dr. Yuan- Ji Day (Lin-Kou Chang Gung Memorial Hospital, Taiwan), and they shared the same background of C57BL/6 mice. The IL-17 knockout mice were originally C57BL/6J and backcrossed to C57BL/6 background for at least eight generations. 33 All mice were housed in a specific pathogen-free area, and female animals at the age of 8 weeks were used in this study. A B16F10-luc-tumor–bearing model was established by subcutaneously injecting the C57BL/6 or IL-17 knockout mice in the center of the back with B16F10-luc cells (3 × 105/mouse). In the shIL-17RA-B16F10-luc model, the C57BL/6 mice were then subcutaneously injected with shIL-17RA-B16F10-luc cells (3 × 105/mouse) or shCTRL-B16F10-luc cells (3 × 105/mouse).
Tumor growth and metastasis in vivo
Tumor size was measured and recorded twice a week. Tumor volume (V) was then calculated according to the following formula: V (mm3) = L × W2/2, where L represented the longest diameter of the tumor, and W represented the diameter at a right angle to the largest diameter. For locating and monitoring spontaneous metastatic lesions, some mice were anesthetized with isoflurane inhalation and subsequently intraperitoneally (i.p.) injected with D-luciferin (150 μg/g of mouse weight; Xenogen, Hopkinton, MA). Bioluminescence imaging was performed with a CCD camera (IVIS, Xenogen) at 5 min after injection. The imaging time was 60 s. Metastasis was measured on days 7, 14, and 21 after cell inoculation. The primary tumor imaging was presented from the back of tumor-bearing mice (Supplementary Fig. S1). The signals indicating metastasis began to appear around the lymph nodes of the groin and subsequently around the abdomen and then the abdomen and chest.
Tumor therapy using adenoviral vectors
The Adeno-X Adenoviral System 3 (Clontech, Mountain View, CA) containing ZsGreen1 fluorescent protein was used to produce an adenoviral vector encoding an shRNA against IL-17RA. Adenoviruses were produced by transfecting 293 cells with pAd-shIL-17RA or scramble plasmid. The recombinant virus, Ad-shIL-17RA or Ad-scramble, was harvested from 293 cells with cytopathic effect, isolated using the freeze-thaw method, and purified on a cesium chloride gradient as described elsewhere. 34 C57BL/6 mice were subcutaneously injected with B16F10-luc tumor cells (3 × 105/mouse). After 7 days (post-tumor inoculation), the average tumor volume was 7.22 ± 2.04 mm3, and the mice were intratumorally injected with Ad-shIL-17RA or Ad-scramble (1010 PFU/mouse). Normal saline was used for the control group. Tumor growth and metastasis were monitored as described above.
Immunohistochemistry and assessment
A tissue array (Pantomics) containing melanoma tissue sections and corresponding normal tissue sections on the same slide was utilized. The tumor sections were fixed in 3.7% formaldehyde (Sigma-Aldrich) and permeabilized with 0.2% Saponin (Sigma-Aldrich). The tumor sections were then labelled with anti-human IL-17RA antibodies (Santa Cruz, TX) and stained with horseradish peroxidase (HRP)-conjugated secondary antibodies (eBioscience, San Diego, CA), and 3-amino-9-ethylcarbazole was used as a chromogen. The slide stained with HRP-conjugated secondary antibodies only served as a control. All slides were stained with hematoxylin and observed under a microscope (Leica, Wetzlar, Germany).
RNA isolation and real-time RT-PCR
Total RNA was collected from tumor tissues, homogenized with beads by TissueLyser (Qiagen, Hilden, Germany), and extracted with TRIzol (Invitrogen) reagent. Total RNA was also extracted from shIL-17RA-B16F10-luc or shCTRL-B16F10-luc cells treated with γmIL-17 (100 ng/mL) using TRIzol reagent according to the manufacturer's instructions. One microgram of RNA was used for reverse transcription. cDNA was synthesized using an iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA) for the following targets: VEGF, HGF, matrix metalloproteinase (MMP)2, MMP9, and IL-17RA (VEGF: 5′ gaccctggctttactgctgta and 3′ gtgaggtttgatccgcatgat; HGF: 5′ aagagtggcatcaagtgccag 3′ ctggattgcttgtgaaacacc; MMP2: 5′ atcgcagactcctggaatg 3′ acttcacgctcttgagactt; MMP9: 5′ cccaaagacctgaaaacctcc 3′ ttctctcccatcatctgggc; IL-17RA: 5′ gcacccaagcaaagtggaaa and 3′ aaacaacgtaggtgccgaagc). β-Actin was used as the control. All real-time PCR reactions were amplified in iQ SYBR Green Supermix (Bio-Rad) using a Bio-Rad detection system. The cycle threshold (Ct) value was determined from duplicate samples to calculate the target gene expression.
Flow cytometry experiments
Phenotypic and intracellular cytokine analysis was performed as published elsewhere 15,35 and was conducted with the following fluorochrome-conjugated monoclonal antibodies (BD Biosciences; eBioscience) against murine antigens: CD4- PerCP, CD8-APC, CD25-APC, Foxp3-PE, IL-17-APC, IFNγ-APC, and IL-17RA-PE. Ten thousand events were collected for each sample, and all data were acquired using CellQuest Pro software on a BD FACS Calibur flow cytometer and analyzed using FCS Express 3 software.
Cytokine measurement
Splenocytes (4 × 106) were co-cultured with B16F10-luc cells for 72 h, and the level of IL-17 in the culture supernatant was determined by an ELISA kit (eBioscience). The level of VEGF in plasma obtained from tumor-bearing mice was measured using an ELISA kit obtained from R&D Systems, Minneapolis, MN, and all protocols were performed according to the manufacturer's manual.
Statistical analyses
All statistical analyses were performed using GraphPad Prism software, version 5.02 (GraphPad Software, Inc.). The statistical significance of the differences between the groups was assessed using one-tailed Student's t tests. The differences with a p value less than 0.05 were considered statistically significant.
Results
IL-17 enhanced proliferation in melanoma tumor cells
Recent evidence has indicated that many human cancers and animal tumor models express increased levels of IL-17. 9 –11,36 An important topic we sought to address is the role of IL-17 in the tumor microenvironment. We hypothesized that tumor cell growth would be promoted by some IL-17–associated mediators. To address this issue, we treated cultured B16F10-luc tumor cells with γmIL-17, VEGF, TGFβ, IL-6, IL-10, and IFNγ and then analyzed tumor cell proliferation using MTT assays. The results showed that the proportion of proliferating B16F10-luc tumor cells was increased more by IL-17 and VEGF than by treatment with medium only (Fig. 1A). Moreover, human melanoma tumor cells were treated with γhIL-17 and then analyzed to determine cell numbers. The results showed that cell numbers were increased by IL-17 (Fig. 1B). To evaluate the effect of IL-17 on cell cycle progression, B16F10-luc tumor cells were further analyzed by flow cytometry. Cells were treated with IL-17 or medium alone, and the percentage of cells in G1, S, and G2 phase were then compared. In B16F10-luc tumor cells treated with IL-17, the percentage of cells in S phase increased (Fig. 1C) from 32.92% to 40.19% after 6 h, compared to the medium-only control group. The number of cells in S phase was then quantified and found to be higher in B16F10-luc tumor cells treated with IL-17 than in those treated with medium alone (Fig. 1C, right). These results suggest that IL-17 enhances proliferation and the proportion of cells in S phase in B16F10-luc tumor cells.
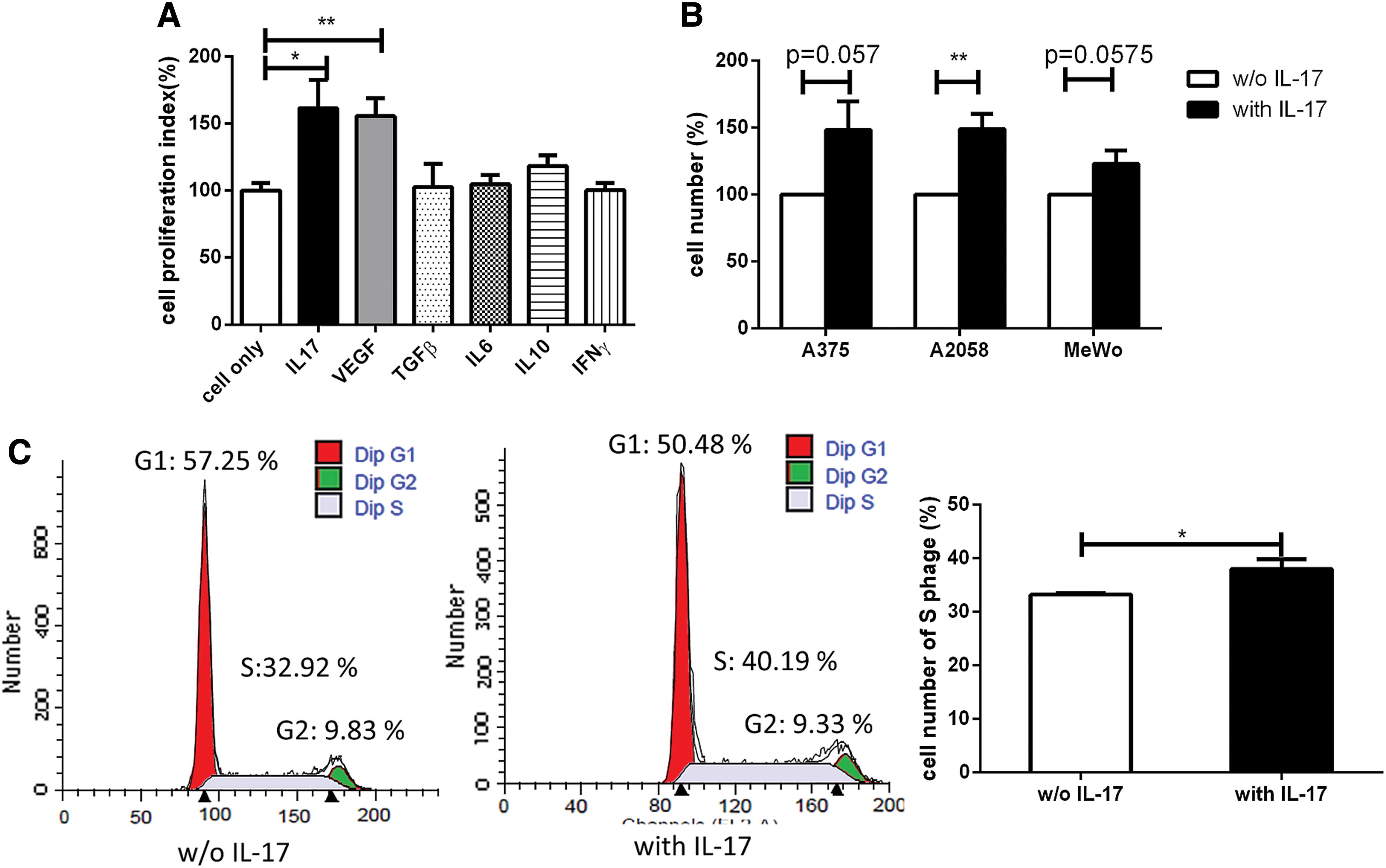
Interleukin (IL)-17 enhanced the proliferation of melanoma tumor cells. Mouse and human melanoma cells were treated with recombinant cytokines or vascular endothelial growth factor (VEGF).
IL-17 deficiency decreased tumor growth and the rate of metastasis
To investigate the association between IL-17 production and tumor development in vivo, we performed experiments in which we injected IL-17 knockout (IL-17 KO) mice with B16F10-luc cells subcutaneously to develop a tumor model. Tumor growth was significantly slower in IL-17–deficient mice than in wild-type (WT) mice (Fig. 2A). The rate of metastasis was also significantly lower in IL-17 KO mice (Fig. 2B) than in WT mice (Fig. 2C). MMP2 and MMP9 play important roles in cancer metastasis, 37 and we therefore next investigated the effect of IL-17 on the expression of these MMPs. The expression levels of MMP2 and MMP9 were compared in tumor tissues by real-time PCR between IL-17 KO and WT mice. The results showed that MMP2 and MMP9 were expressed at higher levels in WT mice (Fig. 2D). IL-17 affects VEGF production in cancer cells 16 , and VEGF is associated with a poor prognosis. 16,27 Hence, we next detected VEGF expression levels in the tumor microenvironment and found that VEGF expression (and that of other proangiogenic factors, such as HGF) was higher in WT mice than in IL-17 KO mice (Fig. 2D). These data indicate that IL-17 may promote tumor growth and the rate of metastasis by enhancing the expression of MMP2, MMP9, VEGF, and HGF.

IL-17 deficiency decreased tumor growth and the rate of metastasis. IL-17 knock-out (KO) and wild-type (WT) mice were challenged with B16F10 melanoma cells.
Knocking down IL-17RA decreased tumor cell growth and the rates of invasion and migration
IL-17 signaling is mediated by the receptor subunits IL-17RA and IL-17RC, which together form a heteromeric complex. 38,39 We therefore knocked down the expression of IL-17RA with an shRNA to explore the effect of IL-17/IL-17RA signaling on tumor immunity. The expression of IL-17RA was detected by real-time PCR and found to be significantly lower in shIL-17RA-B16F10-luc tumor cells (shIL-17RA) (Fig. 3A). We also used flow cytometry to explore the mean fluorescence intensity of cells expressing IL-17RA. The mean fluorescence intensity was significantly lower in shIL-17RA-B16F10-luc tumor cells than in shCTRL-B16F10-luc tumor cells (shCTRL) (Fig. 3B). Cell numbers were increased by IL-17 in shCTRL-B16F10-luc tumor cells but not in shIL-17RA-B16F10-luc tumor cells (Fig. 3C).

Knocking down IL-17 receptor A (IL-17RA) decreased tumor cell growth and the rates of invasion and migration. The shIL-17RA-B16F10-luc and shCTRL-B16F10-luc melanoma cells were treated with IL-17, and the effects on cell proliferation, invasion, and migration were identified.
Next, we explored invasion and migration in shIL-17RA-B16F10-luc tumor cells and found that both were significantly inhibited in IL-17-treated shIL-17RA-B16F10-luc tumor cells (Fig. 3D and 3E). These data suggest that deficiency in IL-17RA inhibits tumor cell proliferation, invasion, and migration.
Knocking down IL-17RA decreased tumor growth and the rate of metastasis
Tumor growth was significantly lower in mice subcutaneously injected with shIL-17RA-B16F10-luc tumor cells than in those injected with control B16F10-luc tumor cells (Fig. 4A). The metastasis rate was also significantly lower in mice injected with shIL-17RA-B16F10-luc tumor cells (Fig. 4B and 4C). Next, to investigate the mechanisms underlying how IL-17RA inhibition suppresses tumor growth, we explored its effect on the tumor microenvironment. VEGF, HGF, MMP2, and MMP9 expression levels were significantly lower in tumor tissues in mice injected with shIL-17RA-B16F10-luc tumor cells than in those injected with control B16F10-luc tumor cells (Fig. 4D). These results indicate that the tumor progression-promoting factors were downregulated in the mice injected with shIL-17RA-B16F10-luc.
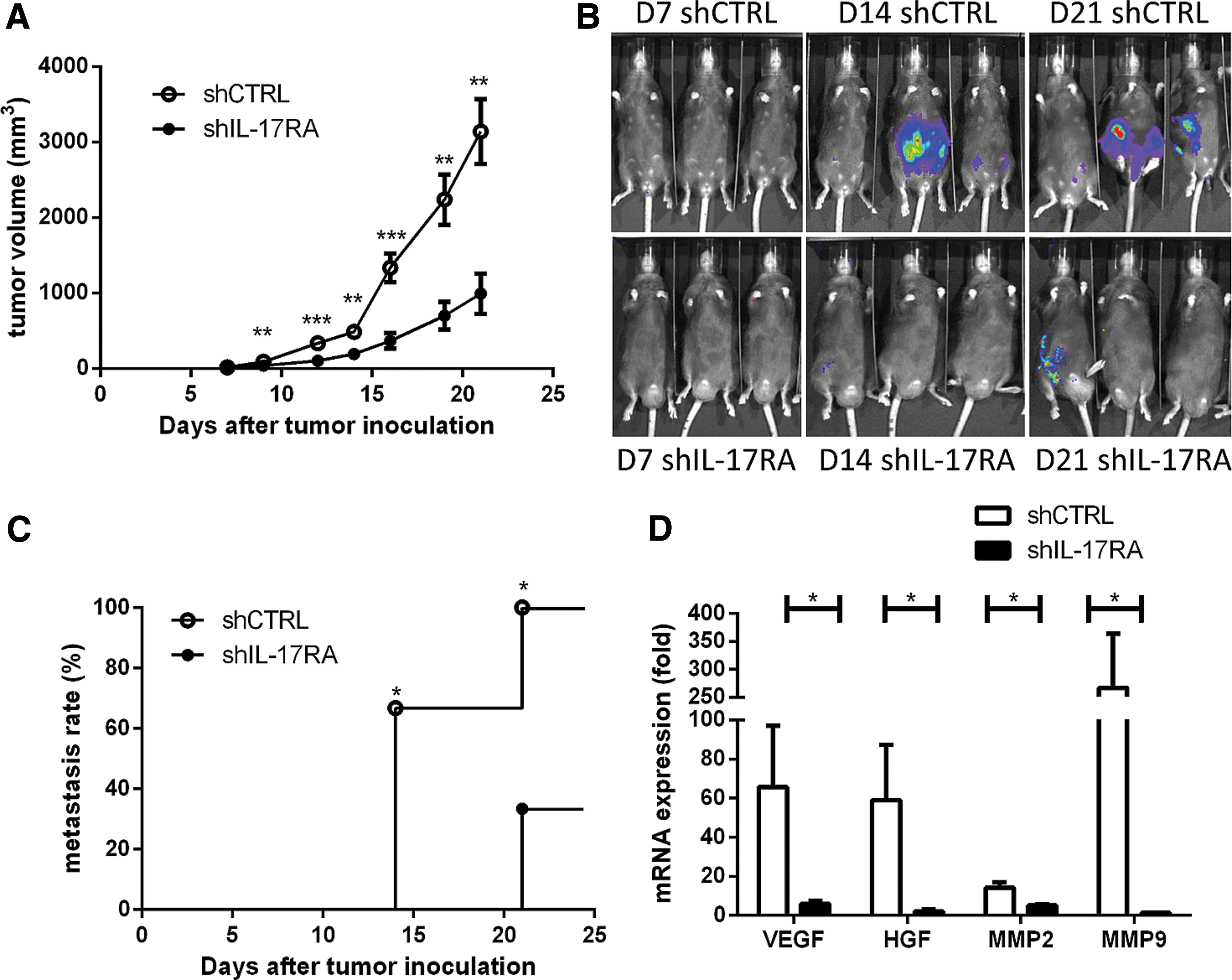
Knocking down IL-17RA decreased tumor growth and the rate of metastasis. The shIL-17RA-B16F10-luc (shIL-17RA) and shCTRL-B16F10-luc (shCTRL) melanoma cells were inoculated in WT mice. The
Local intra-tumor treatment with Ad-shIL-17RA successfully reduced tumor growth
To investigate the role of IL-17RA in tumor tissues, we detected its expression in human melanoma tissues. We used a tissue array and immunohistochemistry to analyze IL-17RA expression in 96 human melanoma specimens, and we found that it was not expressed in normal tissue but positively labelled tumor and metastatic tissues. IL-17RA was expressed at high levels in metastatic tissues and primary tumor tissues (Fig. 5A-B). These results indicate that IL-17RA may play a role in promoting metastasis in melanoma.
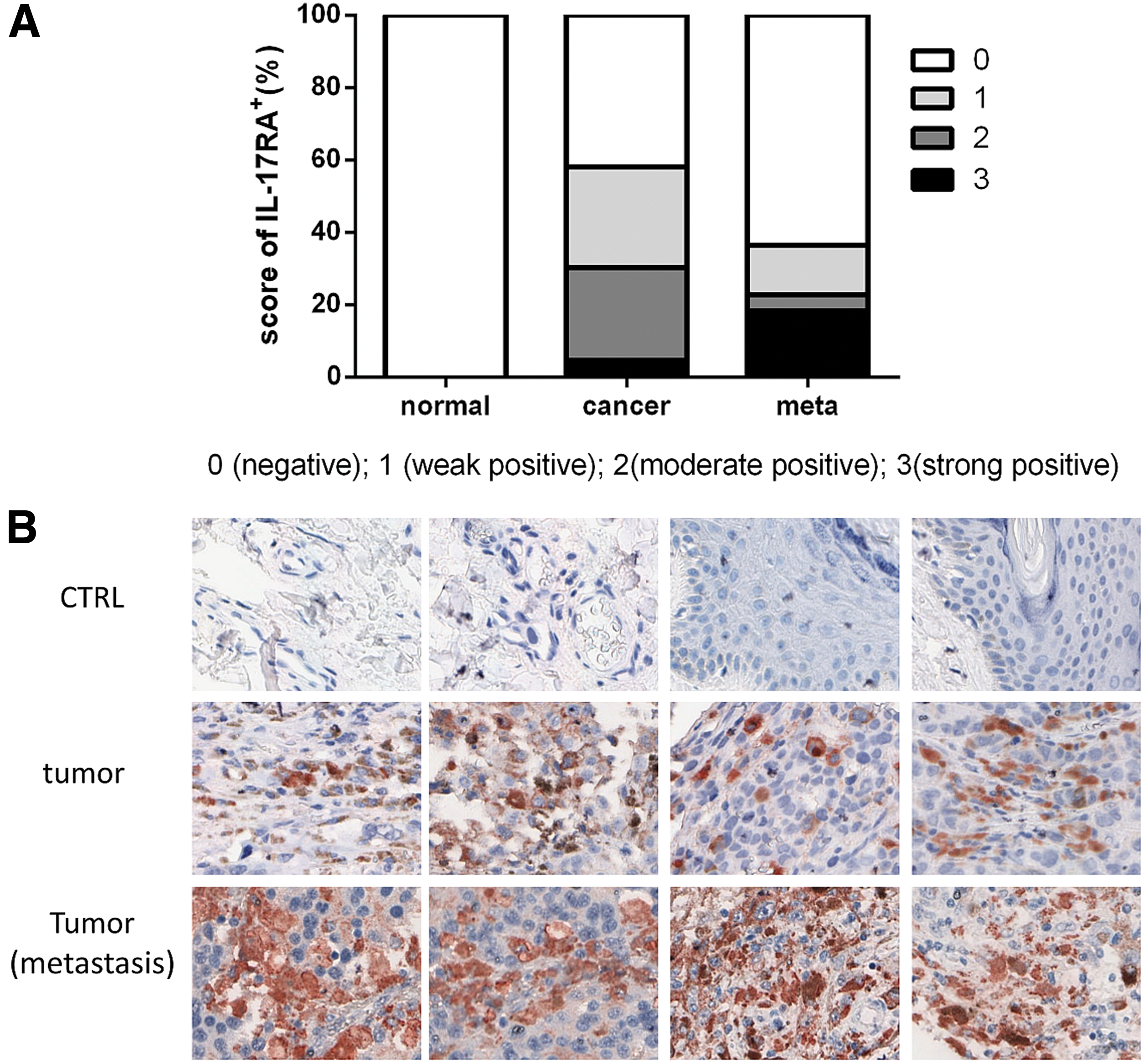
The elicited level of IL-17RA in human melanoma tumor tissues. The expression of IL-17RA was analyzed by immunohistochemistry (IHC) in a tissue array containing human melanoma tissue sections and corresponding normal tissue sections ( × 100 magnification).
To investigate the therapeutic potential of directly blocking the IL-17/IL-17RA axis, we constructed an adenoviral vector encoding an shRNA against IL-17RA. The adenovirus-derived vector treatment for cancer appeared to be a safe and effective strategy in previous research. 40,41 Mice were subcutaneously injected with B16F10-luc tumor cells and then intratumorally injected with Ad-shIL-17RA after 7 days (post-tumor inoculation) (Supplementary Fig. S2). Tumor growth was significantly lower in the Ad-shIL-17RA–injected tumors than in control tumors (Fig. 6A). IL-17 and VEGF expression levels were also lower in the Ad-treated group (Fig. 6B), and VEGF and IL-17 expression levels were positive correlated (Fig. 6C). These data show that Ad-shIL-17RA exerted a potent therapeutic effect against established tumors, possibly by suppressing angiogenesis in the tumor microenvironment. To determine which antitumor immune factor was involved in this effect, we assessed T lymphocytes in tumor-draining lymph nodes. The percentage of IFNγ-producing cells was significantly higher in the IL-17/IL-17RA–deficient group (Fig. 6D). There were also significantly fewer regulatory T cells (Tregs) in the IL-17/IL-17RA–deficient group than in the control group mice (Fig. 5E). Finally, IL-17 expression was significantly lower in the IL-17/IL-17RA–deficient group. Collectively, these data indicate that IL-17/IL-17RA deficiency increased the cytotoxic T lymphocyte (CTL) activity and inhibited suppressor cells.

Local intra-tumor treatment with Ad-shIL-17RA successfully reduced tumor growth. C57BL/6 mice were inoculated with melanoma cells and then treated with/without Ad-shIL-17RA or Ad-scramble, and their tumor growth, the levels of IL-17, and VEGF as well as immune subsets were analyzed.
Discussion
In the present study, we demonstrated that inhibiting IL-17/IL-17RA suppressed tumor growth and metastasis. Experiments performed using different tumor animal models have produced many different results related to the role of IL-17 responses in tumor immunity, and the pathways involved in tumor cell growth differ between animal models. If chronic inflammation is induced by a pathway in tumor cells in an animal model, IL-17 may promote tumor progression. However, if the pathway induces acute inflammation, IL-17 may inhibit tumor growth. With regard for the pathways affecting tumor cells in animals, it appears that animal models involving the induction and subcutaneous injection of tumor cells are associated with chronic inflammation, indicating that IL-17–mediated inflammation has tumor promoting effects. 36,42 –44 Intravenous injection resulted in acute inflammation, indicating that in this kind of animal tumor model, IL-17 may inhibit tumor growth. 45,46 Some studies have indicated that the effect of IL-17 overexpression on tumor tissues depends on enhanced tumor-specific immunity. 47,48 In these studies, the tumor cells were subcutaneously injected into mice, but the tumor cells overexpressed IL-17 in an artificial manner. In this kind of tumor microenvironment, the tumor cells may release much higher levels of IL-17 than are produced in a normal tumor microenvironment. Under these circumstances, IL-17 could immediately participate in the tumor immune response, resulting in acute inflammation. Therefore, in these animal tumor models, IL-17 may inhibit the tumor growth. Most human cancers are associated with chronic inflammation, and the accumulation of IL-17 detected in patients promotes tumor development and is associated with poor survival rates and advanced cancer stages. 9,11,13
IL-17 promotes tumor progression by enhancing inflammation and angiogenesis. 16,27,49 –51 IL-17 overexpression promotes angiogenesis in tumor tissues, and angiogenesis is inhibited in IL-17 knockout mice. 27,50 Tumor angiogenesis contributes to tumor progression, including tumor growth and metastasis. 52 IL-17 upregulates the production of angiogenic factors, such as VEGF, 16 and metastatic factors, such as MMP2 and MMP9. 53 MMP overexpression is an important factor that contributes to tumor invasion and metastasis. 54 MMP2 and MMP9 have been shown to be overexpressed in tumor tissues and associated with tumor invasion and metastasis. 37,55 Inhibiting IL-17 in tumor tissues suppressed microvessel density and decreased the expression of VEGF and MMP9. 44 Our results indicate that inhibiting IL-17/IL-17RA significantly suppressed the expression of VEGF and delayed tumor development. Moreover, inhibiting IL-17/IL-17RA expression decreased the expression of MMP2 and MMP9 and the rate of metastasis. These data suggest that IL-17 might act by upregulating VEGF, MMP2, and MMP9 to promote angiogenesis and metastasis.
IFNγ is a cytokine that plays an important role in preventing tumor development and promoting both innate and adaptive immune responses. 56 Tumor-infiltrating CD4+ and CD8+ T cells mediate immune surveillance and were found to be increased in IL-17 knockout and IL-17 receptor knockout mice, in which more IFNγ was produced. 26,36,57 Studies have also shown that the number of tumor-infiltrating IFNγ+CD4+ cells is increased in IL-17 knockout mice. 26 These increases in T cells and cytokines could suppress tumor-promoting inflammation and tumor progression. Monocyte-produced IL-17 could inhibit cytotoxic T cell functions, 58 and cytotoxic CD8 T cells were activated and Th1 cells increased when IL-17 was inhibited in tumor tissues. 44 The results of these studies indicate that IL-17 suppresses tumor-specific responses by CTLs in the tumor microenvironment, supporting our findings. Our data show that the level of IFNγ was higher in IL-17/IL-17RA-deficient mice. Therefore, the inhibition of IL-17/IL-17RA resulted in the activation of CTL functions and the consequential inhibition of tumor progression.
Many studies have described the roles played by immune suppression cells, such as Tregs and myeloid-derived suppressor cells (MDSCs), in tumor immune responses. 57,59 MDSCs contribute to the negative regulation of immune responses, induce T-cell apoptosis, and inhibit IFNγ, perforin, and granzyme B expression. 57,60 MDSCs are also involved in promoting tumor angiogenesis and metastasis. 60,61 Conversely, Tregs play critical roles in mediating antitumor immune responses and have been found to accumulate in the tumor microenvironment. 17,59,62 Additionally, an increase in Tregs was associated with TNM stage. 17 Depleting tumor-specific Tregs at the tumor site could inhibit tumor development. 63,64 Regarding the relationship between Tregs and IL-17, some studies have indicated that IL-17 could recruit Treg cells to tumor sites by promoting CCL17 and CCL22. 65 CCL22 secretion by tumor cells was positively correlated with Treg infiltration in the tumor microenvironment, and both Tregs and CCL22 were correlated with a poor prognosis in cancer patients. 66 In fact, similar to the recent immunotherapy targeting checkpoints, immunotherapy for suppressing chronic inflammation within the tumor microenvironment has been intensively applied for the treatment of cancers. 67 –69 IL-17 enhances the suppressor functions of Tregs by increasing the expression levels of CD39 and CD73, 65 and inhibiting IL-17 at the tumor site decreased tumor infiltration by Tregs. 44 Both IL-17 and Tregs play important roles in tumor immunity, but the relationship between these two factors needs to be further explored. Indeed, our results showed that blocking IL-17 signaling decreased the numbers of Tregs and some MDSCs (data not shown) and subsequently resulted in the suppression of tumor development. Therefore, inhibiting IL-17/IL-17RA improved antitumor immunity by suppressing Treg cells.
It appeared that the therapeutic efficacy using a single shot of Ad-shIL-17RA fell short, although it did demonstrate alternative therapeutic potential. In fact, the heterologous prime-boost vaccination strategy appears to be more effective than the conventional single-stage vaccination strategy. 70 –72 Recently, several encouraging results on cancer immunotherapy using heterologous prime-boost have been reported. 71,72 For example, Xiang et al. 71 demonstrated that subsequent treatment of DNA and adenoviral vaccines cured mice with metastatic colorectal cancer and induced high-avidity effector CD8+ T cells. Importantly, the first trial using prime-boost vaccination targeting prostatic acid phosphatase showed long-lasting IFNγ- and granzyme B–secreting T cells detected in patients with metastatic castration-resistant prostate cancer, 72 indicating that prime-boost is safe and can augment and diversify the type of immunity elicited with antitumor vaccination. Therefore, the prime-boost strategy consisting of IL-17RA siRNA-expressing DNA and adenoviral vaccines to enhance antitumor efficacy could be further evaluated and investigated to determine whether such heterologous prime-boost vaccination can induce superior antitumor immunity and prevent disease progression.
In conclusion, we found that inducing a blockade against IL-17/IL-17RA suppressed tumor growth and metastasis by reducing angiogenesis and enhanced T-cell–mediated antitumor responses by suppressing Treg cells, resulting in CTL activation. Additionally, we locally delivered shIL-17RA using an adenoviral targeting strategy, and this successfully reduced tumor development, indicating that this may represent a new platform for tumor therapy.
Footnotes
Acknowledgments
The authors gratefully acknowledge the instrument support they received from the Laboratory Animal Center and Core Instrument Center of Chang Gung University, the Microscopy Core Laboratory of Chang Gung Memorial Hospital. We also appreciate our crucial discussions with Dr. Chia-Ning Shen from Academia Sinica, and the generous technical assistance provided by Dr. Fang Pei Chang from Dr. Chia-Ning Shen's group in Academia Sinica and Dr. Yi-Chen Lee from Dr. Ann-Joy Cheng's group in Chang Gung University during the preparation of adenoviral vectors and tumor invasion assays, respectively. This work was supported by the Chang Gung Memorial Hospital [grants number CMRPD3B0053, CORPD1F0031-3, BMRP440], the Ministry of Science and Technology (MOST) [grants number 106-2221-E-131-023-MY3, 107-3017-F-182-001] and the research center for emerging viral infections from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan.
Author Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.